
Theoretical Prediction of CHn Crystal Structures under High Pressures

Abstract
:1. Introduction
2. Materials and Methods
3. Results
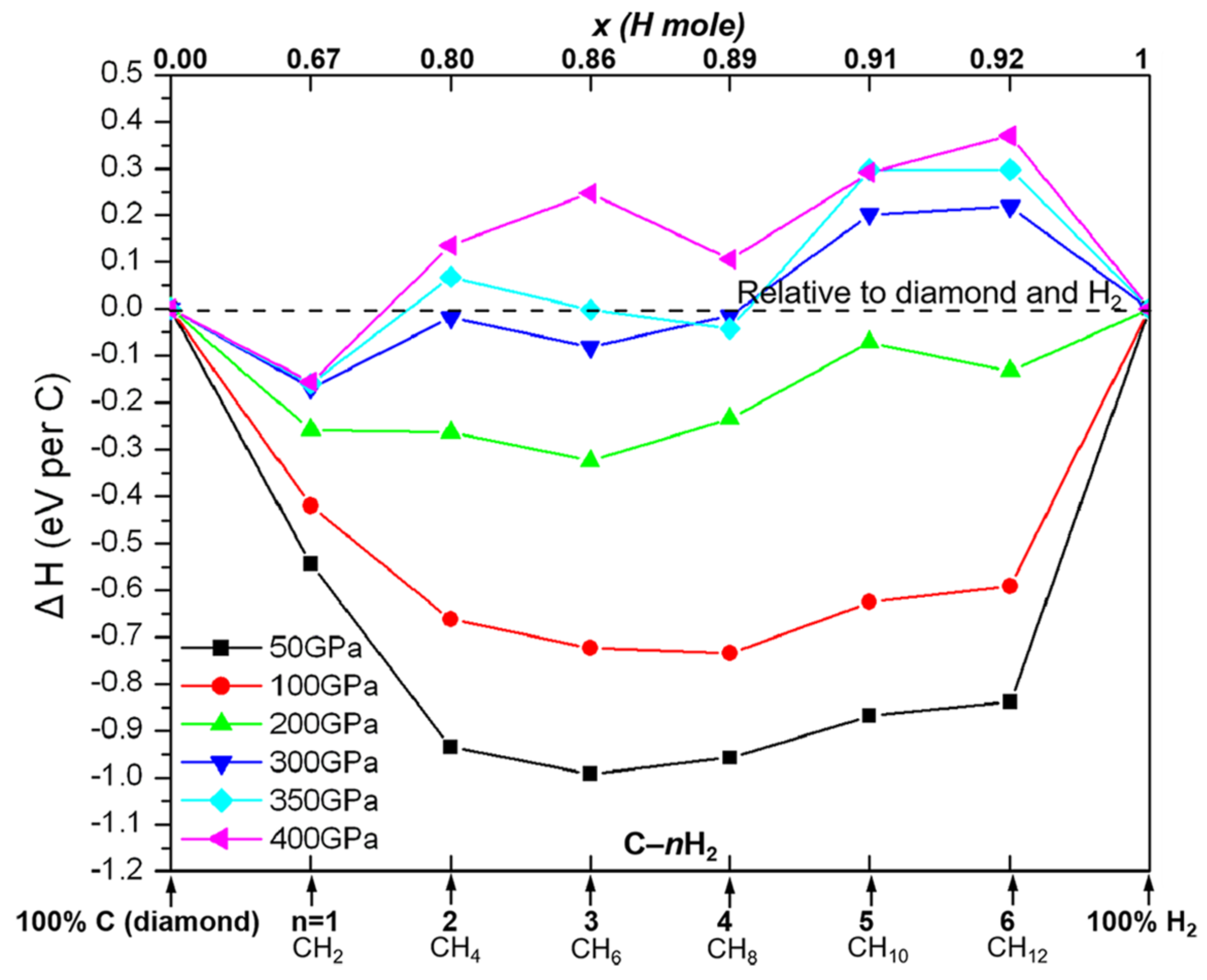
3.1. The Tie Line of CHn Systems
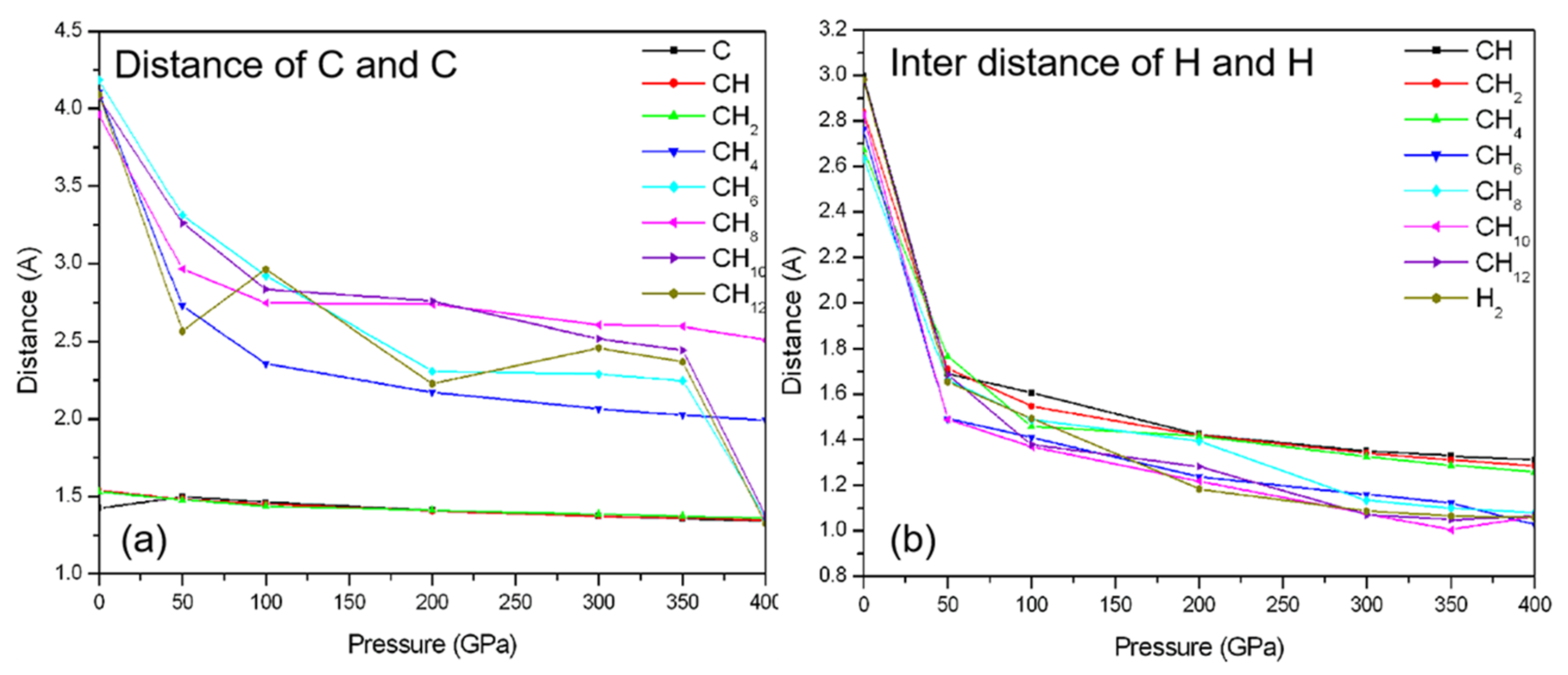
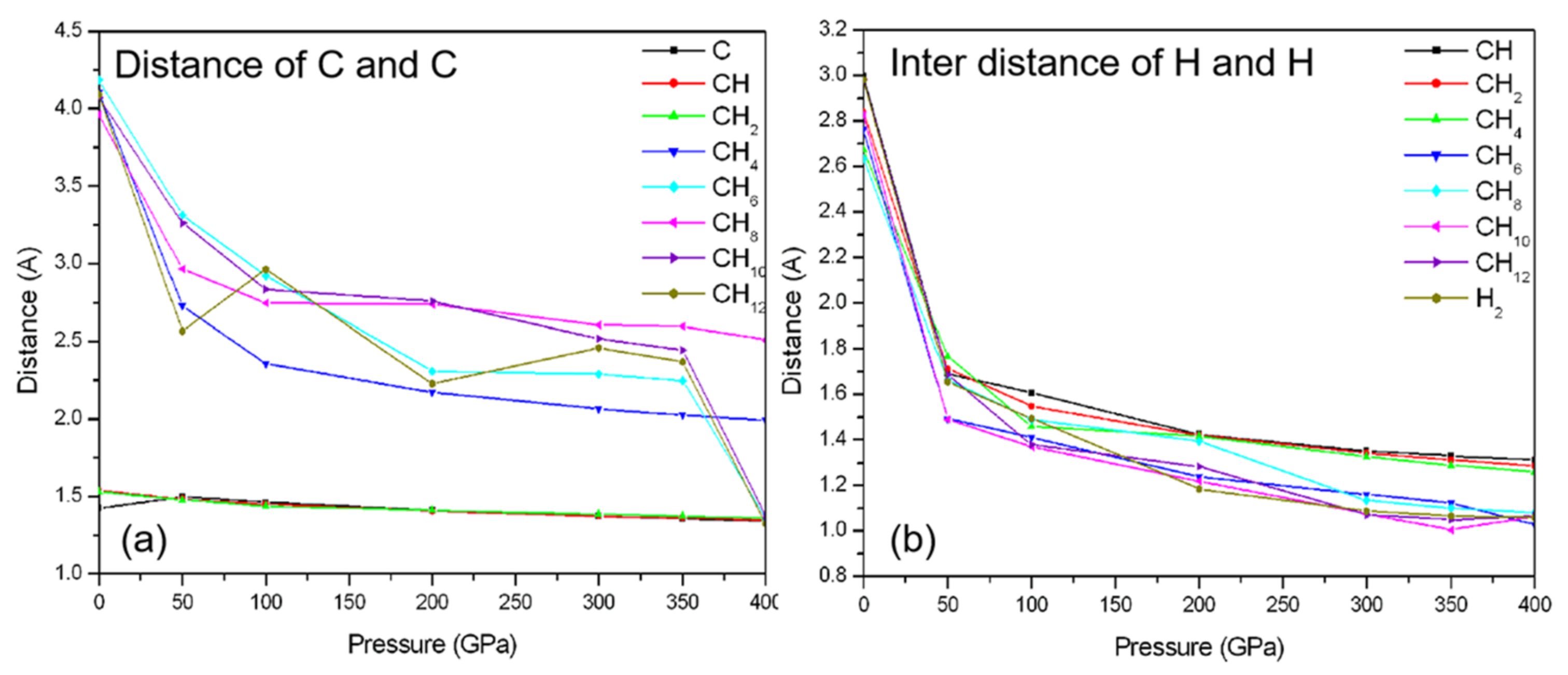
3.2. Compression of CHn
3.3. Electronic Properties of CHn Phase
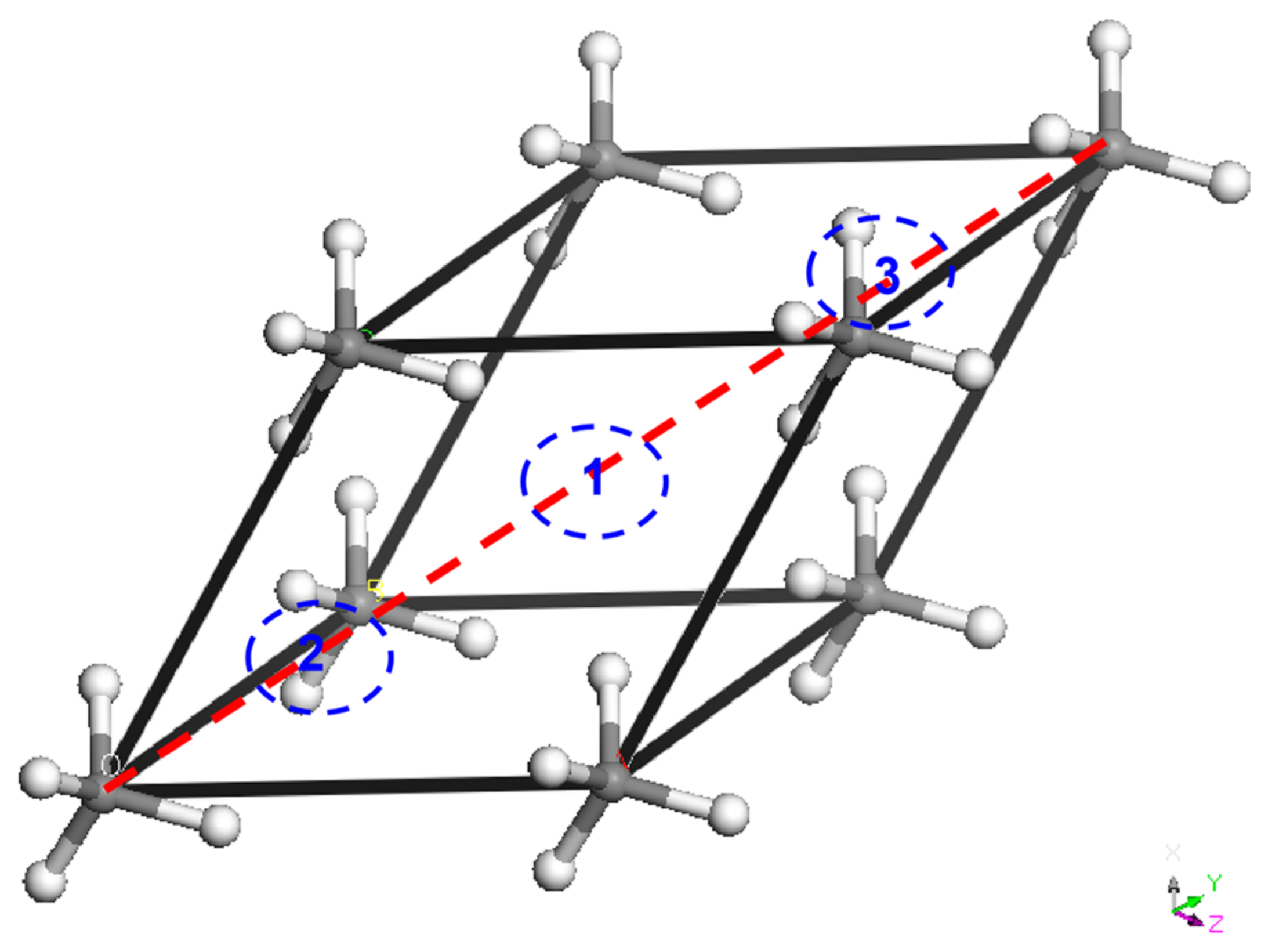
3.4. The Rotational Solid for CHn
3.5. Other Possible Structures for CHn Systems
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashcroft, N.W. Hydrogen dominant metallic alloys: High temperature superconductors? Phys. Rev. Lett. 2004, 92, 187002. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mao, H.K.; Chen, X.J.; Mao, W.L. High pressure chemistry in the H2-SiH4 system. Proc. Natl. Acad. Sci. USA 2009, 106, 14763–14767. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.J.; Struzhkin, V.V.; Song, Y.; Goncharov, A.F.; Ahart, M.; Liu, Z.; Mao, H.K.; Hemley, R.J. Pressure-induced metallization of silane. Proc. Natl. Acad. Sci. USA 2008, 105, 20–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, T.A.; Somayazulu, M.; Hemley, R.J. Novel pressure-induced interactions in silane-hydrogen. Phys. Rev. Lett. 2009, 103, 65701. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Grochala, W.; Jaroń, T.; Hoffmann, R.; Bergara, A.; Ashcroft, N.W. Erratum: Structures and potential superconductivity in SiH4 at high pressure: En route to “metallic hydrogen” (Physical Review Letters (2006) 96 (017006)). Phys. Rev. Lett. 2006, 97, 17006. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Oganov, A.R.; Li, P.; Li, Z.; Wang, H.; Cui, T.; Ma, Y.; Bergara, A.; Lyakhov, A.O.; Iitaka, T.; et al. High-pressure crystal structures and superconductivity of Stannane (SnH4). Proc. Natl. Acad. Sci. USA 2010, 107, 1317–1320. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Oganov, A.R.; Bergara, A.; Martinez-Canales, M.; Cui, T.; Iitaka, T.; Ma, Y.; Zou, G. Superconducting high pressure phase of germane. Phys. Rev. Lett. 2008, 101, 107002. [Google Scholar] [CrossRef] [Green Version]
- Tse, J.S.; Yao, Y.; Tanaka, K. Novel superconductivity in metallic SnH4 under high pressure. Phys. Rev. Lett. 2007, 98, 117004. [Google Scholar] [CrossRef]
- Wen, X.D.; Cahill, T.J.; Hoffmann, R. Exploring group 14 structures: 1D to 2D to 3D. Chem.—A Eur. J. 2010, 16, 6555–6566. [Google Scholar] [CrossRef] [PubMed]
- Mujica, A.; Rubio, A.; Muñoz, A.; Needs, R.J. High-pressure phases of group-IV, III–V, and II–VI compounds. Rev. Mod. Phys. 2003, 75, 863–912. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Canales, M.; Bergara, A. No evidence of metallic methane at high pressure. High Press. Res. 2006, 26, 369–375. [Google Scholar] [CrossRef]
- Somayazulu, M.S.; Finger, L.W.; Hemley, R.J.; Mao, H.K. High-pressure compounds in methane-hydrogen mixtures. Science 1996, 271, 1400–1402. [Google Scholar] [CrossRef]
- Liu, H.; Naumov, I.I.; Hemley, R.J. Dense Hydrocarbon Structures at Megabar Pressures. J. Phys. Chem. Lett. 2016, 7, 4218–4222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Perdew, Burke, and Ernzerhof Reply. Phys. Rev. Lett. 1998, 80, 891. [Google Scholar] [CrossRef]
- Kresse, G. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B—Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W.; Ono, S. High-pressure phases of CaCO3: Crystal structure prediction and experiment. Earth Planet. Sci. Lett. 2006, 241, 95–103. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX-Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Feng, J.; Hennig, R.G.; Ashcroft, N.W.; Hoffmann, R. Emergent reduction of electronic state dimensionality in dense ordered Li-Be alloys. Nature 2008, 451, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Zurek, E.; Hoffmann, R.; Ashcroft, N.W.; Oganov, A.R.; Lyakhov, A.O. A little bit of lithium does a lot for hydrogen. Proc. Natl. Acad. Sci. USA 2009, 106, 17640–17643. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, N.W. Quantum-solid behavior and the electronic structure of the light alkali metals. Phys. Rev. B 1989, 39, 10552–10559. [Google Scholar] [CrossRef] [PubMed]
- Hanfland, M.; Syassen, K.; Christensen, N.E.; Novikov, D.L. New high-pressure phases of lithium. Nature 2000, 408, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Ashcroft, N.W. Structure and bandgap closure in dense hydrogen. Nature 2000, 403, 632–635. [Google Scholar] [CrossRef]
- Hüller, A.; Prager, M.; Press, W.; Seydel, T. Phase III of solid methane: The orientational potential and rotational tunneling. J. Chem. Phys. 2008, 128, 34503. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.D.; Yannoni, C.S.; Dorn, H.C.; Salem, J.R.; Bethune, D.S. C60 rotation in the solid state: Dynamics of a faceted spherical top. Science 1992, 255, 1235–1238. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2 Rotation (eV per H2) | CH4 Rotation (eV per CH4) | |||
|---|---|---|---|---|
| 100 GPa | 300 GPa | 100 GPa | 300 GPa | |
| H2 | 0.014 | 0.04 | – | – |
| CH4 | – | – | 0.70 | 1.70 |
| CH4–H2 by USPEX | 0.10 | 0.27 | 0.75 | 2.00 |
| CH4–2H2 by USPEX | 0.04 | 0.14 | 0.45 | 1.60 |
| CH4–3H2 by USPEX | 0.027 | 0.13 | 1.10 | 1.80 |
| CH4–4H2 by USPEX | 0.012 | 0.09 | 0.60 | 2.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Liu, J.; Liu, X.; Liu, X.; Li, N. Theoretical Prediction of CHn Crystal Structures under High Pressures. Crystals 2021, 11, 1499. https://doi.org/10.3390/cryst11121499
Yang T, Liu J, Liu X, Liu X, Li N. Theoretical Prediction of CHn Crystal Structures under High Pressures. Crystals. 2021; 11(12):1499. https://doi.org/10.3390/cryst11121499
Chicago/Turabian StyleYang, Tao, Jinjia Liu, Xiaotong Liu, Xiulei Liu, and Ning Li. 2021. "Theoretical Prediction of CHn Crystal Structures under High Pressures" Crystals 11, no. 12: 1499. https://doi.org/10.3390/cryst11121499
APA StyleYang, T., Liu, J., Liu, X., Liu, X., & Li, N. (2021). Theoretical Prediction of CHn Crystal Structures under High Pressures. Crystals, 11(12), 1499. https://doi.org/10.3390/cryst11121499