

Synthesis, and Molecular Structure Investigations of a New s-Triazine Derivatives Incorporating Pyrazole/Piperidine/Aniline Moieties

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods




3. Results and Discussion
3.1. Chemistry
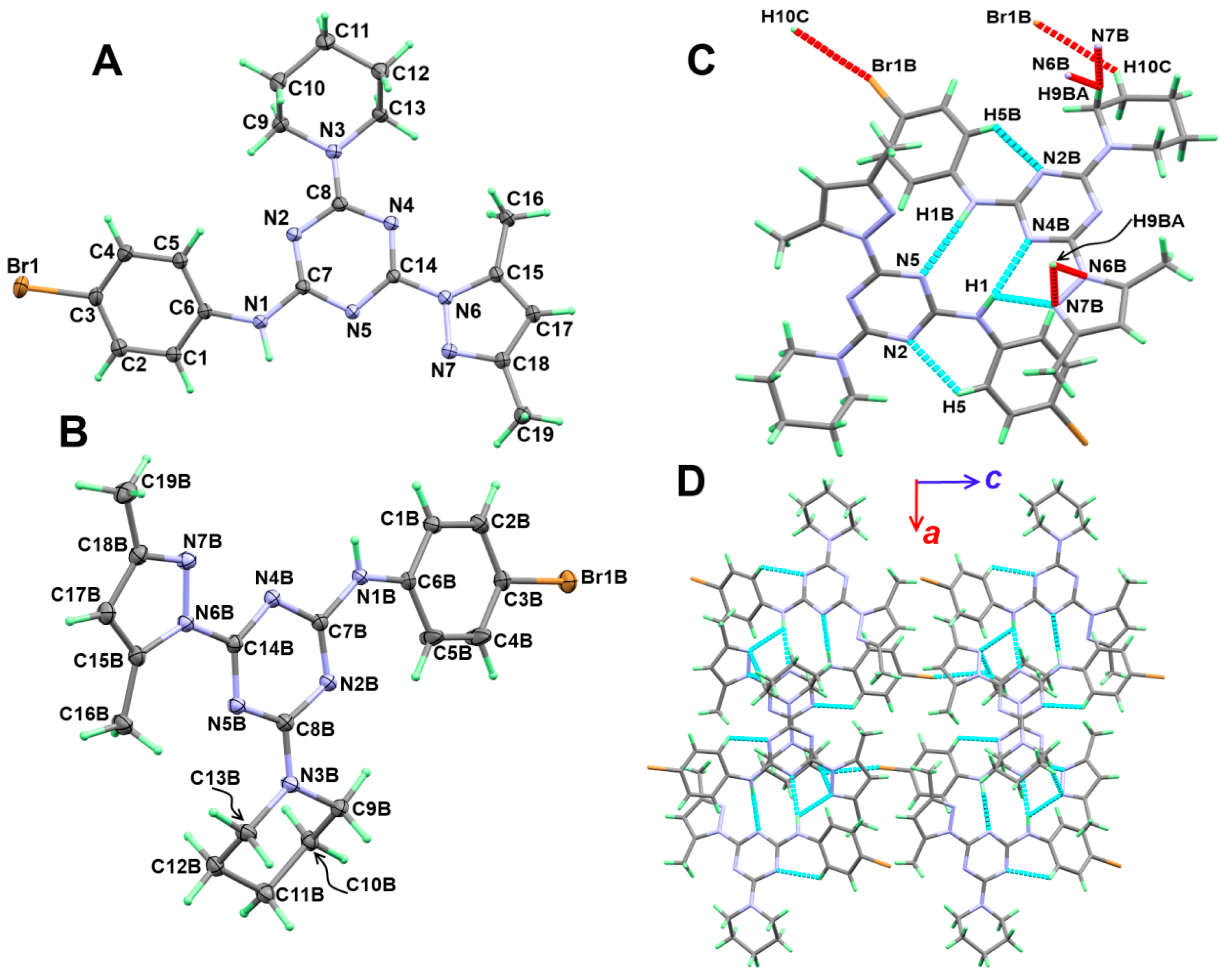
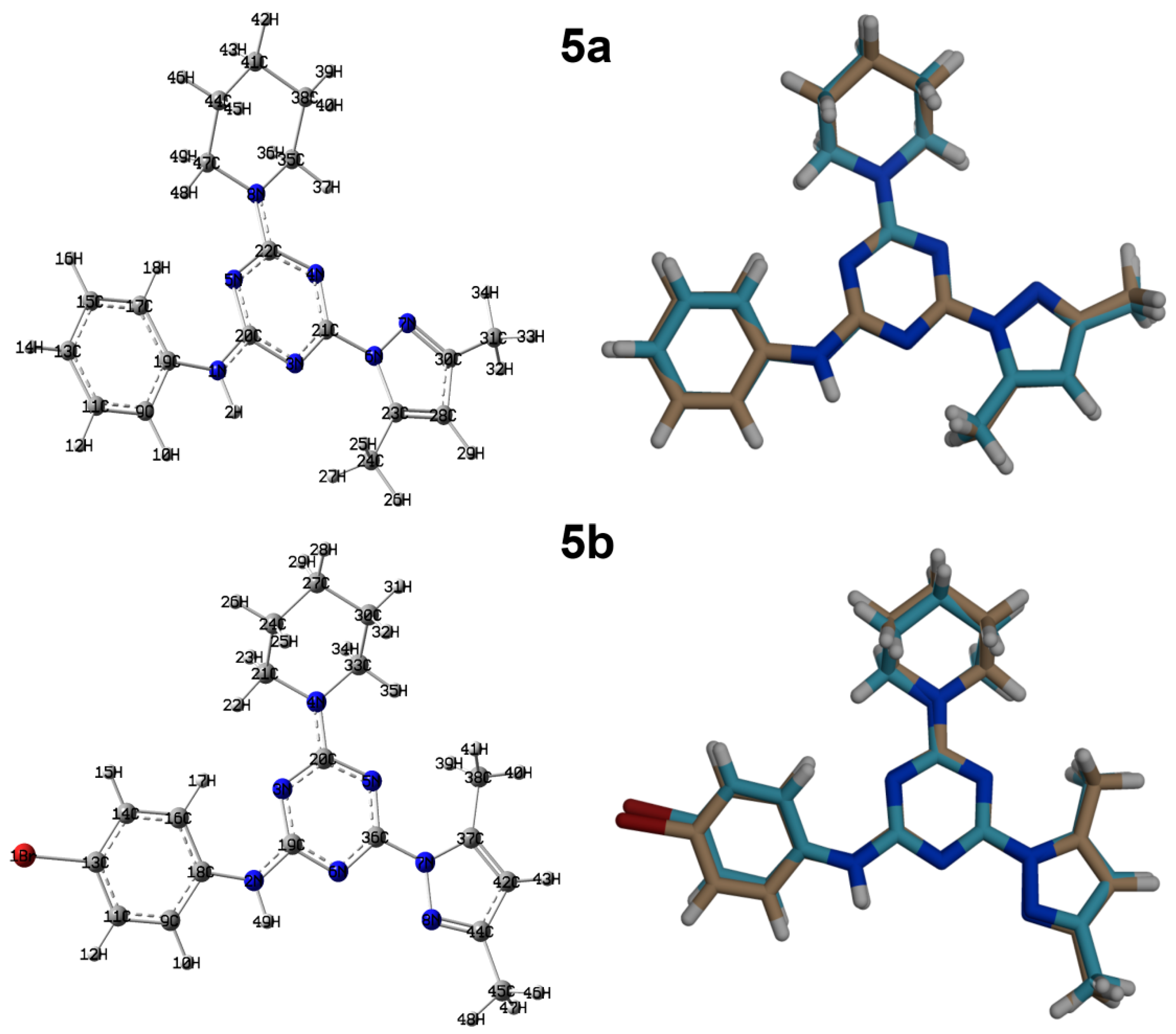
3.2. Crystal Structure Description
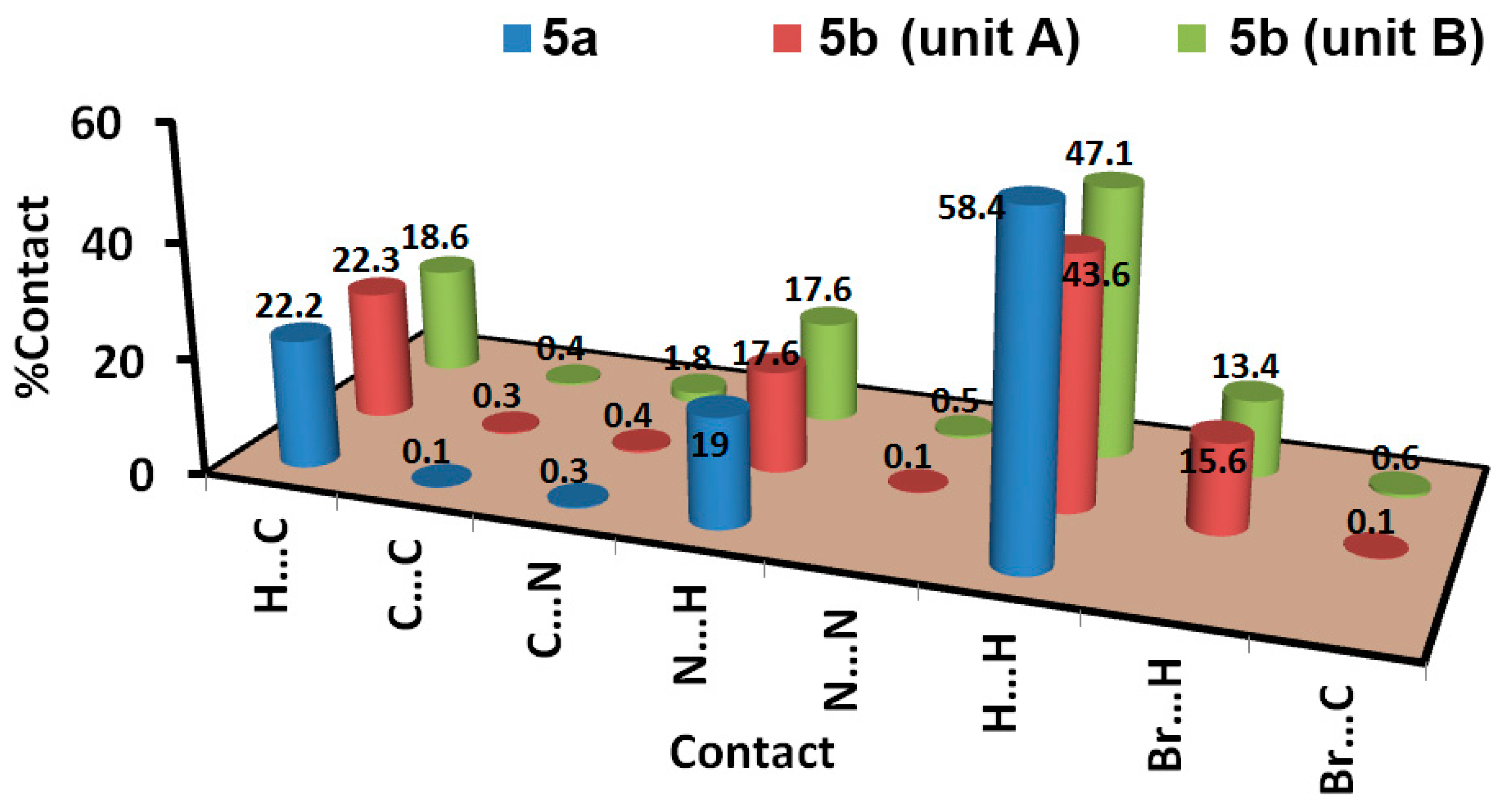
3.3. Analysis of Molecular Packing
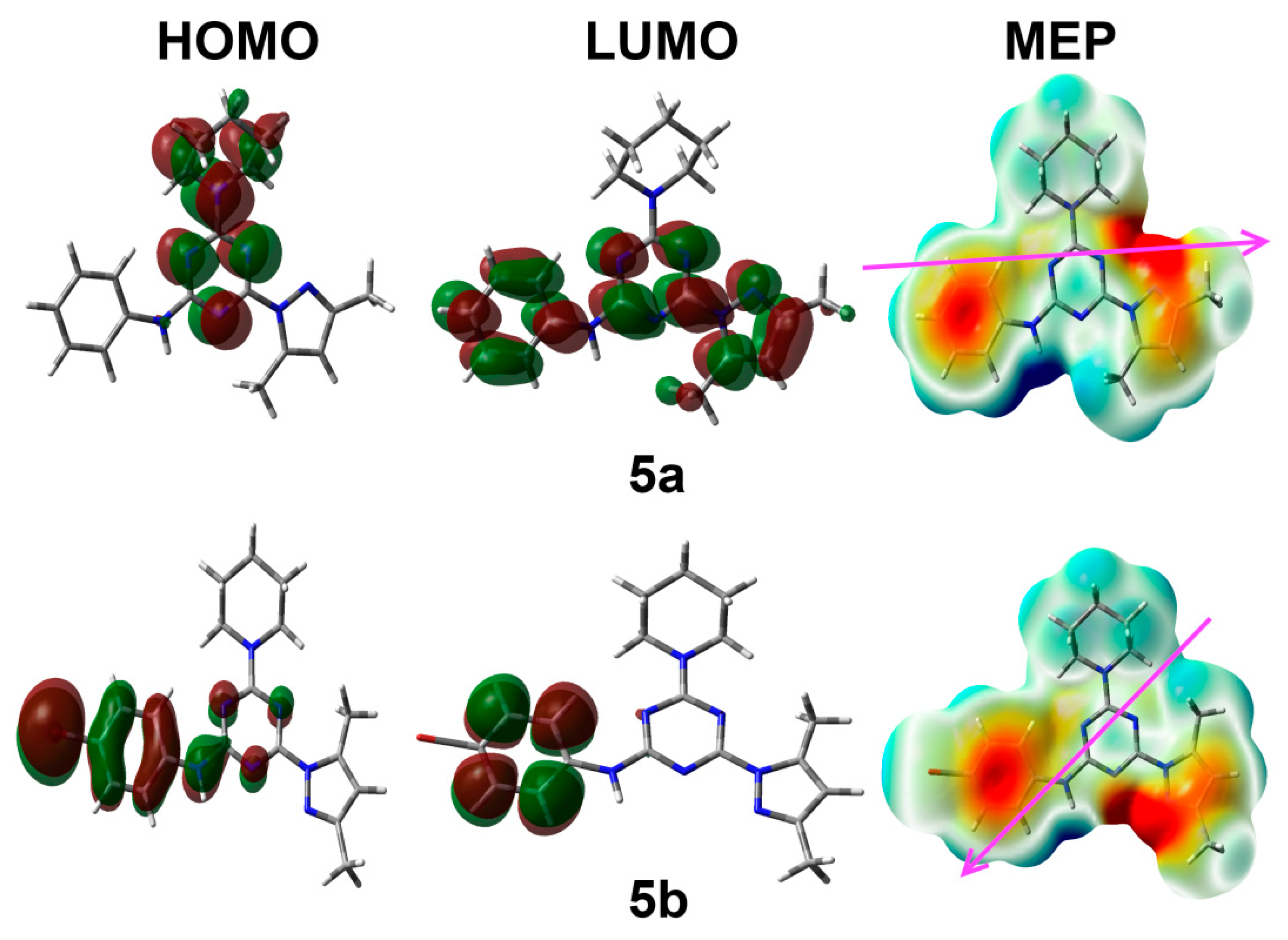
3.4. Electronic Reactivity Parameters
3.5. NBO Analysis
3.6. NMR Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barakat, A.; El-Faham, A.; Haukka, M.; Al-Majid, A.M.; Soliman, S.M. s-Triazine pincer ligands: Synthesis of their metal complexes, coordination behavior, and applications. Appl. Organomet. Chem. 2021, 35, e6317. [Google Scholar] [CrossRef]
- Dugan, J.; Pollyea, D. Enasidenib for the treatment of acute myeloid leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 755–760. [Google Scholar] [CrossRef]
- Kim, E.S. Enasidenib: First global approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Bhat, H.; Gahtori, P. Antifungal activity, SAR and physicochemical correlation of some thiazole-1, 3, 5-triazine derivatives. J. Mycol. Med. 2012, 22, 134–141. [Google Scholar] [CrossRef]
- Shanmugam, M.; Narayanan, K.; Chidambaranathan, V.; Kabilan, S. Synthesis, spectral characterization and antimicrobial studies of novel s-triazine derivatives. Spectrochim. Acta Mol. Biomol. Spectrosc. 2013, 105, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.R.; Modh, R.P.; Chikhalia, K.H. Privileged s-triazines: Structure and pharmacological applications. Future Med. Chem. 2014, 6, 463–477. [Google Scholar]
- Shanmugakala, R.; Tharmaraj, P.; Sheela, C.; Chidambaranathan, N. Transition metal complexes of s-triazine derivative: New class of anticonvulsant, antiinflammatory, and neuroprotective agents. Med. Chem. Res. 2014, 23, 329–342. [Google Scholar] [CrossRef]
- Menicagli, R.; Samaritani, S.; Signore, G.; Vaglini, F.; Dalla Via, L. In vitro cytotoxic activities of 2-Alkyl-4,6-diheteroalkyl-1,3,5-triazines: New molecules in anticancer research. J. Med. Chem. 2004, 47, 4649–4652. [Google Scholar] [CrossRef]
- Viira, B.; Selyutina, A.; García-Sosa, A.T.; Karonen, M.; Sinkkonen, J.; Merits, A.; Maran, U. Design, discovery, modelling, synthesis, and biological evaluation of novel and small, low toxicity s-triazine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. Biorg. Med. Chem. 2016, 24, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.-R.B.; Hafez, H.N. Synthesis of 4-substituted pyrido [2, 3-d] pyrimidin-4 (1H)-one as analgesic and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2009, 19, 3392–3397. [Google Scholar] [CrossRef]
- Behki, R.; Topp, E.; Dick, W.; Germon, P. Metabolism of the herbicide atrazine by Rhodococcus strains. Appl. Environ. Microbiol. 1993, 59, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ghabbour, H.; Khan, S.T.; Beatriz, G.; Albericio, F.; El-Faham, A. Novel pyrazolyl-s-triazine derivatives, molecular structure and antimicrobial activity. J. Mol. Struct. 2017, 1145, 244–253. [Google Scholar] [CrossRef]
- Mikhaylichenko, S.N.; Patel, S.M.; Dalili, S.; Chesnyuk, A.A.; Zaplishny, V.N. Synthesis and structure of new 1, 3, 5-triazine-pyrazole derivatives. Tetrahedron Lett. 2009, 50, 2505–2508. [Google Scholar] [CrossRef]
- Lasri, J.; Haukka, M.; Al-Rasheed, H.H.; Abutaha, N.; El-Faham, A.; Soliman, S.M. Synthesis, Structure and In Vitro Anticancer Activity of Pd(II) Complex of Pyrazolyl-s-Triazine Ligand; A New Example of Metal-Mediated Hydrolysis of s-Triazine Pincer Ligand. Crystals 2021, 11, 119. [Google Scholar] [CrossRef]
- Pathak, P.; Shukla, P.K.; Kumar, V.; Kumar, A.; Verma, A. Quinazoline clubbed 1,3,5-triazine derivatives as VEGFR2 kinase inhibitors: Design, synthesis, docking, in vitro cytotoxicity and in ovo antiangiogenic activity. Inflammopharmacology 2018, 26, 1441–1453. [Google Scholar] [CrossRef]
- Pathak, P.; Rimac, H.; Grishina, M.; Verma, A.; Potemkin, V. Hybrid Quinazoline 1, 3, 5-Triazines as Epidermal Growth Factor Receptor (EGFR) Inhibitors with Anticancer Activity: Design, Synthesis, and Computational Study. ChemMedChem 2021, 16, 822–838. [Google Scholar] [CrossRef]
- Osman, S.M.; Alasmary, F.A.; Kenawy, E.R.; Aly, E.S.A.; Khattab, S.N.; El-Faham, A. Synthesis, characterization and comparative thermal degradation kinetics of s-Triazine based polymers. J. Polym. Res. 2021, 28, 1–12. [Google Scholar] [CrossRef]
- Liao, J.H.; Lai, C.Y.; Yang, C.L. Synthesis, Characterization and Photoluminescence of Lanthanide Metal-organic Frameworks, Constructed from Triangular 4, 4′, 4 ″-s-triazine-1, 3, 5-triyl-p-aminobenzoate Ligands. J. Chin. Chem. Soc. 2014, 61, 1115–1120. [Google Scholar] [CrossRef]
- Gomes, R.; Bhaumik, A. A new triazine functionalized luminescent covalent organic framework for nitroaromatic sensing and CO2 storage. RSC Adv. 2016, 6, 28047–28054. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, J.; Lv, X. Novel hydrazine-bridged covalent triazine polymer for CO2 capture and catalytic conversion. Chinese J. Catal. 2018, 39, 1320–1328. [Google Scholar] [CrossRef]
- Wu, M.-X.; Yang, Y.-W. Applications of covalent organic frameworks (COFs): From gas storage and separation to drug delivery. Chin Chem Lett. 2017, 28, 1135–1143. [Google Scholar] [CrossRef]
- Barakat, A.; El-Senduny, F.F.; Almarhoon, Z.; Al-Rasheed, H.H.; Badria, F.A.; Al-Majid, A.M.; Ghabbour, H.A.; El-Faham, A. Synthesis, X-ray crystal structures, and preliminary antiproliferative activities of new s-triazine-hydroxybenzylidene hydrazone derivatives. J. Chem. 2019, 2019. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, S.M.; El-Faham, A.; Ali, M.; Al-Majid, A.M.; Yousuf, S.; Choudhary, M.I. Three multi-components reaction: Synthesis and X-ray single-crystal of hydroacridinone-based hydrazino-s-triazine derivative as a new class of urease inhibitor. Crystals. 2020, 10, 14. [Google Scholar] [CrossRef]
- Soliman, S.M.; Lasri, J.; Haukka, M.; Elmarghany, A.; Al-Majid, A.M.; El-Faham, A.; Barakat, A. Synthesis, X-ray structure, Hirshfeld analysis, and DFT studies of a new Pd (II) complex with an anionic s-triazine NNO donor ligand. J. Mol. Struct. 2020, 1217, 128463. [Google Scholar] [CrossRef]
- Farooq, M.; Sharma, A.; Almarhoon, Z.; Al-Dhfyan, A.; El-Faham, A.; Abu Taha, N.; Wadaan, M.A.M.; de la Torre, B.G.; Albericio, F. Design and synthesis of mono-and di-pyrazolyl-s-triazine derivatives, their anticancer profile in human cancer cell lines, and in vivo toxicity in zebrafish embryos. Bioorg. Chem. 2019, 87, 457–464. [Google Scholar] [CrossRef]
- Sanders, M.A.; Brahemi, G.; Nangia-Makker, P.; Balan, V.; Morelli, M.; Kothayer, H.; Westwell, A.D.; Shekhar, M.P. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: Design, synthesis, identification, and functional characterization. Mol. Cancer Ther. 2013, 12, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Balaha, M.F.; El-Hamamsy, M.H.; El-Din, N.A.; El-Mahdy, N.A. Synthesis, evaluation and docking study of 1, 3, 5-triazine derivatives as cytotoxic agents against lung cancer. J. Appl. Pharm. Sci. 2016, 6, 28–45. [Google Scholar] [CrossRef]
- Raghu, M.S.; Kumar, C.P.; Prashanth, M.K.; Kumar, K.Y.; Prathibha, B.S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1, 3, 5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. New. J. Chem. 2021, 45, 13909–13924. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Pillai, G.G.; Bhat, H.R.; Verma, A.; Singh, U.P. Design and discovery of novel monastrol-1, 3, 5-triazines as potent anti-breast cancer agent via attenuating epidermal growth factor receptor tyrosine kinase. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef]
- Rikagu Oxford Diffraction. CrysAlisPro; Agilent Technologies Inc.: Yarnton, UK, 2020. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. B. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17; University of Western Australia: 2017. Available online: http://hirshfeldsurface.net (accessed on 17 July 2019).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09; Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- GaussView; Version 4.1; Denningto, R., II, Keith, T., Millam, J., Eds.; Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New Model for Calculation of Solvation Free Energies: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A.; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Hubert Joe, I.; Kostova, I.; Ravikumar, C.; Amalanathan, M.; Pinzaru, S.C. Theoretical and vibrational spectral investigation of sodium salt of acenocoumarol. J. Raman Spectrosc. 2009, 40, 1033–1038. [Google Scholar]
- Sebastian, S.; Sundaraganesan, N.; Spectrochim. The spectroscopic (FT-IR, FT-IR gas phase, FT-Raman and UV) and NBO analysis of 4-Hydroxypiperidine by density functional method. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 941–952. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5a | 5b | |
|---|---|---|
| CCDC | 2113908 | 2113909 |
| empirical formula | C19H23N7 | C19H22BrN7 |
| fw | 349.44 | 428.34 |
| temp (K) | 120(2) | 120(2) |
| λ (Å) | 1.54184 | 1.54184 |
| cryst syst | Orthorhombic | Orthorhombic |
| space group | Pbca | Pbcn |
| a (Å) | 11.49940(10) | 37.4570(3) |
| b (Å) | 14.91530(10) | 8.62930(10) |
| c (Å) | 21.10870(10) | 24.8053(2) |
| V (Å3) | 3620.50(4) | 8017.76(13) |
| Z | 8 | 16 |
| ρcalc (Mg/m3) | 1.282 | 1.419 |
| μ (Mo Kα) (mm−1) | 0.648 | 2.940 |
| No. reflns. | 56200 | 63091 |
| Unique reflns. | 3819 | 8430 |
| Completeness to θ = 67.684° | %100 | %100 |
| GOOF (F2) | 1.028 | 1.038 |
| Rint | 0.0304 | 0.0301 |
| R1a (I ≥ 2σ) | 0.0347 | 0.0321 |
| wR2b (I ≥ 2σ) | 0.0893 | 0.0822 |
| Atoms | Distance | Atoms | Distance |
|---|---|---|---|
| 5a | 5b | ||
| N1-C7 | 1.3611(12) | Br1-C3 | 1.9072(17) |
| N1-C6 | 1.4072(13) | N1-C7 | 1.355(2) |
| N2-C8 | 1.3230(13) | N1-C6 | 1.411(2) |
| N2-C7 | 1.3508(13) | N2-C7 | 1.328(2) |
| N3-C8 | 1.3242(13) | N2-C8 | 1.348(2) |
| N3-C9 | 1.3552(13) | N3-C8 | 1.341(2) |
| N4-C7 | 1.3310(12) | N3-C13 | 1.464(2) |
| N4-C9 | 1.3551(12) | N3-C9 | 1.464(2) |
| N5-N6 | 1.3770(11) | N4-C14 | 1.324(2) |
| N5-C10 | 1.3841(12) | N4-C8 | 1.360(2) |
| N5-C8 | 1.4082(12) | N5-C14 | 1.328(2) |
| N6-C13 | 1.3218(13) | N5-C7 | 1.360(2) |
| N7-C9 | 1.3483(13) | N6-N7 | 1.3772(19) |
| N7-C19 | 1.4653(13) | N6-C15 | 1.388(2) |
| N7-C15 | 1.4660(13) | N6-C14 | 1.404(2) |
| Atoms | Angle | Atoms | Angle |
| C7-N1-C6 | 130.29(8) | C7-N1-C6 | 127.70(14) |
| C8-N2-C7 | 113.12(8) | C7-N2-C8 | 114.69(14) |
| C8-N3-C9 | 112.94(8) | C8-N3-C13 | 122.58(14) |
| C7-N4-C9 | 114.25(8) | C8-N3-C9 | 122.28(14) |
| N6-N5-C10 | 111.63(8) | C13-N3-C9 | 114.33(13) |
| N6-N5-C8 | 119.98(8) | C14-N4-C8 | 113.38(14) |
| C10-N5-C8 | 128.25(8) | C14-N5-C7 | 112.51(13) |
| C13-N6-N5 | 104.77(8) | N7-N6-C15 | 111.75(13) |
| C9-N7-C19 | 122.30(8) | N7-N6-C14 | 118.27(13) |
| C9-N7-C15 | 121.50(8) | C15-N6-C14 | 129.96(14) |
| C19-N7-C15 | 115.29(8) | C18-N7-N6 | 104.93(13) |
| D-H…A | d(D-H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| 5a | ||||
| N1-H1…N6#1 | 0.894(15) | 2.219(15) | 3.0995(12) | 167.9(13) |
| C5-H5…N4 | 0.95 | 2.33 | 2.9366(13) | 121.3 |
| #1 x − 1/2,y, −z + 3/2 | ||||
| 5b | ||||
| C5-H5…N2 | 0.95 | 2.39 | 2.887(2) | 111.9 |
| C5b-H5b…N2B | 0.95 | 2.25 | 2.870(2) | 121.9 |
| C9B-H9BA…N6B#1 | 0.99 | 2.59 | 3.355(2) | 134.4 |
| C9B-H9BA…N7B#1 | 0.99 | 2.54 | 3.501(2) | 165 |
| C10B-H10C…Br1B#2 | 0.99 | 2.97 | 3.7507(17) | 136.7 |
| N1B-H1B…N5 | 0.85(2) | 2.21(2) | 3.0621(19) | 176(2) |
| N1-H1…N4B | 0.92(2) | 2.25(2) | 3.1452(18) | 165(2) |
| N1-H1…N7B | 0.92(2) | 2.43(2) | 3.040(2) | 123.9(19) |
| #1 −x + 1, −y + 1, −z + 1 #2 −x + 1, y, −z + 1/2 | ||||
| Contact | Distance | Contact | Distance | Contact | Distance |
|---|---|---|---|---|---|
| 5a | 5b | ||||
| N6…H1 | 2.108 | H13A…C15 | 2.763 | N7…H1B | 2.627 |
| N4…H12 | 2.476 | H9B…C14 | 2.715 | N6B…H9BA | 2.523 |
| N2…H15b | 2.492 | H1AB…C14 | 2.740 | N7B…H9BA | 2.446 |
| H4…C12 | 2.687 | H12B…C8 | 2.632 | N4B…H13D | 2.561 |
| N7B…H1 | 2.380 | H4B…H4B | 1.795 | ||
| N4B…H1 | 2.162 | Br1B…H10C | 2.902 | ||
| N5…H1B | 2.055 | ||||
| Parameter | 5a | 5b |
|---|---|---|
| EHOMO | −5.6856 | −5.7166 |
| ELUMO | −0.7690 | −0.9129 |
| I | 5.6856 | 5.7166 |
| A | 0.7690 | 0.9129 |
| η | 4.9166 | 4.8037 |
| μ | −3.2273 | −3.3148 |
| ω | 1.0592 | 1.1437 |
| Donor NBO | Acceptor NBO | E(2) | Donor NBO | Acceptor NBO | E(2) |
|---|---|---|---|---|---|
| 5a | 5b | ||||
| σ→σ* | |||||
| BD(1)N3-C20 | BD*(1)N6-C21 | 5.72 | BD(1)N3-C19 | BD*(1)N4-C20 | 4.75 |
| BD(1)N4-C21 | BD*(1)N8-C22 | 4.60 | BD(1)N3-C20 | BD*(1)N2-C19 | 5.55 |
| BD(1)N5-C20 | BD*(1)N8-C22 | 4.71 | BD(1)N5-C20 | BD*(1)N7-C36 | 5.24 |
| BD(1)N5-C22 | BD*(1)N1-C20 | 5.68 | BD(1)N5-C36 | BD*(1)N4-C20 | 4.75 |
| BD(1)N6-N7 | BD*(1)C30-C31 | 4.93 | BD(1)N6-C19 | BD*(1)N7-C36 | 6.05 |
| BD(1)C15-C17 | BD*(1)N1-C19 | 4.76 | BD(1)N6-C36 | BD*(1)N2-C19 | 4.37 |
| BD(1)C23-C28 | BD*(1)N6-C21 | 4.89 | BD(1)N7-N8 | BD*(1)C44-C45 | 4.93 |
| BD(1)C23-C28 | BD*(1)C30-C31 | 5.08 | BD(1)C9-C11 | BD*(1)Br1-C13 | 4.83 |
| BD(1)C28-C30 | BD*(1)C23-C24 | 6.37 | BD(1)C14-C16 | BD*(1)Br1-C13 | 4.87 |
| BD(1)C14-C16 | BD*(1)N2-C18 | 4.49 | |||
| BD(1)C37-C42 | BD*(1)C44-C45 | 5.06 | |||
| BD(1)C42-C44 | BD*(1)C37-C38 | 6.39 | |||
| π→π* | |||||
| BD(2)N3-C20 | BD*(2)N4-C21 | 45.28 | BD(2)N3-C19 | BD*(2)N5-C20 | 38.69 |
| BD(2)N3-C20 | BD*(2)N5-C22 | 4.65 | BD(2)N3-C19 | BD*(2)N6-C36 | 4.66 |
| BD(2)N4-C21 | BD*(2)N3-C20 | 4.93 | BD(2)N5-C20 | BD*(2)N3-C19 | 4.60 |
| BD(2)N4-C21 | BD*(2)N5-C22 | 38.58 | BD(2)N5-C20 | BD*(2)N6-C36 | 44.78 |
| BD(2)N4-C21 | BD*(1)N6-C21 | 5.66 | BD(2)N6-C36 | BD*(2)N3-C19 | 38.96 |
| BD(2)N5-C22 | BD*(2)N3-C20 | 47.40 | BD(2)N6-C36 | BD*(2)N5-C20 | 5.07 |
| BD(2)N5-C22 | BD*(2)N4-C21 | 4.88 | BD(2)N8-C44 | BD*(2)C37-C42 | 11.51 |
| BD(2)N7-C30 | BD*(2)C23-C28 | 11.48 | BD(2)C9-C11 | BD*(2)C13-C14 | 17.56 |
| BD(2)C9-C11 | BD*(2)C13-C15 | 17.18 | BD(2)C9-C11 | BD*(2)C16-C18 | 19.86 |
| BD(2)C9-C11 | BD*(2)C17-C19 | 20.4 | BD(2)C13-C14 | BD*(2)C9-C11 | 21.19 |
| BD(2)C13-C15 | BD*(2)C9-C11 | 22.41 | BD(2)C13-C14 | BD*(2)C16-C18 | 16.81 |
| BD(2)C13-C15 | BD*(2)C17-C19 | 18.66 | BD(2)C16-C18 | BD*(2)C9-C11 | 18.55 |
| BD(2)C17-C19 | BD*(2)C9-C11 | 18.26 | BD(2)C16-C18 | BD*(2)C13-C14 | 23.75 |
| BD(2)C17-C19 | BD*(2)C13-C15 | 21.9 | BD(2)C37-C42 | BD*(2)N8-C44 | 27.09 |
| BD(2)C23-C28 | BD*(2)N7-C30 | 27.29 | |||
| n→π* | |||||
| LP(1)N1 | BD*(2)N3-C20 | 59.13 | LP(1)N4 | BD*(2)N5-C20 | 70.72 |
| LP(1)N1 | BD*(2)C17-C19 | 36.38 | LP(1)N7 | BD*(2)N6-C36 | 51.52 |
| LP(1)N6 | BD*(2)N4-C21 | 51.37 | LP(1)N7 | BD*(2)N8-C44 | 23.23 |
| LP(1)N6 | BD*(2)N7-C30 | 23.44 | LP(1)N7 | BD*(2)C37-C42 | 34.67 |
| LP(1)N6 | BD*(2)C23-C28 | 34.89 | |||
| LP(1)N8 | BD*(2)N5-C22 | 72.58 | |||
| n→σ* | |||||
| LP(1)N3 | BD*(1)N4-C21 | 12.25 | LP(1)N3 | BD*(1)N5-C20 | 12.07 |
| LP(1)N3 | BD*(1)N5-C20 | 12.16 | LP(1)N3 | BD*(1)N6-C19 | 12.90 |
| LP(1)N4 | BD*(1)N3-C21 | 12.99 | LP(1)N4 | BD*(1)C21-H23 | 4.79 |
| LP(1)N4 | BD*(1)N5-C22 | 12.74 | LP(1)N5 | BD*(1)N3-C20 | 12.09 |
| LP(1)N5 | BD*(1)N3-C20 | 13.20 | LP(1)N5 | BD*(1)N6-C36 | 12.63 |
| LP(1)N5 | BD*(1)N4-C22 | 11.88 | LP(1)N6 | BD*(1)N3-C19 | 12.99 |
| LP(1)N7 | BD*(1)N6-C23 | 6.89 | LP(1)N6 | BD*(1)N5-C36 | 12.60 |
| LP(1)N7 | BD*(1)C28-C30 | 6.08 | LP(1)N8 | BD*(1)N7-C37 | 6.87 |
| LP(1)N8 | BD*(1)C44-C47 | 4.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shawish, I.; Soliman, S.M.; Haukka, M.; Aldalbahi, A.; Barakat, A.; El-Faham, A. Synthesis, and Molecular Structure Investigations of a New s-Triazine Derivatives Incorporating Pyrazole/Piperidine/Aniline Moieties. Crystals 2021, 11, 1500. https://doi.org/10.3390/cryst11121500
Shawish I, Soliman SM, Haukka M, Aldalbahi A, Barakat A, El-Faham A. Synthesis, and Molecular Structure Investigations of a New s-Triazine Derivatives Incorporating Pyrazole/Piperidine/Aniline Moieties. Crystals. 2021; 11(12):1500. https://doi.org/10.3390/cryst11121500
Chicago/Turabian StyleShawish, Ihab, Saied M. Soliman, Matti Haukka, Ali Aldalbahi, Assem Barakat, and Ayman El-Faham. 2021. "Synthesis, and Molecular Structure Investigations of a New s-Triazine Derivatives Incorporating Pyrazole/Piperidine/Aniline Moieties" Crystals 11, no. 12: 1500. https://doi.org/10.3390/cryst11121500
APA StyleShawish, I., Soliman, S. M., Haukka, M., Aldalbahi, A., Barakat, A., & El-Faham, A. (2021). Synthesis, and Molecular Structure Investigations of a New s-Triazine Derivatives Incorporating Pyrazole/Piperidine/Aniline Moieties. Crystals, 11(12), 1500. https://doi.org/10.3390/cryst11121500