Abstract
Hydroxysteroid dehydrogenases (HSDHs) are from two superfamilies of short-chain dehydrogenase (SDR) and aldo–keto reductase (AKR). The HSDHs were summarized and classified according to their structural and functional differences. A typical pair of enzymes, 7α–hydroxysteroid dehydrogenase (7α–HSDH) and 7β–hydroxysteroid dehydrogenase (7β–HSDH), have been reported before. Molecular docking of 7-keto–lithocholic acid(7–KLA) to the binary of 7β–HSDH and nicotinamide adenine dinucleotide phosphate (NADP+) was realized via YASARA, and a possible binding model of 7β–HSDH and 7–KLA was obtained. The α side of 7–KLA towards NADP+ in 7β–HSDH, while the β side of 7–KLA towards nicotinamide adenine dinucleotide (NAD+) in 7α–HSDH, made the orientations of C7–OH different in products. The interaction between Ser193 and pyrophosphate of NAD(P)+ [Ser193–OG⋯3.11Å⋯O1N–PN] caused the upturning of PN–phosphate group, which formed a barrier with the side chain of His95 to make 7–KLA only able to bind to 7β–HSDH with α side towards nicotinamide of NADP+. A possible interaction of Tyr253 and C24 of 7–KLA may contribute to the formation of substrate binding orientation in 7β–HSDH. The results of sequence alignment showed the conservation of His95, Ser193, and Tyr253 in 7β–HSDHs, exhibiting a significant difference to 7α–HSDHs. The molecular docking of other two enzymes, 17β–HSDH from the SDR superfamily and 3(17)α–HSDH from the AKR superfamily, has furtherly verified that the stereospecificity of HSDHs was related to the substrate binding orientation.
1. Introduction
Hydroxysteroid dehydrogenases (HSDHs) are a type of nicotinamide adenine dinucleotide (phosphate) NAD(H)/NADP(H)-dependent oxidoreductase that can specifically catalyze the reduction of carbonyl to hydroxy of steroid-skeleton structures such as steroid hormones and bile acids, in addition to a reversible reaction. All HSDHs can be divided into two different protein superfamilies—the short-chain dehydrogenase/reductase (SDR) superfamily and aldo–keto reductase (AKR) superfamily [1,2]. A variety of HSDHs from different species have been reported. Most of them belong to the SDR superfamily, such as 7α–HSDH (EC 1.1.1.159), 7β–HSDH (EC 1.1.1.201), and 11β–HSDH (EC 1.1.1.146), which have been identified in Escherichia coli, Collinsella aerofaciens, and Homo sapiens, respectively [3,4,5], usually with a length of 250–350 amino acids [6]. They all have a core composed of seven parallel β-sheets and several α-helices wraps around the core (Figure 1), which form two typical domains named Rossmann fold (each composed of two βαβ units) for coenzyme binding [6,7]. Aldo–keto reductases (AKRs) catalyze the carbonyl reduction reaction with a wide range of substrate selectivity, including sugar aldehydes, keto-steroids, keto-prostaglandins, and retinals [8]. These enzymes are usually with a length of 320 residues on average, forming an (α/β)8-barrel fold [8,9]. According to the differences of sources, HSDHs such as 3α–HSDH (EC 1.1.1.145), 17α–HSDH (EC 1.1.1.148) and 17β–HSDH (EC 1.1.1.51, EC 1.1.1.357, EC 1.1.1.270, etc.) usually belong to two superfamilies. For example, 3α–HSDHs from Comamonas testosteron [10] and Pseudomonas sp. [11] belong to the SDR superfamily, while from Rattus norvegicus [12] and Homo sapiens [13] belong to the AKR superfamily (Figure 2).
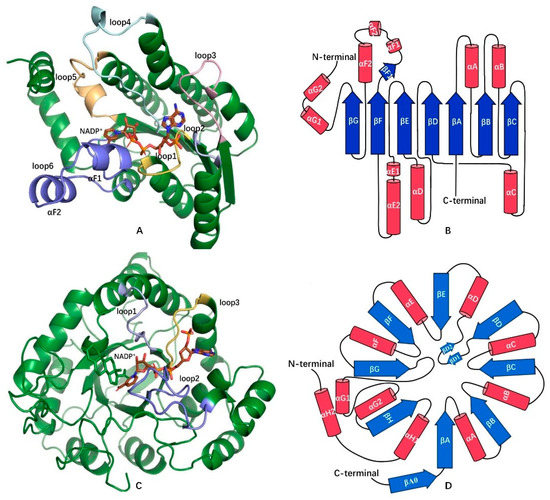
Figure 1.
Comparison of structures (A,C) and folding topologies (B,D) of hydroxysteroid dehydrogenases (HSDHs) from short-chain dehydrogenase/reductase (SDR) superfamily (7β–HSDH(PDB:5GT9)) and aldo–keto reductase (AKR) superfamily (3α–HSDH(PDB:4L1X)). A–helixes are represented as cylinders (in red) and β-sheets are represented as arrows (in blue). Several important loops related to coenzyme or substrate binding between α-helixes and β–sheets are labeled, which are loop1–6 between β(A–F)–α(A–F) in 7β–HSDH and loop1 (βA–αA), loop2 (βG–αG1) and loop3 (βH–αH1) in 3α–HSDH. Figures A and C were created by Pymol [14].
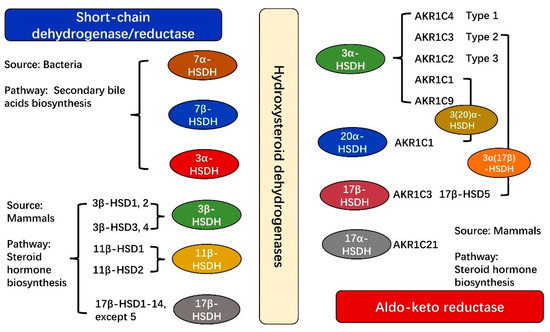
Figure 2.
Classification of common HSDHs from SDR and AKR superfamilies according to their source differences. HSDHs related to secondary bile acids biosynthesis are mainly from bacteria, existing in the SDR superfamily, whereas HSDHs related to steroid hormone biosynthesis are mainly from mammals, existing in both SDR and AKR superfamilies. More detailed information is shown in Table S3.
This article reviews the HSDHs from SDR and AKR superfamilies that were identified in mammals and bacteria and then classifies the HSDHs according to their functions in steroid hormone metabolism or bile acid biosynthesis. The differences of NAD(P)(H)-binding models to enzymes and enzymatic reaction characteristics between SDR and AKR superfamilies were also summarized. A semi-flexible molecular docking was used for further exploration of differences of substrate binding model to enzyme–coenzyme complex between SDR and AKR superfamilies, in addition to the reasons of stereospecificity of substrate or product in HSDHs.
2. Classification and Functions of HSDHs
HSDHs can be classified according to their structural or functional differences. From a structural perspective, HSDHs belong to two protein superfamilies—short-chain dehydrogenase/reductase (SDR) and aldo–keto reductase (AKR) superfamily [1,2]. Most SDRs are active as dimers or tetramers. They usually have a core composed of seven parallel β-sheets, with several α–helixes wrapped around, which form two typical domains named Rossmann fold (each composed of two βαβ units) for coenzyme binding [7]. In contrast, most AKRs are soluble, monomeric proteins and have an architecture of (α/β)8-barrel fold called TIM-barrel, which was named after a prototypical member of AKRs, triosephosphate isomerase (TIM) [15,16].
All HSDHs can specifically catalyze the redox of steroid-skeleton substrates, such as bile acids and a series of steroid hormones [17,18]. According to the differences of reacting positions and hydroxyl orientations of substrates or products, HSDHs can be divided into 11 types at least, which are 3α–HSDH (EC 1.1.1.50, EC 1.1.1.51, EC 1.1.1.53, etc.) and 3β–HSDH (EC1.1.1.145, EC 1.1.1.270, EC 1.1.1.391, etc.), 7α–HSDH (EC 1.1.1.159) and 7β–HSDH (EC 1.1.1.201), 11β–HSDH (EC 1.1.1.146), 12α–HSDH (EC 1.1.1.176) and 12β–HSDH (EC 1.1.1.238), 17α–HSDH (EC 1.1.1.148) and 17β–HSDH (EC 1.1.1.51, EC 1.1.1.357, EC 1.1.1.270, etc.) and 20α–HSDH (EC 1.1.1.149) and 20β–HSDH (EC 1.1.1.53), catalyzing the reduction of carbonyl on C3, C7, C11, C12, C17, and C20 of steroid-backbone substrates (producing hydroxyl with an α or β orientation) and their reverse reactions, respectively. A part of HSDHs only exists in SDR or AKR superfamilies (Table S1 and Table S2). All 7α–HSDHs, 7β–HSDHs, and 11β–HSDHs were reported to exist in the SDR superfamily, while most 17α–HSDHs and 20α–HSDHs belong to AKRs. However, enzymes such as 3α/β–HSDHs and 17β–HSDHs could be found in both superfamilies. Numerous 3α–HSDHs of AKRs have functions of 17α–HSDH, 17β–HSDH or 20α–HSDH [12,19]. In particular, 3α–HSDHs (AKR1C31, AKR1C32, AKR1C33) from Oryctolagus cuniculus (rabbit) have other two functions of 17β–HSDH and 20α–HSDH [20].
2.1. Characteristics and Functions of HSDHs from SDRs
Consisted of more than 163,120 members [21], SDR enzymes are found in different classes of oxidoreductases, isomerases, and lyases [21,22], with multiple functions such as carbonyl–alcohol oxidoreduction, steroid isomerase, enoyl–CoA reduction, decarboxylase, dehalogenase, dehydratase, C=N reduction and epimerase [22,23,24], covering broad substrates such as sugars, steroids, retinoids, lipids, polyols, prostaglandins and androstenedione [24,25]. SDRs could be further classified into six types at least according to their sequence differences, i.e., “Classical,” “Extended,” “Atypical,” “Intermediate,” “Divergent,” “Complex” and some members “Unassigned” (Table 1) [21,22,26]. Sequence alignment was used to analyze structural differences among SDR–HSDHs (HSDHs from SDR superfamily), demonstrating that most SDR–HSDHs are of the “Classical” type except 3β–HSDH, which is more similar to the “Intermediate” type (Table 1, Figure S1). Even though SDR–HSDHs share a low sequence identity of 23–53% (https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Get&RID=3AGHXAGR114 (accessed on 29 January 2021)), all SDR–HSDHs are conservative in structures which composed of a core of Rossmann fold, with several α–helixes wrapped around it (Figure 1). SDR–HSDHs are mainly able to catalyze the dehydrogenation/hydrogenation of substrates with steroid-skeleton, participating in the pathways such as steroid hormone biosynthesis, steroid degradation, and secondary bile acid biosynthesis in organisms (Table S3) [27].

Table 1.
Features of six subfamilies of the SDR superfamily.
2.1.1. HSDHs in Primary or Secondary Bile Acid Biosynthesis
Bile acids serve significant roles in cholesterol metabolism and the digestion of lipids for mammals [28]. The composition of bile acids pools is different in different species. For example, the human bile acids pool is usually composed of hydrophobic bile acids cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), and lithocholic acid (LCA), whereas the mice bile acids pool mainly consists of hydrophilic bile acids, muricholic acids(β–MCA) and ursodeoxycholic acid (UDCA) [17,29,30]. All these bile acids can be divided into two forms-primary bile acids (CA and CDCA) and secondary bile acids (DCA, LCA, UDCA, etc.), whose production is related to the activities of the liver and intestinal microbiota, respectively [31].
A number of SDR–HSDHs related to secondary bile acid synthesis were found in intestinal bacteria. The metabolic pathway of secondary bile acids was shown in Figure 3. 3α–HSDHs found in species Pseudomonas sp. [11], Comamonas testosterone [32], Corynebacterium glutamicum, etc. [33] are able to catalyze the hydrogenation/dehydrogenation of carbonyl and hydroxyl on C3 position of bile acids, participating in the conversation of deoxycholate and lithocholate to isodeoxycholate and isolithocholate (https://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=PWY-7755 (accessed on 29 January 2021)). A pair of epimerases, 7α–HSDH and 7β–HSDH, could specifically catalyze the reduction of C7=O or the oxidation of C7–OH on steroid-skeleton [34,35], with a substrate or product of CDCA(7α–OH) and UDCA(7β–OH). These two epimerases work together for the conversion of CDCA to UDCA in industry, with an intermediate 7-keto–lithocholic acid (7–KLA). UDCA has significant pharmaceutical applications in the treatment of gallstones, biliary cirrhosis, and even liver cancers [36].
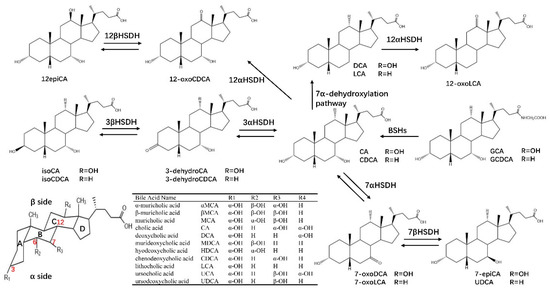
Figure 3.
HSDHs-related bile acids synthesis and metabolism. Primary bile acids (cholic acid (CA) and chenodeoxycholic acid (CDCA)) in the liver are usually conjugated with glycine (G), which can be deconjugated by bile salt hydrolases (BSH). Primary bile acids can be biotransformed to secondary bile acids (deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA), etc.) through the 7α–dehydroxylation pathway and the oxidation and epimerization of 3α–, 7α– and 12α–HSDHs.
2.1.2. HSDHs in Steroid Hormone Biosynthesis
3β–HSDHs (EC 1.1.1.145) have been identified in Homo sapiens (type 1–2), Mus musculus (type 1–6), Rattus norvegicus (type 1–4), Mycobacterium tuberculosis, etc., usually with activities of dehydrogenase and isomerase (Δ5–Δ4) [37]. Both Human type 1 3β–HSD1/isomerase and type 2 3β–HSD2/isomerase are NAD(H)-dependent enzymes, catalyzing the conversion of 3β-hydroxy-5-ene-steroids to 3-oxo-4-ene-steroids with two sequential enzymatic reactions. Firstly, 3β–HSDH catalyzes the dehydrogenation of 3β-hydroxysteroid to a 3-oxo intermediate, and then the Δ5-form intermediate was isomerized to Δ4-form via their isomerase activity (Figure 4). This catalytic function of 3β–HSDH is usually related to the conversation of dehydroepiandrosterone (DHEA) to androstenedione, 17α–hydroxypregnenolone to 17α–hydroxyprogesterone and pregnenolone to progesterone [38,39,40]. In humans, 3β–HSD1 (type 1) and 3β–HSD2 (type 2) are encoded by two distinct genes which are expressed in a tissue-specific pattern [41]. 3β–HSD1 is expressed in peripheral tissues such as breast, placenta, prostate, and endometrium, with roles of production of some downstream steroids such as testosterone, dihydrotestosterone (DHT), and estrogens [42]. The testosterone and estradiol will promote the growth of breast tumors [43] and prostate tumors [44]; therefore, human 3β–HSD1 is a potential target for the inhibition of estradiol production in breast tumors and testosterone production in prostate tumors. 3β–HSD2 is expressed in adrenals, ovaries, and testis, which are related to the synthesis of steroid precursors required for the production of cortisol, aldosterone, and the female and male sex hormones [17,29,31].
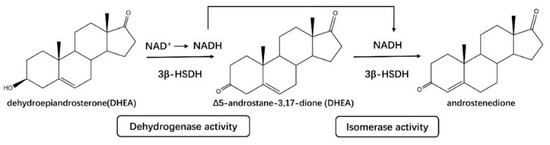
Figure 4.
3β–HSDHs (3β–HSD1 and 3β–HSD2) from the SDR superfamily have both activities of dehydrogenase and isomerase, catalyzing the oxidation of 3β–hydroxy and the isomerization of Δ5 to Δ4-form.
11β–HSDHs (EC 1.1.1.146) in humans can be generally divided into two types, 11β–HSD1 and 11β–HSD2, working together on glucocorticoid metabolism to regulate the availability of active glucocorticoids [45]. 11β–HSD1 is NADP(H)-dependent enzyme, facilitating the conversion of inactive glucocorticoids, cortisone in human and 11-deoxycorticosterone in rodents, to their active 11-keto metabolites, cortisol, and corticosterone, respectively [46] (Figure 5). 11β–HSD1 is mainly expressed in liver, adipose tissues, brain and skeletal muscles [47], whose function associated with insulin resistance, dyslipidemia, obesity, hypertension, and other features of mineralocorticoid excess; hence, the inhibition of 11β–HSD1 is a potential method for the treatment of glucocorticoid (active)-related disorders [48,49]. 11β–HSD2 was found primarily in mineralocorticoid target tissues, such as kidneys, sweat glands, salivary glands, and colonic mucosa [50], catalyzing the conversion of glucocorticoids to inactive 11-ketosteroids in a NAD(H)-dependent activity, in contrast to 11β–HSD1 [51]. Another type of 11β–HSDH, 11β–HSD3, has been identified in zebrafish, fathead minnow, and some paralogs amphioxus [52].
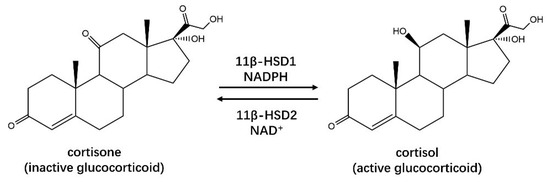
Figure 5.
Human 11β–HSD1 from the SDR superfamily is able to catalyze the activation of glucocorticoids, while 11β–HSD2 catalyzes a reverse reaction.
17β–HSDHs are encoded by non-homologous genes with different subcellular localizations, coenzymes, and substrate preferences [53]. Up to now, over 14 different subtypes of 17β–HSDHs (17β–HSD1–14) have been identified, most of them belong to the SDR superfamily except 17β–HSD5, which is a member of the AKR superfamily [54,55]. All 17β–HSDHs are able to catalyze the reduced or oxidized reactions at position C17 of C18– or C19–steroids, involved in the activation and inactivation of steroid hormones (Figure 6) [54]. 17β–HSD1 could catalyze the activation of estrone to estradiol in humans. However, the production of estradiol is linked to the development of diseases such as breast cancer, ovarian tumor, endometriosis, endometrial hyperplasia, and uterine leiomyoma [56]. It was demonstrated that estradiol can be converted to genotoxic metabolites in breast tissue. Consequently, the inhibition of 17β–HSD1 is one of the treatments for breast cancer in clinical [57]. Meanwhile, the decreasing of estradiol will lead to osteoporosis. 17β–HSD2 have opposing actions to 17β–HSD1, catalyzing the inactivation of estradiol to estrone, which means the inhibition of 17β–HSD2 is one of the therapies for osteoporosis [58]. Otherwise, 17β–HSD1 could catalyze the conversion of dehydroepiandrosterone (DHEA) to Δ5–androstane–3β, 17β–diol while 17β–HSD2 catalyze a reverse reaction. Δ5–androstane-3β, 17β–diol can be converted to estradiol with functions of 3β–HSDH and aromatase [59]. 17β–HSD3 exists predominantly in testis and catalyzes the conversion of Δ4–androsenedione to testosterone. It was confirmed that its mRNA was overexpressed in prostate cancer tissue. Testosterone is responsible for cell proliferation in androgen-dependent diseases, and the inhibition of 17β–HSD3 is effective for the treatment of diseases such as prostate cancer [60]. The functions of other subtypes of 17β–HSDH were described in Table S3.
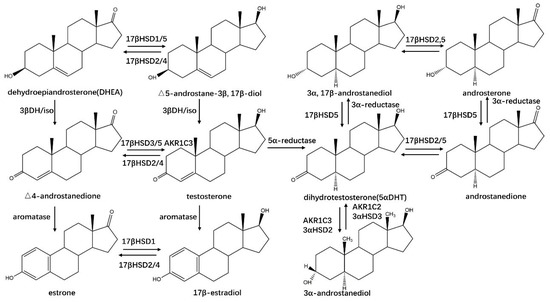
Figure 6.
HSDHs-related steroid hormones synthesis and metabolism. “3βDH/iso” signalizes the dehydrogenase and isomerase activities of 3β–HSDH.
2.2. Characteristics and Functions of HSDHs from AKRs
Most aldo–keto reductases (AKRs) are soluble monomeric NAD(P)(H) oxidoreductases that catalyze a NAD(P)H-dependent reduction of aldehydes and ketones, yielding primary and secondary alcohols [61]. Over 190 members belonging to AKRs have been reported, with a high sequence identity of 40–60% [2,61,62]. The (α/β)8–TIM barrel provides a common architecture for a NAD(P)(H)-dependent catalytic activity [63]. AKRs have broad substrate specificity, which was determined by the variation of loops on the C-terminal. Their functions were related to the metabolism of sugar, lipid aldehydes, steroids, and prostaglandins, etc. [61]. All AKRs can be further classified into 16 families (AKR1–AKR16) [61,62] and HSDHs reported from AKRs (AKR–HSDHs) belong to AKR1 [63]. Up to now, four isoforms of human 3α–HSDHs and one isoform of rat 3α–HSDH belonging to AKRs have been reported, which are AKR1C1 (human 3α(20α)–HSDH), AKR1C2 (human type 3), AKR1C3 (human type 2), AKR1C4 (human type 1) and AKR1C9 (rat liver), respectively [64,65,66,67]. The human type 2 3α–HSDH (AKR1C3) is also known as type 5 17β–HSDH, while type 3 (AKR1C2) is also known as bile acid-binding protein [64,65,66].
Human aldo–keto reductase AKR1C3 (type 2 3α–HSDH/type 5 17β–HSDH) is also known as prostaglandin F synthase [68] and possesses activities in the reduction of Δ4–androstene-3,17-dione to testosterone, 5α–dihydrotestosterone(5α–DHT) to 3α– and 3β–androstanediol, and estrone to 17β–estradiol, respectively (Figure 6) [69], which is expressed in sex hormone-dependent tissues (testis, breast, endometrium, and prostate) and sex hormone-independent tissues (kidney and urothelium) [70], whose functions are associated with breast cancer, endometrial cancer and the development of endometriosis and dysmenorrhea [71]. AKR1C2 is another AKR1C isozyme that is highly expressed in the prostate and catalyzes the oxidation of 3α–androstanediol to the active hormone 5α–DHT, opposite to AKR1C3 [66,72].
AKR1C4 can work in concert with steroid 5β–reductase to produce the 5β,3α–tetrahydrocholestanes in the liver, play a pivotal role in the bile acid biosynthesis, and steroid hormone metabolism [66]. AKR1C1 is usually expressed in lung, liver, testis, mammary gland, endometrium, brain, kidney, adipose cells, skin, osteoblasts, and optic nerve head astrocytes [73,74], exhibiting both 3α/β– and 20α–HSD activities and mainly reducing progesterone and 5α–pregnan–3α–ol–20-one to their corresponding inactive metabolite 20a-hydroxyprogesterone with a function of 20-ketosteroid reductase [73,74]. Progesterone plays a crucial role in preventing the onset of premature birth, blunting the estrogenic effects in endometriosis and endometrial cancer [75,76]. Thus, the inhibition of AKR1C1 is a potential agent for the treatment of these disorders [77,78].
3. Coenzyme Binding Modes of HSDHs
Nearly 150,000 different sequences were annotated as or predicted to be NAD(H)/NADP(H)-dependent oxidoreductases/reductases [79]. The binding model of NAD(H)/NADP(H) in the enzyme is significant for the proton transfer. Several structures were reported for coenzyme binding, which are Rossmann fold, (α/β)8–TIM–barrel, dihydroquinoate synthase-like, and FAD/NAD-binding folds [79,80,81].
3.1. Coenzyme Binding Modes of the HSDHs from SDRs
The sequence alignment of HSDHs in the SDR superfamily shows a low sequence identity of 23–53% (https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Get&RID=3AGHXAGR114 (accessed on 29 January 2021)). However, all SDR–HSDHs share similar and conservative motifs for NAD(P)(H) binding. The basic structure of SDRs is composed of two Rossman folds, which is stable and no loss of function occurs even with mutations at some sites of the folding and that is why SDRs retain similar catalytic functions even with such low sequence identities [82,83,84].
The binding sites of the adenosine ring of coenzyme to enzyme are on the loop1, loop2, and loop3 (Figure 1), close to the C-terminal part of SDRs, while the nicotinamide-binding region is on loop 4 and loop 6, close to the N-terminal part. A conserved glycine-rich pattern GxxxGxG between the first β–strand and α–helix is usually interacted with pyrophosphate [84,85,86]. The individual subfamilies of the SDR superfamily differ regarding the spacing of the three glycine residues [87]. For example, it was demonstrated that 3β–HSDHs have a motif of X1–Gly–X2–X3–Gly–X4–X5–Gly (Figure S1), which is similar to the “Intermediate” type [88], while most SDR–HSDHs have a motif of X1–Gly–X2–X3–X4–Gly–X5–Gly and belong to the “Classical” type. In the meantime, the sequence alignment of SDR–HSDHs from different sources showed that the residue X1 in 7β–HSDHs is L/C, which is obviously different from the residue T/S in other SDR–HSDHs (Figure S1). The features among different subfamilies of SDRs were described in Table 1. The motif YxxxK on αE-helix has two functions of coenzyme binding and catalysis [89], showing differences among different types of SDRs as well, which will be described in detail below.
There is no doubt that NAD(H)/NADP(H) can act as a proton donor or acceptor during redox reactions [90] and can participate in the remodeling of the substrate-binding pocket. The common belief is that the broad substrate specificity of HSDHs is related to the substrate-binding loop in previous studies, but are the interactions between NAD(P)(H) and the enzyme also responsible for the substrate specificity, especially the stereospecificity? The distance between C6 of adenine and C2 of nicotinamide in a syn conformation is frequently used to express the degree of extension of coenzyme molecule [91], which is 13.4 Å, 13.7 Å, 14.1 Å, and 14.3Å in 7α–HSDH (PDB: 1FMC) [92], 7β–HSDH (PDB: 5GT9) [93], 11β–HSDH (PDB: 3G49) and 17β–HSDH (PDB: 3QWH), respectively, showing little difference (Figure 7). The dihedral angles of PA–O3–PN–O5D (pyrophosphate group), C4B–C2B–C1B–N9A (adenosine ring), and C4D–C2D–C2N–C5N (nicotinamide ring) were used for a further study on conformation change of NAD(P)(H) (Table 2). In the pyrophosphate binding region, Ile23 and Thr194 of 7α–HSDH, Val20 and Thr191 of 7β–HSDH, Ile21 and Thr195 of 11β–HSDH, in addition to Ile30 and Thr202 of 3α(17β)–HSDH, are equivalent in function. However, the dihedral angle PA–O3–PN–O5D (–80.6°) in the 7β–HSDH binary is apparently different compared with others, which may lead to the difference of substrate binding pocket. Another crystal structure of 7α–HSDH from Clostridium absonum was obtained as a tetramer in the study by Lou et al., which exhibited a coenzyme-dependent of NADP(H) [94]. The dihedral angle PA–O3–PN–O5D in the pyrophosphate region is −143.4° (Figure 8), which is similar to 7α–HSDH–NAD+ complex (–147.8°) from E. coli, although the coenzymes of these two crystals are completely different. Both of these two dihedral angles PA–O3–PN–O5D are different from 7β–HSDH–NADP+ complex (–80.6°), and this is probably able to prove that the stereospecificity of substrate/product is related to the coenzyme conformation.
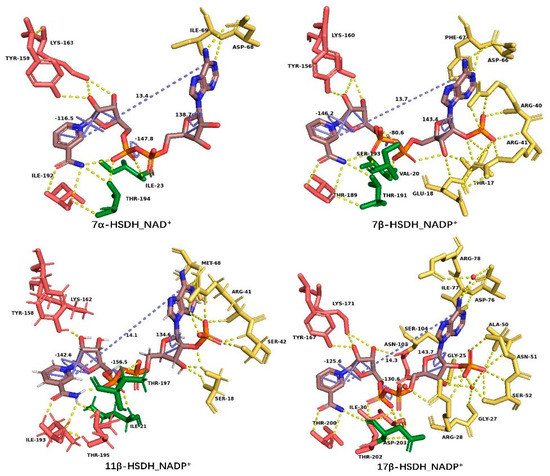
Figure 7.
Residues that interacted with NAD(P)(H) in the SDR superfamily, in which the residues in yellow, green, and red color interact with the adenosine region, pyrophosphate region, and nicotinamide region of coenzyme, respectively. The distance between C6 of adenine and C2 of nicotinamide and the dihedral angles of PA–O3–PN–O5D (pyrophosphate group), C4B–C2B–C1B–N9A (adenosine ring), and C4D–C2D–C2N–C5N (nicotinamide ring) in blue color are also shown. This figure was created by Pymol [14].
Table 2.
Residues that interacted with NAD(P)(H) in the SDR superfamily and the NAD(P)(H)-conformation differences among them.
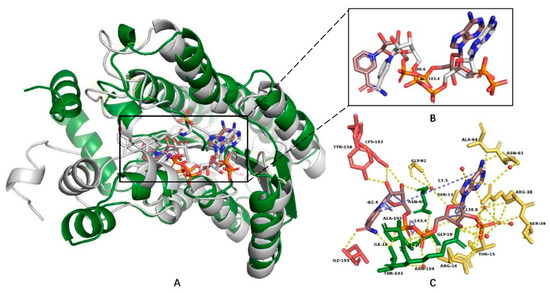
Figure 8.
The structural alignment (A) of 7α–HSDH (PDB: 5EPO) and 7β–HSDH (PDB: 5GT9) and the NADP(H) conformation differences between them (dihedral angles of PA–O3–PN–O5D of pyrophosphate group) (B). Figure (C) exhibits the NADP(H)-interacted residues in the 7α–HSDH complex. This figure was created by Pymol [14].
The pyrophosphate-interacted residues, Ser193 in 7β–HSDH, Thr197 in 11β–HSDH and Ser11 in 3α(17β)–HSDH, are quite special compared with 7α–HSDH ternary. The interaction [Thr194–OG1⋯2.79 Å⋯O1N–PN] in 7α–HSDH ternary was replaced by [Ser193–OG⋯3.11Å⋯O1N–PN] in 7β–HSDH, leading to a rotation of the phosphate group PN to the outside, which made the coenzyme conformation different (Figure 7).
In the adenosine ring binding region, the existence of O2′ ribose phosphate group in NADP(H) makes the interactions between the adenosine ribose region and β–HSDHs complicated. A hydrogen-bond network between coenzyme, residues, and soluble molecules maintains the adenosine ribose conformation of NADP+ in binary or ternary complexes.
These residues in 7β–HSDH are Arg40–Arg41, in 11β–HSDH are Arg41–Ser42, and in 3α(17β)–HSDH are Ala50–Asn51–Ser52 (Table 2). In the nicotinamide ring binding region, the interactions between nicotinamide ribose and residues seem to be conserved. Motifs Tyr–X–X–X–Lys [89,90,91,92,93,94] and Thr/Ile–X–Thr have conserved interactions with NAD(P)(H) (Figure 7 and Table 2). The hydrogen bonds existed among PN–phosphate group of pyrophosphate region, nicotinamide, and Thr contribute to maintaining the orientation of pro-S side of nicotinamide towards catalytic sites, for example, the interactions [Thr189–N⋯2.9 Å⋯O7N–Nicotinamide], [Thr189–O⋯3.2 Å⋯N7N–Nicotinamide], [Thr189–O⋯2.9 Å⋯N–Thr191], [Thr191–OG1⋯3.0 Å⋯N7N–Nicotinamide], [Thr191–OG1⋯2.7 Å⋯O2N–PN], and [Nicotinamide–O7N⋯2.9 Å⋯O2N–PN] in the holo-form of 7β–HSDH, which making the happen of reaction possible.
3.2. NAD(H)/NADP(H) Binding Modes of the HSDHs from AKRs
HSDHs from AKRs (AKR–HSDHs) are not only conserved in structure, but also share a high sequence identity of 64–93% (https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Get&RID=3AH34HGD114 (accessed on 29 January 2021)). The basic structure of AKRs is composed of eight β-sheets. At least one α–helix exists between every two β-sheets, which surround the barrel around outside [95,96]. This structure was named after TIM-barrel fold, consisted of an eightfold repeat of (α/β) units ((α/β)8), was first seen in triosephosphate isomerase (TIM) [97,98].
Loops between β-sheets and α-helixes are used for the binding of the coenzyme and substrate. In contrast with SDR–HSDHs, the residues that interacted with NADP(H) adenosine are closer to the N-terminal, whereas residues that interacted with NADP(H) nicotinamide are closer to the C-terminal [99]. The NAD(P)H binds AKRs in an extended anti-conformation [61], stretching through the channel formed by Van der Waals contacts [100], which composed of loop 1(βA–αA, residues 22–31), loop 2(βG–αG, residues 216–235), and loop 3(βH-αH, residues 269–274) (Figure 1).
The interactions between AKR–HSDHs and coenzymes in the binary or ternary structures of 3α–HSDH (PDB: 1J96), 3(17)α–HSDH (PDB: 2P5N), 17β–HSDH (PDB: 1ZQ5), and 20α–HSDH (PDB: 1Q5M) were analyzed using Pymol (distributed by Schrödinger, New york, NY, US) (Figure 9) [14], to explore the conformation changes of NADP(H) after binding to the enzyme and whether these changes are conservative in all AKR–HSDHs. The conformation of NADP(H) is highly conserved in AKRs, showing little difference. The distance between C6 of adenine and C2 of nicotinamide is 17.0 Å, which means the occurrence of structural extension of NADPH (15.6 Å) due to the interactions between residues and coenzyme, such as hydrogen bonds, π–π interactions, and hydrophobic interactions [101], and it is definitely different from SDR–HSDHs, which is roughly 14 Å. Other structural changes and interacted residues are described in Table S4, which demonstrated high similarity and conservation in 3α–HSDH, 3(17)α–HSDH, 3α(17β)–HSDH, 17β–HSDH, and 20α–HSDH.
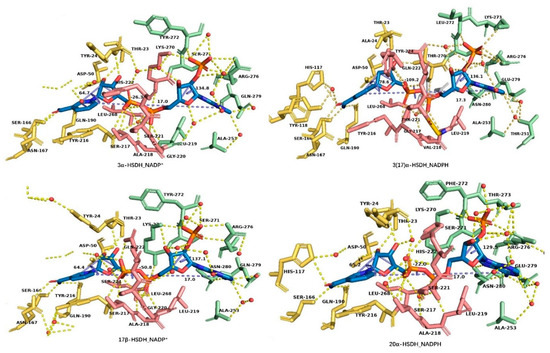
Figure 9.
Residues that interacted with NAD(P)(H) in AKR superfamily, in which the residues in green, red, and yellow color interact with the adenosine region, pyrophosphate region, and nicotinamide region, respectively. The distance between C6 of adenine and C2 of nicotinamide and the dihedral angles of PA–O3–PN–O5D (pyrophosphate group), C4B–C2B–C1B–N9A (adenosine ring), and C4D–C2D–C2N–C5N (nicotinamide ring) in blue color are also shown. This figure was created by Pymol [14].
Taking the 3α–HSDH holo-form (PDB: 1J96), for example, the adenine ribose group in AKR–HSDHs is embedded between αG1-helix and αH-helix, interacted with residues of Ala253 from αG1-helix, Arg276, and Gln279 from αH-helix, while the O2’ ribose phosphate group was contained in the cavity formed by Lys70–Ser271–Tyr272 on the loop 1 and Arg276 on αH-helix. These residues and the water molecules formed a hydrogen bond network with the adenosine ribose group, which helps to fix the adenosine ribose group at the N-terminal. All these interactions are highly conserved in AKR–HSDHs, especially the interactions between Ala253, Arg276, and coenzymes, for example, the interactions [Ala253–N⋯3.1Å⋯H2O⋯3.1Å⋯N1A–adenosine] and [Arg276–NH2⋯3.0Å⋯O2X–Phosphate group] [102,103,104].
The pyrophosphate group crosses through the tunnel composed of loop 1, loop 2, and loop 3, connecting the adenosine ribose at the outside of the barrel and nicotinamide ribose in the center. On the inner wall of the tunnel, the residues Ser217–Ala218–Leu219–Ser221 on loop 2, Leu268, Lys270 on loop 3, and Tyr24 on loop 1 formed a hydrogen bond network with water molecules and pyrophosphate groups and maintained the conformation of this region (Figure 9). Among them, Ser/Thr217–Ala218–Leu219 and Leu268 are highly conserved. The residue Tyr24 has hydrogen-bond linkage to nicotinamide ribose while Leu219 has hydrogen-bond linkage to adenosine ribose, respectively.
4. Catalytic Mechanism of HSDHs
4.1. Catalytic Mechanism of HSDHs from SDRs
It is recognized that the motif Tyr–X–X–X–Lys is associated with the catalytic function of SDRs. This conserved motif attracted much attention as early as 1981 [26,105], and its function was reported in the studies of 3α(20β)–HSDH from Streptomyces hydrogenans [106] and 7α–HSDH from E. coli [92]. However, the conservative motif shows differences in different types of SDRs. The most common motif Tyr–X–X–X–Lys exists in “Classical” SDRs, while the motif in the “Divergent” type is Tyr–x–x–Met–x–x–x–Lys [26], the features of these six types of SDRs were shown in Table 1. Tyr is strictly conserved even in the whole SDR superfamily, while Lys and Ser are only conserved in most SDRs. With the assistance of Lys–NH2 to decreases the pKa of Tyr–OH, the side chain phenolic group of Tyr acts as a general acid/base catalyst. The hydride of the substrate is released to the pro-S side of the nicotinamide group of NAD(P)(H) in an oxidated reaction [107]. Apart from being a proton donor or accepter, NAD(P)(H) plays a vital role in the formation and stability of the substrate-binding pocket. A strict binding order of substrate and coenzyme to drosophilid alcohol dehydrogenase (DADH) was reported [108]. DADH could convert alcohol to aldehyde/ketone. In an oxidated reaction, the coenzyme is bound to a free enzyme to form a binary complex firstly and then the substrate binds to the binary complex and the rate-limiting step is the release of the coenzyme product (NADH) [109,110,111]. This mechanism is similar to the “bi–bi” mechanism of AKRs in which coenzyme binds first and leaves last [16] and probably applies to all SDRs.
The catalytic mechanism (Figure 10) of SDRs was investigated through X-ray crystallographic structure determination of 3α(20β)–HSDH in a study by Ghosh et al. [106]. It was proposed that the catalytic function of 3α(20β)–HSDH was realized by a triad Ser–Tyr–Lys. Tyr–OH acts as a proton donor applying for an electrophilic attack on the 20-keto oxygen in the reduction of 20-keto [106]. Lys–NH3+ is close to Tyr, facilitating the transformation of the proton. Ser participates in catalysis through direct interaction with Tyr–OH or just as part of a proton-relay network [106]. Tanaka et al. proposed a three-step catalytic mechanism for 7α–HSDH and confirmed it through the mutation of Tyr159, Lys163, and Ser146 [92]. Firstly, the Lys163 binds to NAD(H) and lowers the pKa value of Tyr159 and then Tyr159 was deprotonated and interacted with the C7 position of 7-oxoglycochenodeoxycholic acid (7-oxo–GCDCA), the intermediate was stabilized by Ser146 with a hydrogen bond to C7–O. Secondly, the deprotonated Tyr acts as a catalytic base and extracts the hydrogen from C7–OH. NAD+ accepts another hydrogen released from position C7 in the same time [92,112]. In the studies of DADH, the theoretical calculations suggested that the proton was released from a coupled irregular ionization of the active site groups Tyr151 and Lys155 instead of a single residue Tyr151 [91,108].
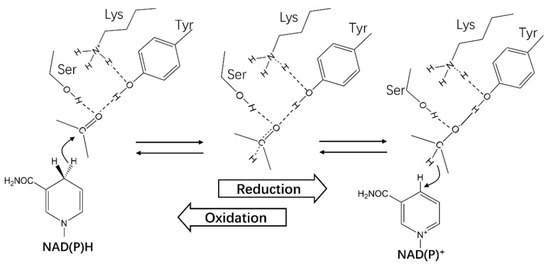
Figure 10.
The catalytic mechanism of HSDHs from the SDR superfamily.
Other amino acid residues participating in the catalytic reactions were proposed. Lou et al. elaborated the mechanism in the study of 7α–HSDH from Clostridium absonum [94], it was found that the serine in Ser–Tyr–Lys triad was replaced by threonine, without a complete activity loss of 7α–HSDH. In the study of 3(20)β–HSDH, it was found that the Asp participated in the catalysis, which formed an active site tetrad Ser–Tyr–Lys–Asp.
4.2. Catalytic Mechanism of HSDHs from AKRs
All HSDHs catalyze the oxidations of hydroxyl to ketone/aldehyde on steroid-skeleton substrates or their reversible reaction, with the assistance of NAD(P)(H) and sharing a similar catalytic mechanism no matter in SDR or in AKR. However, unlike in SDRs, it was a tetrad Asp–Tyr–Lys–His instead of a triad Ser–Tyr–Lys play a catalytic role in AKRs. AKR–HSDHs accept 4-pro-R hydride from A-face of NADPH, while SDR–HSDHs accept 4-pro-S hydride from B-face NAD(P)H [61,113,114]. In the verification experiment of these sites, no activity of the enzyme was detected with a mutation of Y48F, while 1460-fold less active in K77M mutation and 8000-folds increasing of Km in H110N mutation was observed in aldose reductase (EC 1.1.1.21) [115,116,117].
An ordered “bi–bi” kinetic mechanism of AKRs was proposed, in which coenzyme binds first and leaves last [16,118]. Cooper et al. described the mechanism detailed in the structure of rat 3α–HSDH (AKR1C9), which contains nine discrete steps in the kinetic mechanism and is associated with 16 rate constants. Firstly, the coenzyme binds to the enzyme and forms a loose structure of the coenzyme factor complex, and then the complex undergoes two conformational changes to make the binding tighter. It was found that an 86-fold and 110-fold increase of binding affinity for NADPH and NADP+ was realized after successive conformational changes, via the verification of steady-state, transient state kinetics, and kinetic isotope effects [103,117].
A “push–pull” catalytic mechanism was proposed to explanate the reduced and oxidated reactions of AKRs (Figure 11) [119]. In a reduction direction at pH 6.0, the side chain amino group(–NH2) of Lys was protonated, which was then interacted with the phenolic group (–OH) of Tyr. In the meantime, the imidazole group of His attracted hydrogen at 3-N position, which was also interacted with the phenolic group (–OH) of Tyr. These two interactions ultimately reduce the pKa value of Tyr and promote the transfer of protons. Meanwhile, the Asp also participates in the reaction through a salt bridge to Lys. In an oxidation direction, the interaction between –COO– of Asp and –NH2 of Lys promotes the abstraction of Tyr phenolate anion to hydrogen from the steroid alcohol [113,114,115,116,117,118,119,120].
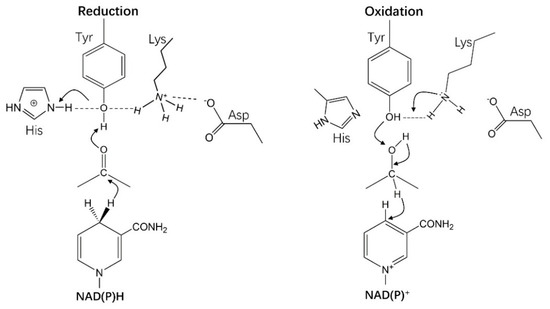
Figure 11.
The catalytic mechanism of HSDHs from the AKR superfamily.
5. Stereospecificity Study of HSDHs through Molecular Docking
In the reduction direction of HSDHs, the carbonyl is reduced to α–OH or β–OH. The stereoisomeric differences at different positions will lead to functional divergences of steroid molecules. For example, both UDCA and CDCA are reduction products of 7-keto-LCA (7–KLA), with an α–OH in CDCA and β–OH in UDCA. However, UDCA exhibits much better performance than CDCA in dissolving cholesterol, with less toxicity. In the process of UDCA synthesis with a substrate of CDCA, 7–KLA (C7=O) usually occurs as an intermediate. The two-step process including the oxidization of 7α–OH firstly and then the reduction of 7-keto group [121,122,123]. Both reactions require NAD(P)(H) as a proton acceptor or donor to participate. Other crystal structures of HSDHs were reported, such as 3α–HSDH, 3β–HSDH, 17α–HSDH, 17β–HSDH, etc. In the stereospecificity study of 7β–HSDH, Savino et al. proposed a possibility that the substrates were bound to 7α–HSDH and 7β–HSDH in opposite orientations, which caused opposite stereoselectivity of products, based on a structural alignment of 7β–HSDH (apo-form) [124] and 7α–HSDH (holo-form) [92].
Other SDRs, such as tropinone reductase (EC 1.1.1.236) TR-I and TR-II, are able to stereospecifically reduce the 3-keto group of tropinone to stereoisomeric forms the enantiomers tropine (α-hydroxy) and pseudotropine (β-hydroxy). It is believed that the reaction stereospecificities of TR-I and TR-II are related to the electrostatic environment of the substrate-binding pocket, which is caused by differently charged residues (positively charged His112 in TR-I while negatively charged Glu156 in TR-II). The substrate-binding directions are determined by charged residues in the pocket, which lead to the stereospecificity of reduction products [125,126]. (R)-hydroxypropyl-coenzyme M dehydrogenase (R-HPCDH) and (S)-hydroxypropyl-coenzyme M dehydrogenase (S-HPCDH) existing in epoxide metabolism of Xanthobacter autotrophicus Py2, usually catalyze the oxidization of (R)- and (S)-enantiomers of 2-hydroxypropyl coenzyme to the achiral product 2-ketopropyl-coenyme M, respectively. A negatively charged sulfonate group, participating in the reaction as a convenient “handle” for substrate binding, was considered related to the substrate binding orientation [127,128]. Other enzymes, such as naphthalene dioxygenase (NDO) from Pseudomonas sp. are able to catalyze cis-dihydroxylation of multiple substrates, whose stereospecificity is related to Phe-352 [129,130,131].
5.1. The Methods of Molecular Docking
In order to explore the stereoisomeric characteristics of HSDHs, the holo-form of 7β–HSDH (PDB: 5GT9), 17β–HSDH (PDB: 3QWH) [132] from the SDR superfamily and 3(17)α–HSDH (PDB: 2P5N) [133] from AKR superfamily were used as receptors for molecular docking. The 7β–HSDH (PDB: 5GT9) structure contains two monomers (A and B), with each one binding one NADP+ molecule. The 17β–HSDH (PDB: 3QWH) structure contains four monomers (A, B, C, and D), and each one is complexed with a NADP+, 3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one and di(hydroxyethyl)ether. The 3(17)α–HSDH (PDB: 2P5N) contains two crystallo-graphically independent monomers, with each one binding an NADPH and (4S)-2-methyl-2,4-pentanediol (Protein Data Bank: https://www.rcsb.org/structure/3QWH (accessed on 29 January 2021) and https://www.rcsb.org/structure/2P5N (accessed on 29 January 2021)).
For simplifying the docking, we used a single subunit as the receptor. In 5GT9, the conformations of NADP+ in subunits A and B are quite different, and subunit B was finally used as receptor after structure alignment or comparison with 7α–HSDH (PDB:1FMC) and 11β–HSDH (PDB:3G49), for they share similar NAD(P)(H) conformation. In 3QWH and 2P5N, the NADP(H) conformations are similar between subunits, and other molecules except NADP(H) were removed. Two ligands, 7–KLA and androstenedione, were downloaded from ZINC (http://zinc.docking.org/substances/ZINC000004428526/ (accessed on 29 January 2021) and http://zinc.docking.org/substances/ZINC000013547326/ (accessed on 29 January 2021)). Both ligands and receptors were processed for energy minimization before docking. A suitable cell around the substrate-binding pocket of receptors was set. The files should be named X_receptor and X_ligand (X represents the same letters or numbers) and the files were placed in the same folder.
The docking was realized via YASARA (distributed by YASARA Biosciences GmbH, Vienna, VIE, AUT. http://www.yasara.org/ (accessed on 29 January 2021)) under pH 7.4 with a forcefield of AMBER03. In order to make the substrate bind the binding pocket better, several flexible residues were set, which in 5GT9 are Val 20, His 95, Tyr156, Lys160, Thr189, Thr191, Ser193, and Tyr253; in 3QWH are Val107, Phe109, Ser153, Asn154, Phe159, Tyr167, Pro197, Gly198, Gly199, Thr200, Phe205, His206, Glu207, Val208, Ser209, Tyr212, Ile213, Pro214, Met227, Ala228, Ala231, Asp266, and Ala269; in 2P5N are Lys31, Tyr55, His117, Tyr118, Phe219, Tyr224, Gly225 Gly226, and Trp227. Setting files X_receptor and X_ligand as the targets through YASARA, the docking was realized by dock_run.mcr, and then dock_play. mcr was used for further analysis.
5.2. Results and Discussion of Molecular Docking
A crystal structure of the ternary complex of 7α–HSDH, NAD+ and 7-oxo–GCDCA was obtained in a study by Tanaka N et al. [92]. In the crystal structure, 7-oxo–GCDCA is bound to 7α–HSDH–NADH complex in the orientation of α side toward catalytic residue Tyr159 and β side towards NADH. A hydrogen-bond network was constructed among the C24=O of 7-oxo–GCDCA, Gly97, the adenosine ribose region, and the pyrophosphate region of NADH as well as water molecules for substrate binding, with a binding direction of substrate C24=O towards C-terminal and C3–OH toward N-terminal of 7α–HSDH. Hydrogen bonds directed or mediated by water molecules exist among C3–OH, Asn151, and Glu253. At the active sites, hydrogen-bond connections of [Ser146–OH⋯2.6 Å⋯O–C7] and [Tyr163–OH⋯2.8 Å⋯O–C7] were generated among C7=O of substrate and side chains of Ser146 and Tyr159. The structure binding model is analogous when in a tetramer crystal of 7α–HSDH from Clostridium absonum, even though with a coenzyme of NADP+ and a substrate of taurochenodeoxycholic acid (TCDCA); even the Ser of Tyr–Lys–Ser was changed by Thr [94].
The semi-flexible molecular docking was realized using the holo-form structure of 7β–HSDH–NADP+ as a receptor, and the 7–KLA as a ligand. The residues Val 20, His 95, Tyr156, Lys160, Thr189, Thr191, Ser193, and Tyr253 were set as flexible residues, and the results are shown in Figure 12. The substrate 7–KLA binds to the binary 7β–HSDH–NADP+ in the orientation of β side toward catalytic residue Tyr156 and α side toward NADP+, with a distance of 4.0 Å and 3.8 Å between C7–O and Tyr156–OH and C4–nicotinamide group ([Tyr163–OH⋯3.1 Å⋯O–C7] and [C7–O⋯3.4 Å⋯C4–nicotinamide]), respectively, which is opposite to 7α–HSDH. The binding energy is 7.79 kcal/mol. Three key residues His95, Ser193, and Tyr253 are probably related to the orientation of substrate binding, which conserved in7β–HSDH and definitely different from 7α–HSDHs (Figure 13). An obstruction formed by the side chain of His95, Ser193 and the pyrophosphate group of NADP+ makes the substrate only able to bind to the pocket of the nicotinamide region for the happening of reaction, which make the β side of the substrate toward the NADP+ impossible. The orientation, with C3–OH toward C-terminal (or the adenosine region of NADP+) and C24–O towards N-terminal (or nicotinamide region of NADP+), helped to the formation of this binding model.
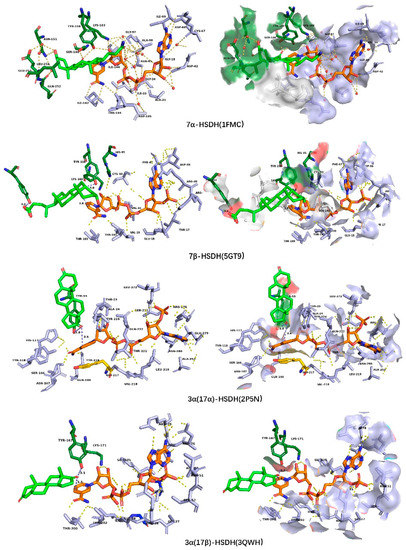
Figure 12.
The substrate-binding model in 7α–HSDH (PDB: 1FMC) and the molecular docking results of 7–KLA to 7β–HSDH (PDB: 5GT9), androstenedione to 17β–HSDH (PDB: 3QWH) and 3(17)α–HSDH (PDB: 2P5N), respectively. The distances between the reaction position C7=O (in 7α–HSDH and 7β–HSDH) and Tyr–OH, C4 of nicotinamide and the reaction position C17=O (in 17β–HSDH and 3(17)α–HSDH) and Tyr–OH, C4 of nicotinamide were measured using Pymol and shown in blue color.
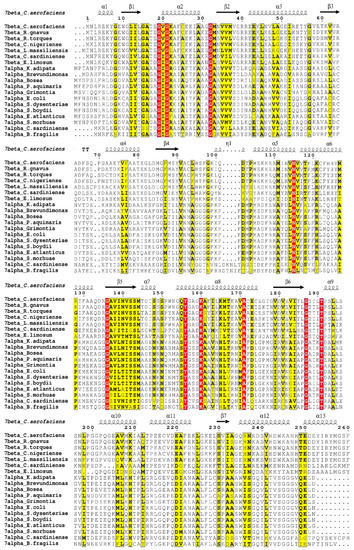
Figure 13.
Sequence alignment of 7α–HSDHs and 7β–HSDHs. The accession number are: 7β–HSDH from Collinsella aerofaciens, UniProtKB/Swiss-Prot A4ECA9.1; from Ruminococcus gnavus, UniProtKB/Swiss-Prot A7B4V1.1; from Clostridium sardiniense, GenBank AET80684.1; from Ruminococcus torques, GenBank CBL26204.1; from Eubacterium limosum, WP_038354045.1; from Clostridium nigeriense, WP_066892209.1; from Libanicoccus massiliensis, WP_073294202.1. 7α–HSDH from Escherichia coli, UniProtKB/Swiss-Prot P0AET8.1; from Clostridium sardiniense, UniProtKB/Swiss-Prot G9FRD7.1; from Shigella dysenteriae, GenBank ABB61947.1; from Bacteroides fragilis, GenBank AAD49430.2; from Shigella boydii, GenBank ABB66134.1; from Bosea sp., GenBank OYW60865.1; from Kaistia adipate, WP_029074103.1; from Grimontia sp., WP_046304034.1; from Brevundimonas, WP_046653274.1; from Erythrobacter atlanticus, WP_048884278.1; from Shewanella morhuae, WP_076500293.1; from Pseudoruegeria aquimaris, WP_085868047.1. This figure was created by ESPript on line [134].
The differences of substrate binding between SDRs and AKRs were analyzed through a semi-flexible molecular docking of ligand androstenedione to 17β–HSDH-NADP+ (PDB: 3QWH) from SDR superfamily and 3α(17α)–HSDH–NADP+ (PDB: 2P5N) from AKR superfamily, with flexible residues Val107, Phe109, Ser153, Asn154, Phe159, Tyr167, Pro197, Gly198, Gly199, Thr200, Phe205, His206, Glu207, Val208, Ser209, Tyr212, Ile213, Pro214, Met227, Ala228, Ala231, Asp266, and Ala269 in 17β–HSDH, as well as Lys31, Tyr55, His117, Tyr118, Phe219, Tyr224, Gly225 Gly226, and Trp227 in 3α(17α)–HSDH. The results were shown in Figure 12. In AKRs, the substrate-binding pocket is composed of loops between αA-βA (residues 21–30), αB–βB (residues 52–60), and αG–βG (residues 220–228). The binding directions of substrate and coenzyme are vertical in space, while it is parallel in SDRs, which may be the reason why HSDHs with a catalytic function at positions 7, 11, and 12 are mainly present in SDRs. The relative position of substrate binding pocket and coenzyme binding pocket makes it difficult for the positions 7, 11, and 12 of the steroid-skeleton substrates to approach the catalytic center, which impacts the proton transfer among substrate, catalytic residues, and nicotinamide group of NAD(P)(H).
The catalytic residues in 17β–HSDH and 3α(17α)–HSDH are Ser–Tyr–Lys and Asp–Tyr–Lys–His, respectively. The hydrogen of hydroxyl (–OH) at substrate reaction position was from the catalytic residue Tyr when in a reduction direction. Androstenedione is bound to 17β–HSDH–NADP+ binary with an orientation of β side towards catalytic residue Tyr and α side towards B-face of nicotinamide group, with a distance of 3.1 Å and 3.4 Å between C17=O and Tyr–OH and C4–nicotinamide group in 17β–HSDH. The ligand androstenedione binds to 3α(17α)–HSDH–NADP+ binary in an opposite orientation to 17β–HSDH, with a distance of 3.8 Å and 3.6 Å between C17=O and Tyr–OH and C4–nicotinamide group in 3α(17α)–HSDH. A docking result of C3=O of the ligand toward catalytic residue Tyr and α side toward A-face of nicotinamide group was obtained as well, with a distance of 6.2 Å and 3.4 Å between C3=O and Tyr–OH and C4–nicotinamide group. Further determination of the binding method may require molecular dynamics simulation.
The catalytic mechanism of 3α(17α)–HSDH (AKR1C21) for different reactions on positions C3 and C17 has been studied through molecular docking. 3-keto/3α-hydroxysteroids and the 17-keto steroids were used for docking to 3α(17α)–HSDH. C3 and C17 steroid substrates exhibit different binding orientations due to hydrogen-bonding interactions affected by residues Lys31, Gly225, and Gly226 in the substrate-binding pocket. Human HSDHs of AKR1C1 and AKR1C2 usually act as 3α/3β–HSDHs in the reduction of 5α-dihydrotestosterone (5α–DHT), catalyzing the reduction of 5α–DHT to produce 3α– and 3β–androstanediol. The molecular docking was realized using 5α–DHT as a ligand and AKR1C1 and AKR1C2 as receptors. It was found that the A-ring of steroid 5α–DHT presented its β-face to the 4-pro-R hydrogen when docked into AKR1C2(3β–HSDHs),
Whereas A-ring of 5α–DHT presented its α-face to the 4-pro-R hydrogen of coenzyme when docked into AKR1C1(3α–HSDHs). The different orientations may be related to different residues of the steroid-binding pockets, which is Leu of AKR1C1 while Val in AKR1C2 [100,133,135].
6. Conclusions
In this article, the HSDHs from the SDR and AKR superfamilies were summarized and classified according to their structural and functional characteristics. The different NAD(P)(H)-binding models of these two types of HSDHs were analyzed, with the pro-S side of nicotinamide group towards catalytic residues in SDR superfamily while pro-R side of nicotinamide group towards catalytic residues in AKR superfamily. It impacts the catalytic mechanisms of SDR–HSDHs and AKR–HSDHs. AKR–HSDHs accept 4-pro-R hydride from NADPH, while SDR–HSDHs accept 4-pro-S hydride from NAD(P)H. The majority of AKR–HSDHs are NADPH-dependent and conserved in coenzyme conformations. However, the conformations of the NAD(P)(H) show large differences in the SDR superfamily, especially between α–HSDHs and β–HSDHs. The different conformation in the pyrophosphate region of NAD(P)(H) between 7α–HSDH and 7β–HSDH attracted much attention. The dihedral angle (PA–O3–PN–O5D) of the pyrophosphate region is roughly –140° in two structures of 7α–HSDH with different coenzymes, while it is –80.6° in 7β–HSDH. A typical pair of stereoisomeric enzymes, 7α–HSDH and 7β–HSDH, were used to analyze the stereospecificity of HSDHs. A semi-flexible molecular docking was realized using the holo-form of 7β–HSDH–NADP+ as a receptor and 7–KLA as a ligand via YASARA. A possible binding mode of 7β–HSDH and 7–KLA was obtained. The result was confirmed by comparison to a ternary complex of 7α–HSDH. A hydrophilic face (α side) of 7–KLA toward NADP+ in 7β–HSDH made the orientation of C7–OH different in the product, while a hydrophobic face (β side) towards NAD+ in 7α–HSDH. One reason leading to the different substrate orientations is the different conformation of the pyrophosphate region of coenzymes caused by Ser193 in 7β–HSDH. An interaction toward pyrophosphate [Ser193–OG⋯3.11 Å⋯O1N–PN] caused the upturning of PN–phosphate, which formed a barrier with the side chain of His95 to prevent 7–KLA from passing through this region and could only be combined with NADP+ as α side towards nicotinamide group. A possible interaction existing in the Tyr253 of 7β–HSDH and C24–O of 7–KLA may contribute to the formation of substrate binding orientation. The results of sequence alignment showed the conservative of these three residues (His95, Ser193, Tyr253) in 7β–HSDH, which had a significant difference to 7α–HSDH. The molecular docking of the other two enzymes, 17β–HSDH from the SDR superfamily and 3(17)α–HSDH from the AKR superfamily, has further verified the relation between stereospecificity of HSDHs and substrate binding orientation. However, the results still need a further mutation to confirm in future studies.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/11/3/224/s1. Figure S1: Sequence alignment of HSDHs from SDR superfamily, Table S1: HSDHs identified belonging to SDR superfamily, Table S2: HSDHs identified belonging to AKR superfamily, Table S3: Functions of HSDHs and their related metabolic pathways, Table S4: Residues that interact with NAD(P)(H) in AKR superfamily and their conformation differences of coenzyme.
Author Contributions
Conceptualization, L.L. and K.N.; methodology, M.G. and H.X.; validation, M.Q., L.L. and M.G.; resources, H.X.; data curation, L.L. and M.G.; writing—original draft preparation, L.L. and M.G.; writing—review and editing, L.L. and M.Q.; supervision, F.W.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 21978017, 21978020, 21861132017).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kisiela, M.; Skarka, A.; Ebert, B.; Maser, E. Hydroxysteroid dehydrogenases (HSDs) in bacteria—A bioinformatic perspective. J. Steroid Biochem. Mol. Biol. 2012, 129, 31–46. [Google Scholar] [CrossRef]
- Penning, T.M. Human hydroxysteroid dehydrogenases and pre-receptor regulation: Insights into inhibitor design and evaluation. J. Steroid Biochem. Mol. Biol. 2011, 125, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Higashi, H.; Kanatani, A.; Lin, X.S.; Nagai, H.; Oyama, H.; Kurazono, K.; Tsuru, D. Cloning and sequencing of the 7 alpha-hydroxysteroid dehydrogenase gene from Escherichia coli HB101 and characterization of the expressed enzyme. J. Bacteriol. 1991, 173, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Masuda, N. Characterization of NADP-dependent 7 beta-hydroxysteroid dehydrogenases from Peptostreptococcus productus and Eubacterium aerofaciens. Appl. Environ. Microbiol. 1982, 43, 1057–1063. [Google Scholar] [CrossRef]
- Tannin, G.M.; Agarwal, A.K.; Monder, C.; New, M.I.; White, P.C. The human gene for 11 beta-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. J. Biol. Chem. 1991, 266, 16653–16658. [Google Scholar] [CrossRef]
- Jörnvall, H.; Höög, J.-O.; Persson, B. SDR and MDR: Completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett. 1999, 445, 261–264. [Google Scholar] [CrossRef]
- Jörnvall, H.; Persson, B.; Krook, M.; Atrian, S.; Gonzalez-Duarte, R.; Jeffery, J.; Ghosh, D. Short-chain dehydrogenases/reductases (SDR). Biochemistry 1995, 34, 6003–6013. [Google Scholar] [CrossRef]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef]
- Jez, J.M.; Bennett, M.J.; Schlegel, B.P.; Lewis, M.; Penning, T.M. Comparative anatomy of the aldo–keto reductase superfamily. Biochem. J. 1997, 326, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Möbus, E.; Maser, E. Molecular Cloning, Overexpression, and Characterization of Steroid-inducible 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase from Comamonas testosteroni. J. Biol. Chem. 1998, 273, 30888–30896. [Google Scholar] [CrossRef]
- Suzuki, K.; Ueda, S.; Sugiyama, M.; Imamura, S. Cloning and expression of a Pseudomonas 3α-hydroxy steroid dehydrogenase-encoding gene in Escherichia coli. Gene 1993, 130, 137–140. [Google Scholar] [CrossRef]
- Bennett, M.J.; Schlegel, B.P.; Jez, J.M.; Penning, T.M.; Lewis, M. Structure of 3α-Hydroxysteroid/Dihydrodiol Dehydrogenase Complexed with NADP+. Biochemistry 1996, 35, 10702–10711. [Google Scholar] [CrossRef]
- Deyashiki, Y.; Ogasawara, A.; Nakayama, T.; Nakanishi, M.; Miyabe, Y.; Sato, K.; Hara, A. Molecular cloning of two human liver 3 α-hydroxysteroid/dihydrodiol dehydrogenase isoenzymes that are identical with chlordecone reductase and bile-acid binder. Biochem. J. 1994, 299, 545–552. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMol Molecular Graphics System. Proteins 2002, 30, 442–454. [Google Scholar]
- Bashton, M.; Chothia, C. The geometry of domain combination in proteins 1 1Edited by J. Thornton. J. Mol. Biol. 2002, 315, 927–939. [Google Scholar] [CrossRef]
- Hyndman, D.; Bauman, D.R.; Heredia, V.V.; Penning, T.M. The aldo-keto reductase superfamily homepage. Chem. Interact. 2003, 144, 621–631. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Schiffer, L.; Barnard, L.; Baranowski, E.S.; Gilligan, L.C.; Taylor, A.E.; Arlt, W.; Shackleton, C.H.; Storbeck, K.-H. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2019, 194, 105439. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Kamiya, T.; Usami, N.; Shintani, S.; Shiraishi, H.; Ishikura, S.; Yamamoto, I.; Hara, A. Molecular Characterization of Two Monkey Dihydrodiol Dehydrogenases. Drug Metab. Pharmacokinet. 2002, 17, 348–356. [Google Scholar] [CrossRef][Green Version]
- Endo, S.; Matsunaga, T.; Arai, Y.; Ikari, A.; Tajima, K.; El-Kabbani, O.; Yamano, S.; Hara, A.; Kitade, Y. Cloning and Characterization of Four Rabbit Aldo-Keto Reductases Featuring Broad Substrate Specificity for Xenobiotic and Endogenous Carbonyl Compounds: Relationship with Multiple Forms of Drug Ketone Reductases. Drug Metab. Dispos. 2014, 42, 803–812. [Google Scholar] [CrossRef]
- Persson, B.; Kallberg, Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs). Chem. Interact. 2013, 202, 111–115. [Google Scholar] [CrossRef]
- Maser, E. Xenobiotic carbonyl reduction and physiological steroid oxidoreduction. Biochem. Pharmacol. 1995, 49, 421–440. [Google Scholar] [CrossRef]
- Kallberg, Y.; Oppermann, U.; Persson, B. Classification of the short-chain dehydrogenase/reductase superfamily using hidden Markov models. FEBS J. 2010, 277, 2375–2386. [Google Scholar] [CrossRef]
- Roth, S.; Stockinger, P.; Steff, J.; Steimle, S.; Sautner, V.; Tittmann, K.; Pleiss, J.; Müller, M. Crossing the Border: From Keto- to Imine Reduction in Short-Chain Dehydrogenases/Reductases. ChemBioChem 2020, 21, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.R.; Kaserer, T.; Schuster, D.; Odermatt, A. Virtual screening applications in short-chain dehydrogenase/reductase research. J. Steroid Biochem. Mol. Biol. 2017, 171, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Maser, E. Carbonyl Reductases and Pluripotent Hydroxysteroid Dehydrogenases of the Short-chain Dehydrogenase/reductase Superfamily. Drug Metab. Rev. 2007, 39, 87–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lukacik, P.; Kavanagh, K.L.; Oppermann, U. SDR-type human hydroxysteroid dehydrogenases involved in steroid hormone activation. Mol. Cell. Endocrinol. 2007, 265, 71–76. [Google Scholar] [CrossRef]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Fern, L.A.; Ismail, D.S.N.R.P.H.; Chaiyasut, C. The influence of probiotics on bile acids in diseases and aging. Biomed. Pharmacother. 2020, 128, 110310. [Google Scholar] [CrossRef]
- Oppermann, U.C.T.; Maser, E. Characterization of a 3alpha-Hydroxysteroid Dehydrogenase/Carbonyl Reductase from the Gram-Negative Bacterium Comamonas testosteroni. JBIC J. Biol. Inorg. Chem. 1996, 241, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, S. Comparative analysis of Corynebacterium glutamicum genomes: A new perspective for the industrial production of amino acids. BMC Genom. 2017, 18, 940. [Google Scholar] [CrossRef]
- Liu, L.; Aigner, A.; Schmid, R.D. Identification, cloning, heterologous expression, and characterization of a NADPH-dependent 7β-hydroxysteroid dehydrogenase from Collinsella aerofaciens. Appl. Microbiol. Biotechnol. 2010, 90, 127–135. [Google Scholar] [CrossRef]
- Chen, X.; Cui, Y.; Feng, J.; Wang, Y.; Liu, X.; Wu, Q.; Zhu, D.; Ma, Y. Flavin Oxidoreductase-Mediated Regeneration of Nicotinamide Adenine Dinucleotide with Dioxygen and Catalytic Amount of Flavin Mononucleotide for One-Pot Multi-Enzymatic Preparation of Ursodeoxycholic Acid. Adv. Synth. Catal. 2019, 361, 2497–2504. [Google Scholar] [CrossRef]
- He, X.-L.; Wang, L.-T.; Gu, X.-Z.; Xiao, J.-X.; Qiu, W.-W. A facile synthesis of ursodeoxycholic acid and obeticholic acid from cholic acid. Steroids 2018, 140, 173–178. [Google Scholar] [CrossRef]
- Simard, J.; Ricketts, M.-L.; Gingras, S.; Soucy, P.; Feltus, F.A.; Melner, M.H. Molecular Biology of the 3β-Hydroxysteroid Dehydrogenase/Δ5-Δ4 Isomerase Gene Family. Endocr. Rev. 2005, 26, 525–582. [Google Scholar] [CrossRef]
- Thomas, J.L.; Mack, V.L.; Sun, J.; Terrell, J.R.; Bucholtz, K.M. The functions of key residues in the inhibitor, substrate and cofactor sites of human 3β-hydroxysteroid dehydrogenase type 1 are validated by mutagenesis. J. Steroid Biochem. Mol. Biol. 2010, 120, 192–199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, J.L.; Duax, W.L.; Addlagatta, A.; Brandt, S.; Fuller, R.R.; Norris, W. Structure/Function Relationships Responsible for Coenzyme Specificity and the Isomerase Activity of Human Type 1 3β-Hydroxysteroid Dehydrogenase/Isomerase. J. Biol. Chem. 2003, 278, 35483–35490. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.L.; Bose, H.S. Regulation of human 3-beta-hydroxysteroid dehydrogenase type-2 (3βHSD2) by molecular chaperones and the mitochondrial environment affects steroidogenesis. J. Steroid Biochem. Mol. Biol. 2015, 151, 74–84. [Google Scholar] [CrossRef]
- Pletnev, V.Z.; Thomas, J.L.; Rhaney, F.L.; Holt, L.S.; Scaccia, L.A.; Umland, T.C.; Duax, W.L. Rational proteomics V: Structure-based mutagenesis has revealed key residues responsible for substrate recognition and catalysis by the dehydrogenase and isomerase activities in human 3β-hydroxysteroid dehydrogenase/isomerase type 1. J. Steroid Biochem. Mol. Biol. 2006, 101, 50–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hettel, D.; Sharifi, N. HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nat. Rev. Urol. 2017, 15, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Yue, W.; Wang, J.-P. Estrogen metabolites and breast cancer. Steroids 2015, 99, 61–66. [Google Scholar] [CrossRef]
- Duarsa, G.W.K.; Sari, Y.A.; Oka, A.A.G.; Santosa, K.B.; Yudiana, I.W.; Tirtayasa, P.M.W.; Pramana, I.B.P.; Kloping, Y.P. Serum testosterone and prostate-specific antigen levels are major risk factors for prostatic volume increase among benign prostatic hyperplasia patients. Asian J. Urol. 2020, 1–9. [Google Scholar] [CrossRef]
- Damiani, F.; Makieva, S.; Rinaldi, S.F.; Hua, L.; Marcolongo, P.; Petraglia, F.; Norman, J.E. 11β-hydroxysteroid dehydrogenase type 1 and pregnancy: Role in the timing of labour onset and in myometrial contraction. Mol. Cell. Endocrinol. 2017, 447, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Dammann, C.; Stapelfeld, C.; Maser, E. Expression and activity of the cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 is tissue and species-specific. Chem. Interact. 2019, 303, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Valeur, E.; Christmann-Franck, S.; Lepifre, F.; Carniato, D.; Cravo, D.; Charon, C.; Bozec, S.; Musil, D.; Hillertz, P.; Doare, L.; et al. Structure-based design of 7-azaindole-pyrrolidine amides as inhibitors of 11β-hydroxysteroid dehydrogenase type I. Bioorgan. Med. Chem. Lett. 2012, 22, 5909–5914. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, F.W.; Dossetter, A.G.; Scott, J.S.; Robb, G.R.; Boyd, S.; Groombridge, S.D.; Kemmitt, P.D.; Sjögren, T.; Gutierrez, P.M.; Deschoolmeester, J.; et al. Optimization of Brain Penetrant 11β-Hydroxysteroid Dehydrogenase Type I Inhibitors and in Vivo Testing in Diet-Induced Obese Mice. J. Med. Chem. 2014, 57, 970–986. [Google Scholar] [CrossRef]
- Siu, M.; Johnson, T.O.; Wang, Y.; Nair, S.K.; Taylor, W.D.; Cripps, S.J.; Matthews, J.J.; Edwards, M.P.; Pauly, T.A.; Ermolieff, J.; et al. N-(Pyridin-2-yl) arylsulfonamide inhibitors of 11β-hydroxysteroid dehydrogenase type 1: Discovery of PF-915275. Bioorgan. Med. Chem. Lett. 2009, 19, 3493–3497. [Google Scholar] [CrossRef]
- Sandeep, T.C.; Walker, B.R. Pathophysiology of modulation of local glucocorticoid levels by 11β-hydroxysteroid dehydrogenases. Trends Endocrinol. Metab. 2001, 12, 446–453. [Google Scholar] [CrossRef]
- Zhu, Q.; Ge, F.; Dong, Y.; Sun, W.; Wang, Z.; Shan, Y.; Chen, R.; Sun, J.; Ge, R.-S. Comparison of flavonoids and isoflavonoids to inhibit rat and human 11β-hydroxysteroid dehydrogenase 1 and 2. Steroids 2018, 132, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E. Evolution of 11β-hydroxysteroid dehydrogenase-type 1 and 11β-hydroxysteroid dehydrogenase-type 3. FEBS Lett. 2010, 584, 2279–2284. [Google Scholar] [CrossRef] [PubMed]
- Mindnich, R.; Möller, G.; Adamski, J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2004, 218, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Multifunctionality of human 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2006, 248, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2009, 301, 7–19. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef]
- Gangloff, A.; Shi, R.; Nahoum, V.; Lin, S. Pseudo-symmetry of C19-steroids, alternative binding orientations and multispecificity in human estrogenic 17β-hydroxysteroid dehydrogenase. FASEB J. 2002, 17, 274–276. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Calvo, E.-L.; Yang, C.-Q.; Liu, J.; Sang, X.-Y.; Lin, S.-X. Transcriptome of 17β-hydroxysteroid dehydrogenase type 2 plays both hormone-dependent and hormone-independent roles in MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2019, 195, 105471. [Google Scholar] [CrossRef]
- Abdelsamie, A.S.; Salah, M.; Siebenbürger, L.; Hamed, M.M.; Börger, C.; Van Koppen, C.J.; Frotscher, M.; Hartmann, R.W. Development of potential preclinical candidates with promising in vitro ADME profile for the inhibition of type 1 and type 2 17β-Hydroxysteroid dehydrogenases: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2019, 178, 93–107. [Google Scholar] [CrossRef]
- Ning, X.; Yang, Y.; Deng, H.; Zhang, Q.; Huang, Y.; Su, Z.; Fu, Y.; Xiang, Q.; Zhang, S. Development of 17β-hydroxysteroid dehydrogenase type 3 as a target in hormone-dependent prostate cancer therapy. Steroids 2017, 121, 10–16. [Google Scholar] [CrossRef]
- Mindnich, R.D.; Penning, T.M. Aldo-keto reductase (AKR) superfamily: Genomics and annotation. Hum. Genom. 2009, 3, 362–370. [Google Scholar] [CrossRef]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef]
- Rižner, T.L.; Penning, T.M. Role of aldo–keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef]
- Rižner, T.L.; Penning, T.M. Aldo-keto reductase 1C3—Assessment as a new target for the treatment of endometriosis. Pharmacol. Res. 2020, 152, 104446. [Google Scholar] [CrossRef]
- Penning, T.M.; Jin, Y.; Heredia, V.V.; Lewis, M. Structure–function relationships in 3α-hydroxysteroid dehydrogenases: A comparison of the rat and human isoforms. J. Steroid Biochem. Mol. Biol. 2003, 85, 247–255. [Google Scholar] [CrossRef]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.-F.; Lin, H.-K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1‒AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef]
- Wan, R.; Kong, X.; Yang, Y.; Tao, S.; Chen, Y.; Teichmann, A.T.; Wieland, F.H. Role of human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C3) in the extrahepatic metabolism of the steroidal aromatase inactivator Formestane. J. Steroid Biochem. Mol. Biol. 2020, 198, 105527. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 2019, 489, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Steckelbroeck, S.; Fung, K.-M.; Jones, A.N.; Penning, T.M. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids 2004, 69, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Lin, H.-K.; Rogers, K.A.; Brame, L.S.; Yeh, M.M.; Yang, Q.; Fung, K.-M. Expression of aldo-keto reductase family 1 member C3 (AKR1C3) in neuroendocrine tumors & adenocarcinomas of pancreas, gastrointestinal tract, and lung. Int. J. Clin. Exp. Pathol. 2013, 6, 2419–2429. [Google Scholar]
- Šmuc, T.; Rižner, T.L. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol. Cell. Endocrinol. 2009, 301, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Rižner, T.L.; Lin, H.K.; Penning, T.M. Role of human type 3 3α-hydroxysteroid dehydrogenase (AKR1C2) in androgen metabolism of prostate cancer cells. Chem. Interact. 2003, 144, 401–409. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Dhagat, U.; Hara, A. Inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1). J. Steroid Biochem. Mol. Biol. 2011, 125, 105–111. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Scammells, P.J.; Day, T.; Dhagat, U.; Endo, S.; Matsunaga, T.; Soda, M.; Hara, A. Structure-based optimization and biological evaluation of human 20α-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. Eur. J. Med. Chem. 2010, 45, 5309–5317. [Google Scholar] [CrossRef]
- Lima, M.A.; Silva, S.V.; Jaeger, R.G.; Freitas, V.M. Progesterone decreases ovarian cancer cells migration and invasion. Steroids 2020, 161, 108680. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, G.C.; Tosto, V.; Tsibizova, V. Progesterone: History, facts, and artifacts. Best Pr. Res. Clin. Obstet. Gynaecol. 2020, 69, 2–12. [Google Scholar] [CrossRef]
- Brožič, P.; Cesar, J.; Kovač, A.; Davies, M.; Johnson, A.; Fishwick, C.; Rižner, T.L.; Gobec, S. Derivatives of pyrimidine, phthalimide and anthranilic acid as inhibitors of human hydroxysteroid dehydrogenase AKR1C1. Chem. Interact. 2009, 178, 158–164. [Google Scholar] [CrossRef]
- Beranič, N.; Gobec, S.; Rižner, T.L. Progestins as inhibitors of the human 20-ketosteroid reductases, AKR1C1 and AKR1C3. Chem. Interact. 2011, 191, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta BBA Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Huang, R.; Wei, X.; Zhu, Z.; Zhang, Y.-H.P. Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular mechanism of metabolic NAD(P)H-dependent electron-transfer systems: The role of redox cofactors. Biochim. Biophys. Acta BBA Bioenerg. 2019, 1860, 233–258. [Google Scholar] [CrossRef]
- Filling, C.; Berndt, K.D.; Benach, J.; Knapp, S.; Prozorovski, T.; Nordling, E.; Ladenstein, R.; Jörnvall, H.; Oppermann, U. Critical Residues for Structure and Catalysis in Short-chain Dehydrogenases/Reductases. J. Biol. Chem. 2002, 277, 25677–25684. [Google Scholar] [CrossRef]
- Kavanagh, K.L.; Jornvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef]
- Bhatia, C.; Oerum, S.; Bray, J.; Kavanagh, K.L.; Shafqat, N.; Yue, W.; Oppermann, U. Towards a systematic analysis of human short-chain dehydrogenases/reductases (SDR): Ligand identification and structure–activity relationships. Chem. Interact. 2015, 234, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.E.; Marsden, B.D.; Oppermann, U. The human short-chain dehydrogenase/reductase (SDR) superfamily: A bioinformatics summary. Chem. Interact 2009, 178, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Filling, C.; Wu, X.; Shafqat, N.; Hult, M.; Mårtensson, E.; Shafqat, J.; Oppermann, U.C. Subcellular targeting analysis of SDR-type hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2001, 171, 99–101. [Google Scholar] [CrossRef]
- Duax, W.L.; Ghosh, D.; Pletnev, V. Steroid dehydrogenase structures, mechanism of action, and disease. Vitam. Horm. 2000, 58, 121–148. [Google Scholar] [CrossRef]
- Grant, G.A. Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch. Biochem. Biophys. 2012, 519, 175–185. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, S.; Zhang, C.; Zhu, X.; Hammad, M.A.; Zhang, X.; Christian, M.; Zhang, H.; Liu, P. Hydroxysteroid dehydrogenase family proteins on lipid droplets through bacteria, C. elegans, and mammals. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2018, 1863, 881–894. [Google Scholar] [CrossRef]
- Sherbet, D.P.; Papari-Zareei, M.; Khan, N.; Sharma, K.K.; Brandmaier, A.; Rambally, S.; Chattopadhyay, A.; Andersson, S.; Agarwal, A.K.; Auchus, R.J. Cofactors, redox state, and directional preferences of hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2007, L, 83–88. [Google Scholar] [CrossRef]
- Wushur, I.; Sylte, I.; Winberg, J.-O. The catalytic reaction mechanism of drosophilid alcohol dehydrogenases. Perspect. Sci. 2015, 4, 46–54. [Google Scholar] [CrossRef][Green Version]
- Tanaka, N.; Nonaka, T.; Tanabe, T.; Yoshimoto, T.; Tsuru, D.; Mitsui, Y. Crystal Structures of the Binary and Ternary Complexes of 7α-Hydroxysteroid Dehydrogenase from Escherichia coli. Biochemistry 1996, 35, 7715–7730. [Google Scholar] [CrossRef]
- Wang, R.; Wu, J.; Jin, D.K.; Chen, Y.; Lv, Z.; Chen, Q.; Miao, Q.; Huo, X.; Wang, F. Structure of NADP+-bound 7β-hydroxysteroid dehydrogenase reveals two cofactor-binding modes. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 246–252. [Google Scholar] [CrossRef]
- Lou, D.; Wang, B.; Tan, J.; Zhu, L.; Cen, X.; Ji, Q.; Wang, Y. The three-dimensional structure of Clostridium absonum 7α-hydroxysteroid dehydrogenase: New insights into the conserved arginines for NADP(H) recognition. Sci. Rep. 2016, 6, 22885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhu, D.-W.; Hu, X.-J.; Zhou, M.; Shang, P.; Lin, S.-X. Human 3-alpha hydroxysteroid dehydrogenase type 3 (3α-HSD3): The V54L mutation restricting the steroid alternative binding and enhancing the 20α-HSD activity. J. Steroid Biochem. Mol. Biol. 2014, 141, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-F.; Legrand, P.; Cantin, L.; Labrie, F.; Luu-The, V.; Breton, R. Loop Relaxation, A Mechanism that Explains the Reduced Specificity of Rabbit 20α-Hydroxysteroid Dehydrogenase, A Member of the Aldo-Keto Reductase Superfamily. J. Mol. Biol. 2004, 339, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ratnam, K.; Penning, T.M. Mutation of Nicotinamide Pocket Residues in Rat Liver 3α-Hydroxysteroid Dehydrogenase Reveals Different Modes of Cofactor Binding. Biochemistry 2000, 39, 102–109. [Google Scholar] [CrossRef]
- Wierenga, R. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef]
- Banfield, M.J.; Salvucci, M.E.; Baker, E.N.; Smith, C.A. Crystal structure of the NADP(H)-dependent ketose reductase from Bemisia argentifolii at 2.3 Å resolution. J. Mol. Biol. 2001, 306, 239–250. [Google Scholar] [CrossRef]
- Jin, Y.; Penning, T.M. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): Insights into positional and stereochemical preferences. Steroids 2006, 71, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Bennett, M.J.; Smith-Hoog, S.; Schlegel, B.P.; Jez, J.M.; Lewis, M. Structure and function of 3α-hydroxysteroid dehydrogenase. Steroids 1997, 62, 101–111. [Google Scholar] [CrossRef]
- Khan, M.S.; Qais, F.A.; Rehman, T.; Ismail, M.H.; Alokail, M.S.; Altwaijry, N.; Alafaleq, N.O.; Alajmi, M.F.; Gaber, N.S.; Alqhatani, R. Mechanistic inhibition of non-enzymatic glycation and aldose reductase activity by naringenin: Binding, enzyme kinetics and molecular docking analysis. Int. J. Biol. Macromol. 2020, 159, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.C.; Jin, Y.; Penning, T.M. Elucidation of a Complete Kinetic Mechanism for a Mammalian Hydroxysteroid Dehydrogenase (HSD) and Identification of All Enzyme Forms on the Reaction Coordinate. J. Biol. Chem. 2007, 282, 33484–33493. [Google Scholar] [CrossRef] [PubMed]
- Borhani, D.; Harter, T.; Petrash, J. The crystal structure of the aldose reductase NADPH binary complex. J. Biol. Chem. 1992, 267, 24841–24847. [Google Scholar] [CrossRef]
- Persson, B.; Kallberg, Y.; Oppermann, U.; Jörnvall, H. Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs). Chem. Interact. 2003, 144, 271–278. [Google Scholar] [CrossRef]
- Ghosh, D.; Wawrzak, Z.; Weeks, C.M.; Duax, W.L.; Erman, M. The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure 1994, 2, 629–640. [Google Scholar] [CrossRef]
- Gani, O.A.B.S.M.; Adekoya, O.A.; Giurato, L.; Spyrakis, F.; Cozzini, P.; Guccione, S.; Winberg, J.-O.; Sylte, I. Theoretical Calculations of the Catalytic Triad in Short-Chain Alcohol Dehydrogenases/Reductases. Biophys. J. 2008, 94, 1412–1427. [Google Scholar] [CrossRef]
- Koumanov, A.; Benach, J.; Atrian, S.; Gonzàlez-Duarte, R.; Karshikoff, A.; Ladenstein, R. The catalytic mechanism of Drosophilaalcohol dehydrogenase: Evidence for a proton relay modulated by the coupled ionization of the active site Lysine/Tyrosine pair and a NAD+ ribose OH switch. Proteins Struct. Funct. Bioinform. 2003, 51, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Winberg, J.-O.; Brendskag, M.K.; Sylte, I.; Lindstad, R.I.; McKinley-Mckee, J.S. The catalytic triad in Drosophila alcohol dehydrogenase: pH, temperature and molecular modelling studies. J. Mol. Biol. 1999, 294, 601–616. [Google Scholar] [CrossRef]
- Hovik, R.; Winberg, J.-O.; McKinley-Mckee, J.S. Drosophila melanogaster alcohol dehydrogenase. Insect Biochem. 1984, 14, 345–351. [Google Scholar] [CrossRef]
- Wuxiuer, Y.; Morgunova, E.; Cols, N.; Popov, A.; Karshikoff, A.; Sylte, I.; Gonzàlez-Duarte, R.; Ladenstein, R.; Winberg, J.-O. An intact eight-membered water chain in drosophilid alcohol dehydrogenases is essential for optimal enzyme activity. FEBS J. 2012, 279, 2940–2956. [Google Scholar] [CrossRef]
- Tanabe, T.; Tanaka, N.; Uchikawa, K.; Kabashima, T.; Ito, K.; Nonaka, T.; Mitsui, Y.; Tsuru, M.; Yoshimoto, T. Roles of the Ser146, Tyr159, and Lys163 Residues in the Catalytic Action of 7-Hydroxysteroid Dehydrogenase from Escherichia coli. J. Biochem. 1998, 124, 634–641. [Google Scholar] [CrossRef]
- Penning, T.M.; Drury, J.E. Human aldo–keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007, 464, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Flynn, T.G.; Green, N.C.; Bhatia, M.B.; El-Kabbani, O. Structure and Mechanism of Aldehyde Reductase. Adv. Exp. Med. Biol. 1995, 372, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Grimshaw, C.E.; Lai, C.J.; Harrison, D.H.; Ringe, D.; Petsko, G.A.; Gabbay, K.H. Tyrosine-48 Is the Proton Donor and Histidine-110 Directs Substrate Stereochemical Selectivity in the Reduction Reaction of Human Aldose Reductase: Enzyme Kinetics and Crystal Structure of the Y48H Mutant Enzyme. Biochemistry 1994, 33, 2021–2032. [Google Scholar] [CrossRef]
- Tarle, I.; Borhani, D.W.; Wilson, D.K.; Quiocho, F.A.; Petrash, J.M. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J. Biol. Chem. 1993, 268, 25687–25693. [Google Scholar] [CrossRef]
- Bohren, K.; Grimshaw, C.; Gabbay, K. Catalytic effectiveness of human aldose reductase. Critical role of C-terminal domain. J. Biol. Chem. 1992, 267, 20965–20970. [Google Scholar] [CrossRef]
- Grimshaw, C.E.; Bohren, K.M.; Lai, C.-J.; Gabbay, K.H. Human Aldose Reductase: Rate Constants for a Mechanism Including Interconversion of Ternary Complexes by Recombinant Wild-Type Enzyme. Biochemistry 1995, 34, 14356–14365. [Google Scholar] [CrossRef]
- Schlegel, B.P.; Jez, J.M.; Penning, T.M. Mutagenesis of 3α-Hydroxysteroid Dehydrogenase Reveals a “Push−Pull” Mechanism for Proton Transfer in Aldo−Keto Reductases. Biochemistry 1998, 37, 3538–3548. [Google Scholar] [CrossRef]
- Penning, T. Molecular determinants of steroid recognition and catalysis in aldo-keto reductases. Lessons from 3α-hydroxysteroid dehydrogenase. J. Steroid Biochem. Mol. Biol. 1999, 69, 211–225. [Google Scholar] [CrossRef]
- Eggert, T.; Bakonyi, D.; Hummel, W. Enzymatic routes for the synthesis of ursodeoxycholic acid. J. Biotechnol. 2014, 191, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Arai, H.; Nakamura, Y.; Fukiya, S.; Wada, M.; Yokota, A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid Res. 2013, 54, 3062–3069. [Google Scholar] [CrossRef]
- Zheng, M.-M.; Chen, F.-F.; Li, H.; Li, C.-X.; Xu, J.-H. Continuous Production of Ursodeoxycholic Acid by Using Two Cascade Reactors with Co-immobilized Enzymes. ChemBioChem 2018, 19, 347–353. [Google Scholar] [CrossRef]
- Savino, S.; Ferrandi, E.E.; Forneris, F.; Rovida, S.; Riva, S.; Monti, D.; Mattevi, A. Structural and biochemical insights into 7β-hydroxysteroid dehydrogenase stereoselectivity. Proteins Struct. Funct. Bioinform. 2016, 84, 859–865. [Google Scholar] [CrossRef]
- Nakajima, K.; Hashimoto, T.; Yamada, Y. Two tropinone reductases with different stereospecificities are short-chain dehydrogenases evolved from a common ancestor. Proc. Natl. Acad. Sci. USA 1993, 90, 9591–9595. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Yamashita, A.; Akama, H.; Nakatsu, T.; Kato, H.; Hashimoto, T.; Oda, J.; Yamada, Y. Crystal structures of two tropinone reductases: Different reaction stereospecificities in the same protein fold. Proc. Natl. Acad. Sci. USA 1998, 95, 4876–4881. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, A.M.; Nocek, B.P.; Clark, D.D.; Ensign, S.A.; Peters, J.W. Structural Basis for Stereoselectivity in the (R)- and (S)-Hydroxypropylthioethanesulfonate Dehydrogenases. Biochemistry 2006, 45, 8831–8840. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, D.A.; Krishnakumar, A.M.; Peters, J.W.; Ensign, S.A. Molecular Basis for Enantioselectivity in the (R)- and (S)-Hydroxypropylthioethanesulfonate Dehydrogenases, a Unique Pair of Stereoselective Short-Chain Dehydrogenases/Reductases Involved in Aliphatic Epoxide Carboxylation. Biochemistry 2010, 49, 3487–3498. [Google Scholar] [CrossRef]
- Chen, D.; Wu, S.; Xue, H.; Jiang, J. Stereoselective catabolism of compounds by microorganisms: Catabolic pathway, molecular mechanism and potential application. Int. Biodeterior. Biodegrad. 2020, 146, 104822. [Google Scholar] [CrossRef]
- Parales, R.E.; Lee, K.; Resnick, S.M.; Jiang, H.; Lessner, D.J.; Gibson, D.T. Substrate Specificity of Naphthalene Dioxygenase: Effect of Specific Amino Acids at the Active Site of the Enzyme. J. Bacteriol. 2000, 182, 1641–1649. [Google Scholar] [CrossRef]
- Parales, R.E.; Resnick, S.M.; Yu, C.-L.; Boyd, D.R.; Sharma, N.D.; Gibson, D.T. Regioselectivity and Enantioselectivity of Naphthalene Dioxygenase during Arene cis-Dihydroxylation: Control by Phenylalanine 352 in the α Subunit. J. Bacteriol. 2000, 182, 5495–5504. [Google Scholar] [CrossRef]
- Cassetta, A.; Stojan, J.; Krastanova, I.; Kristan, K.; Švegelj, M.B.; Lamba, D.; Rižner, T.L. Structural basis for inhibition of 17β-hydroxysteroid dehydrogenases by phytoestrogens: The case of fungal 17β-HSDcl. J. Steroid Biochem. Mol. Biol. 2017, 171, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Steckelbroeck, S.; Jin, Y.; Oyesanmi, B.; Kloosterboer, H.J.; Penning, T.M. Tibolone Is Metabolized by the 3α/3β-Hydroxysteroid Dehydrogenase Activities of the Four Human Isozymes of the Aldo-Keto Reductase 1C Subfamily: Inversion of Stereospecificity with a Δ5(10)-3-Ketosteroid. Mol. Pharmacol. 2004, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Dhagat, U.; Carbone, V.; Chung, R.P.-T.; Schulze-Briese, C.; Endo, S.; Hara, A.; El-Kabbani, O. Structure of 3(17)α-hydroxysteroid dehydrogenase (AKR1C21) holoenzyme from an orthorhombic crystal form: An insight into the bifunctionality of the enzyme. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 825–830. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).