
The Photovoltaic Performance of Sensitizers for Organic Solar Cells Containing Fluorinated Chalcones with Different Halogen Substituents

 and
and
Abstract
:
1. Introduction
2. Methodology
2.1. Synthesis of Halogenated Chalcone Derivatives
2.2. Spectroscopy Analysis
2.3. X-ray Crystallography Analysis
2.4. Hirshfeld Surface Analysis
2.5. Computational Analysis
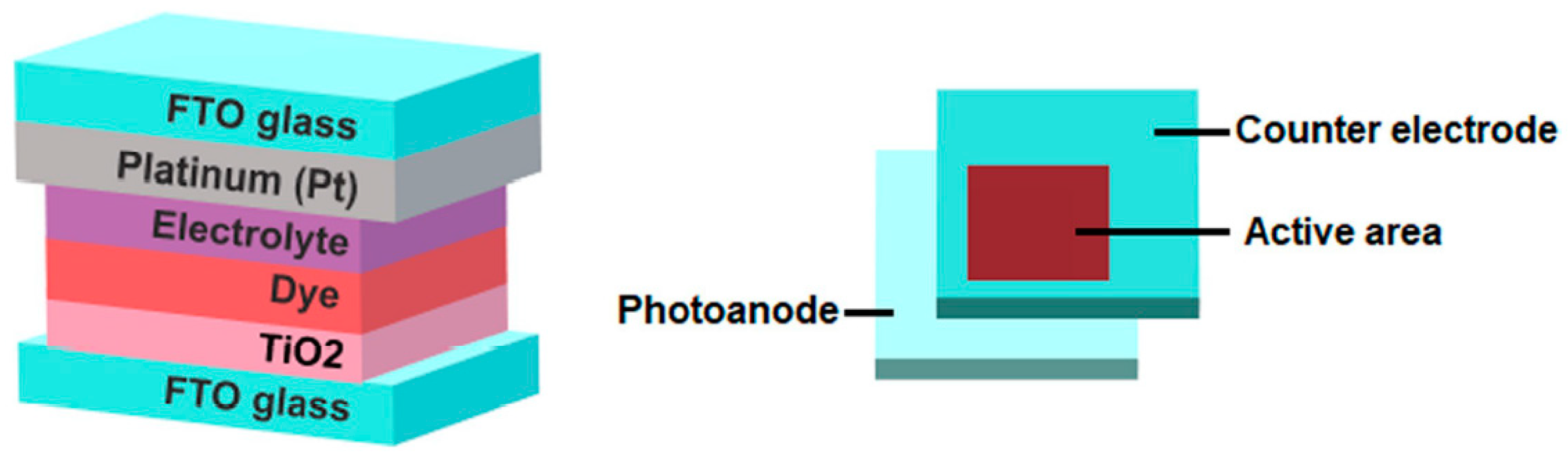
2.6. Dye-Synthesized Solar Cell (DSSC) Fabrication
2.6.1. Preparation of Halogenated Chalcones as Organic Dye Sensitizers
2.6.2. Preparation of Ruthenium Standard Dye as a Reference
2.6.3. Preparation of DSSC Fabrication
3. Results and Discussion
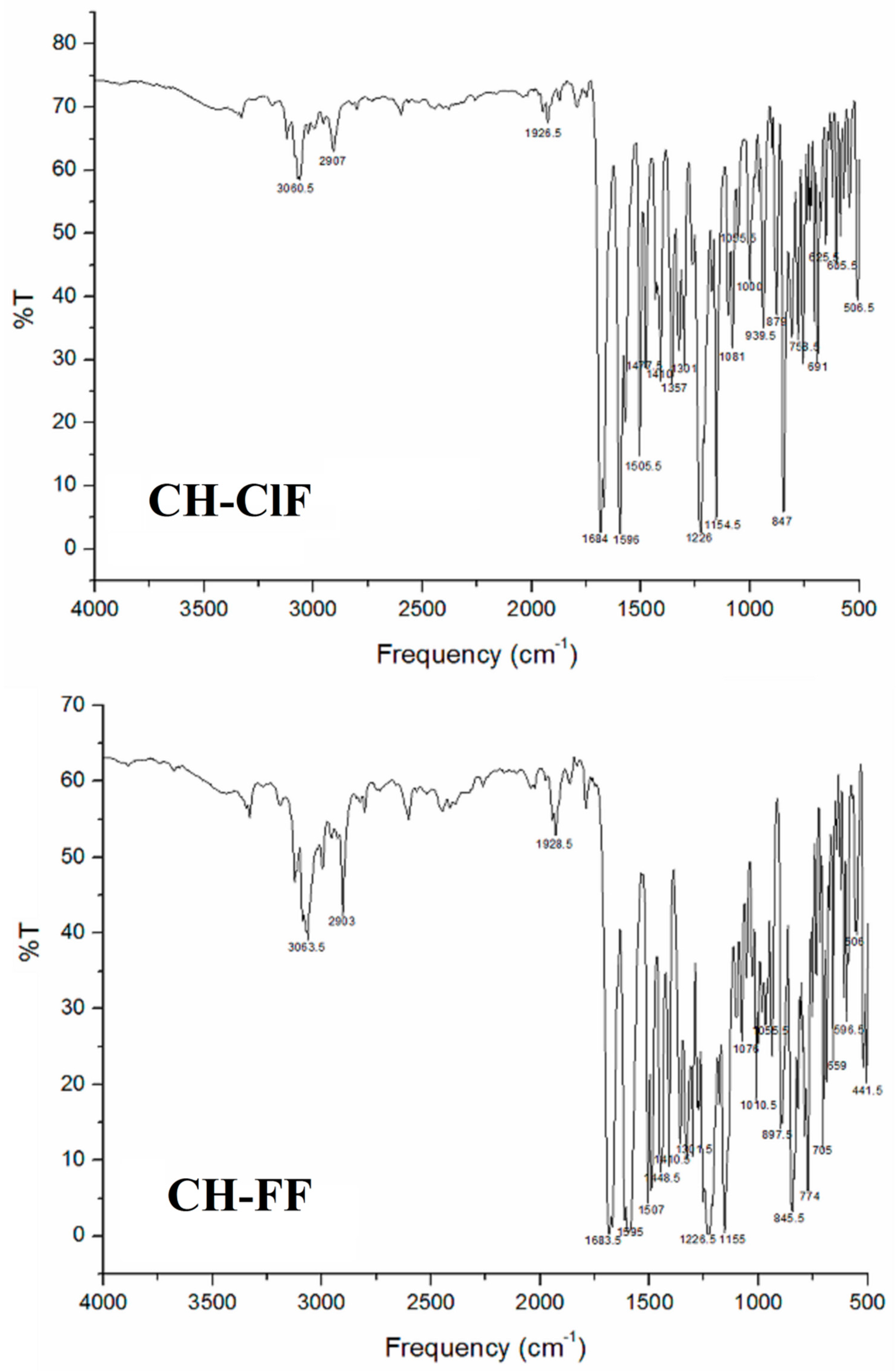
3.1. Fourier Transform Infrared Spectroscopy (FTIR) Studies
3.1.1. C–H Vibrations
3.1.2. C = O Vibrations
3.1.3. C = C and C—C Vibrations
3.1.4. C—X Vibrations
3.2. Nuclear Magnetic Resonance (NMR) Analysis
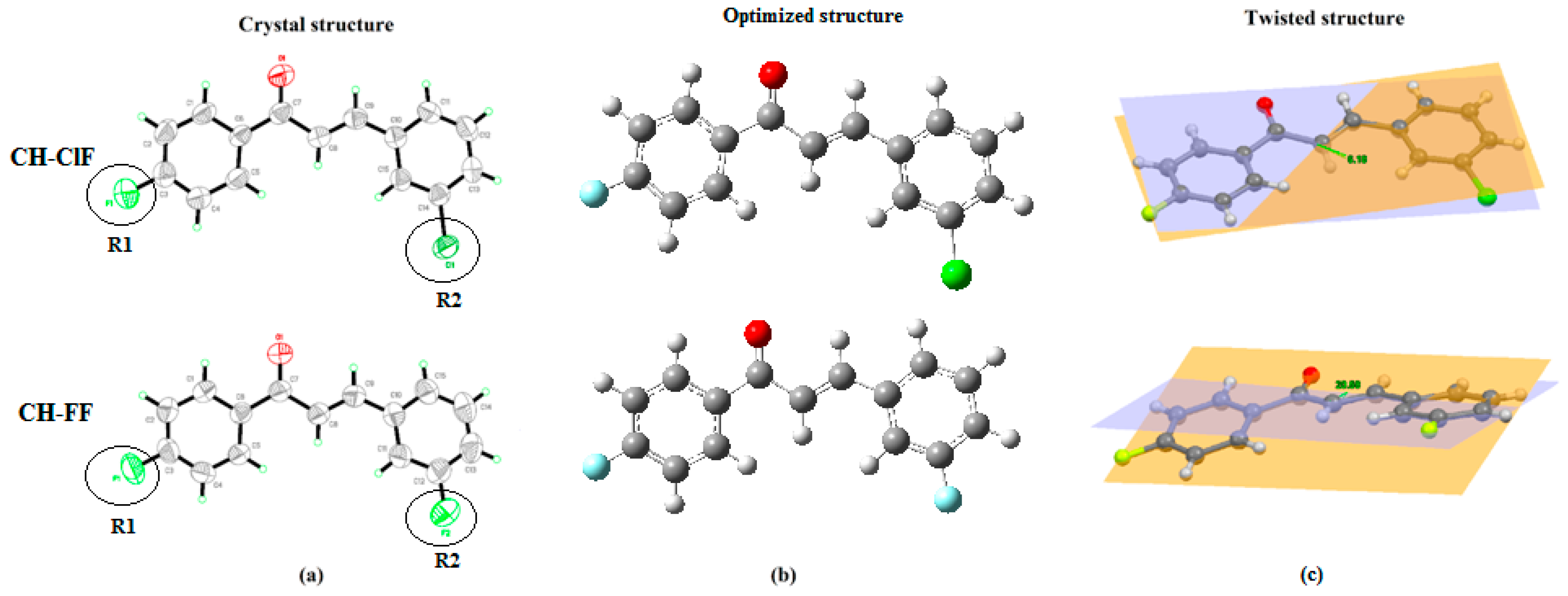
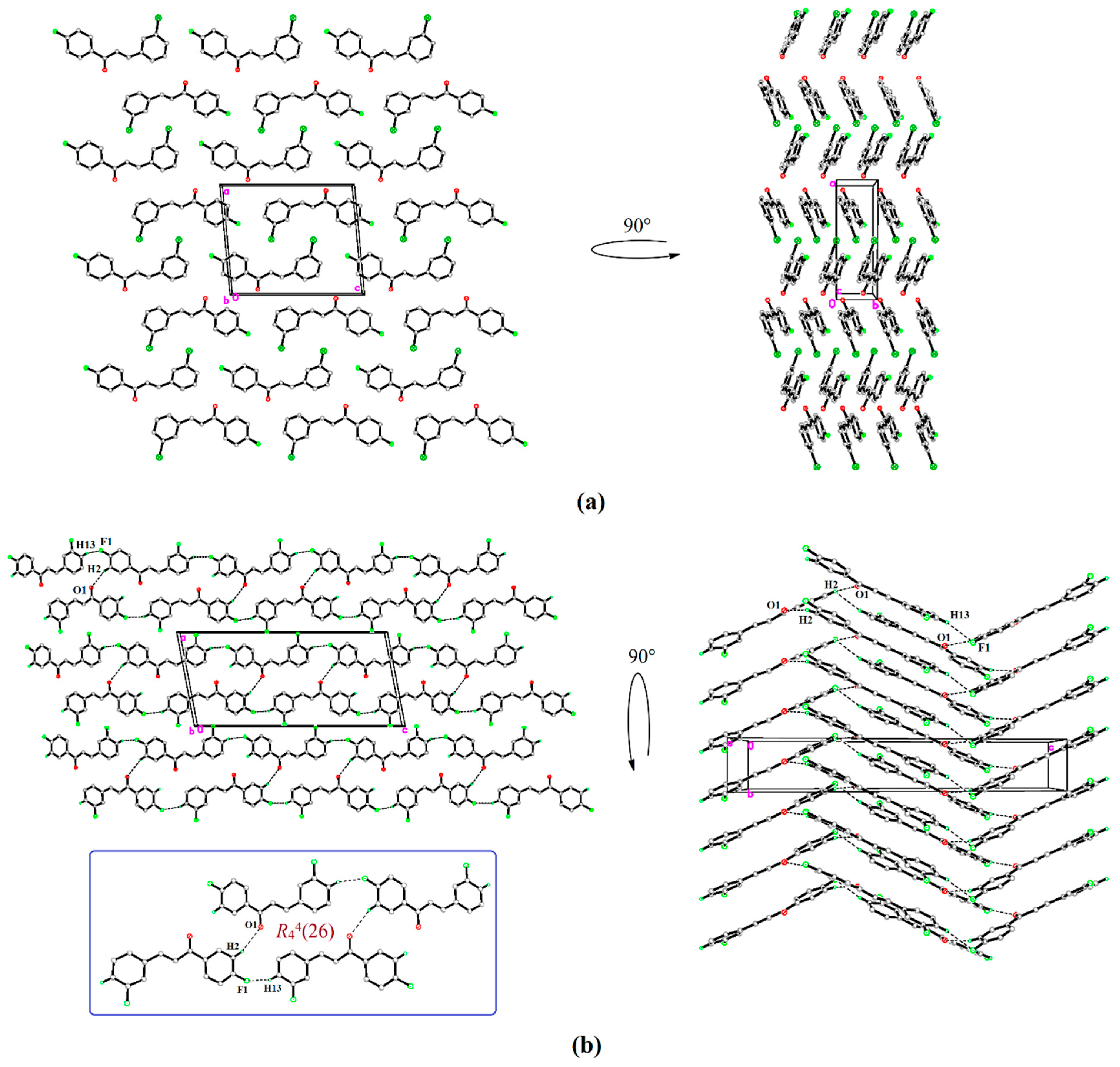
3.3. Molecular and Crystal Structural Analysis
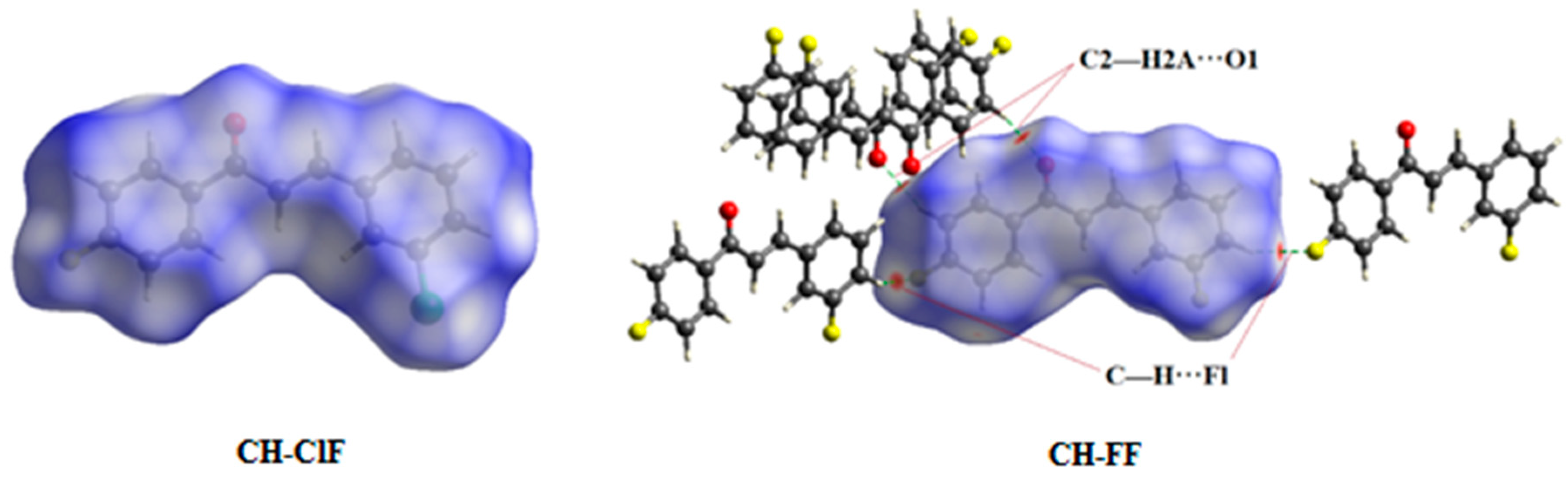
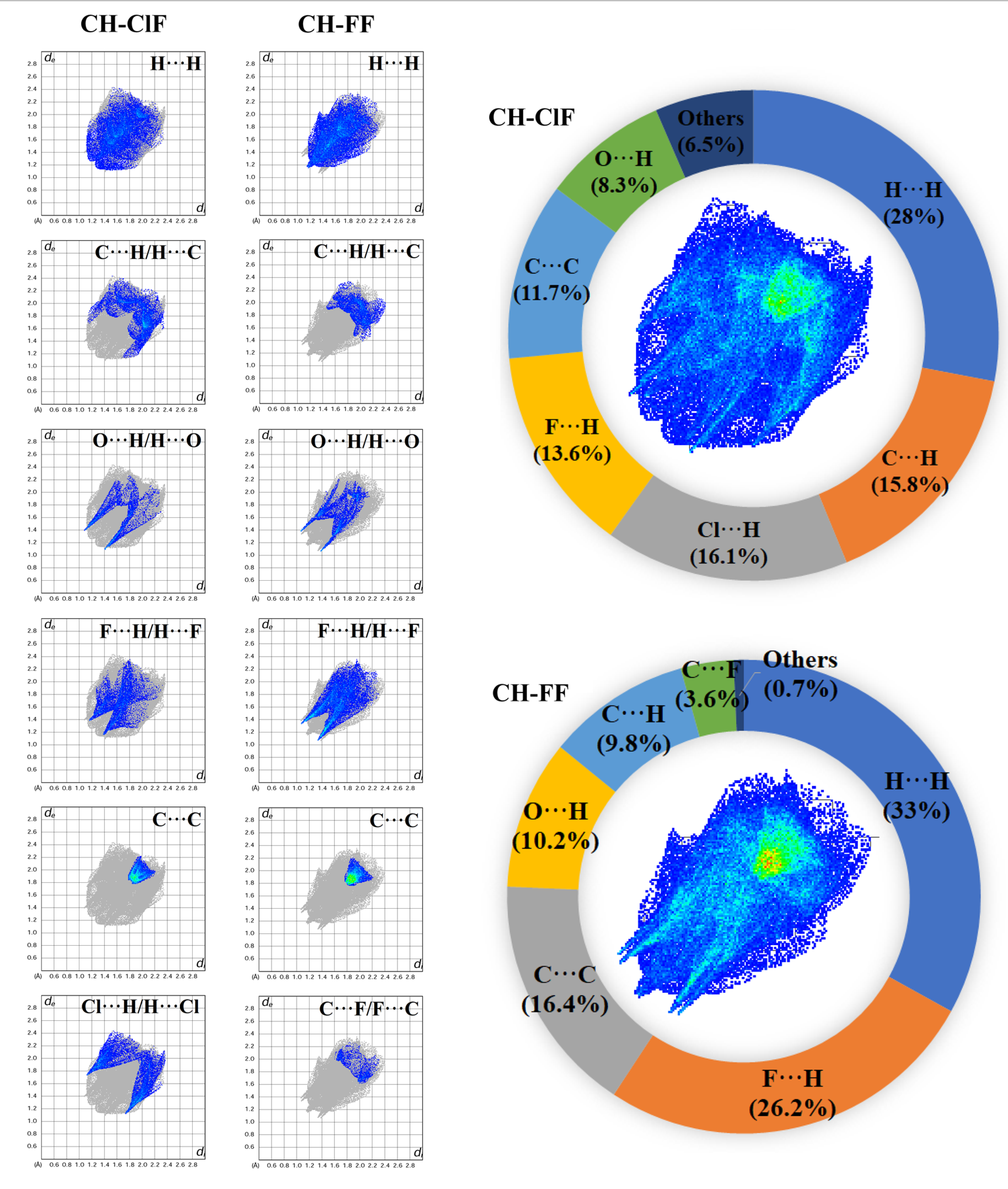
3.4. Hirshfeld Surface Calculations
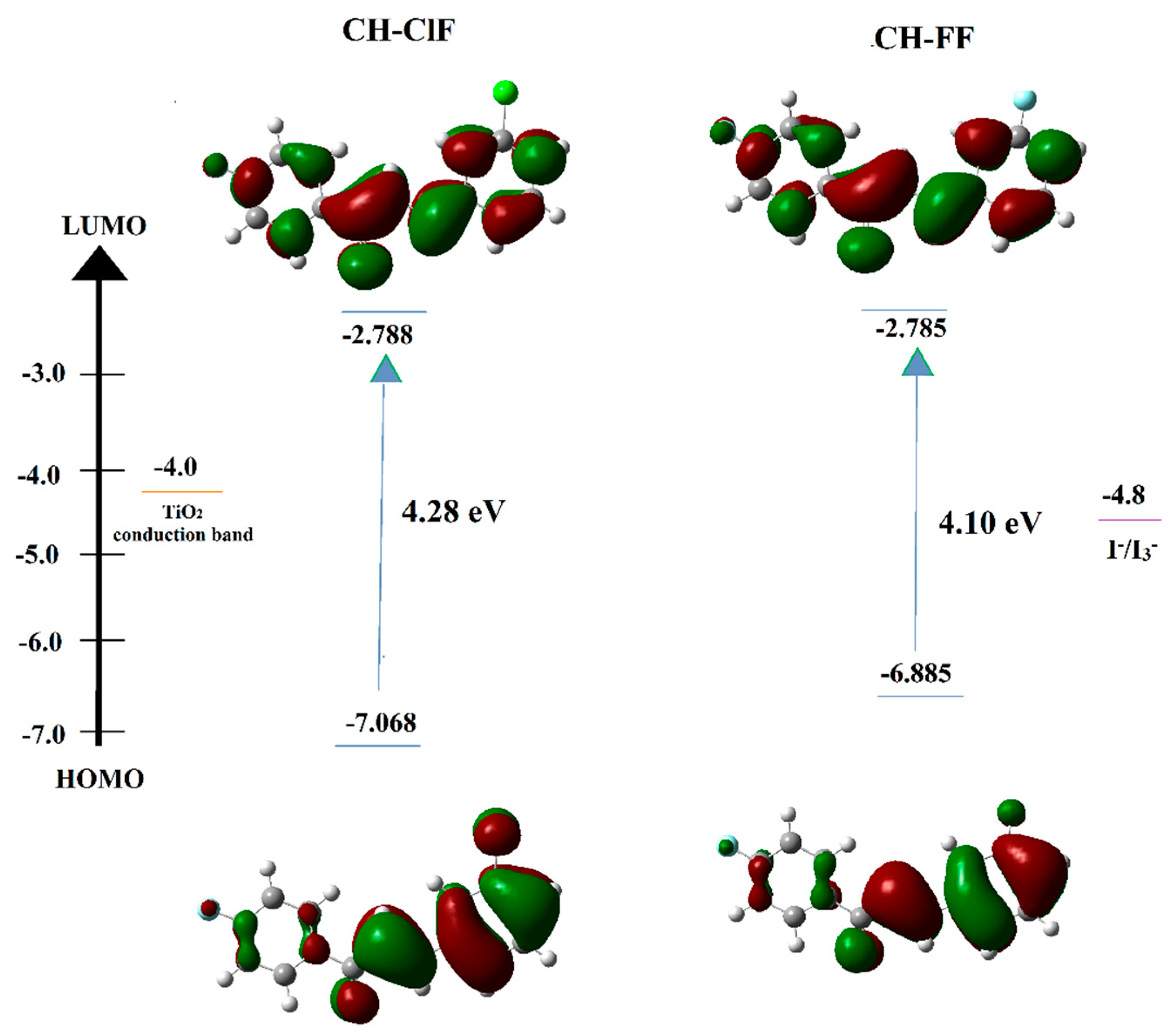
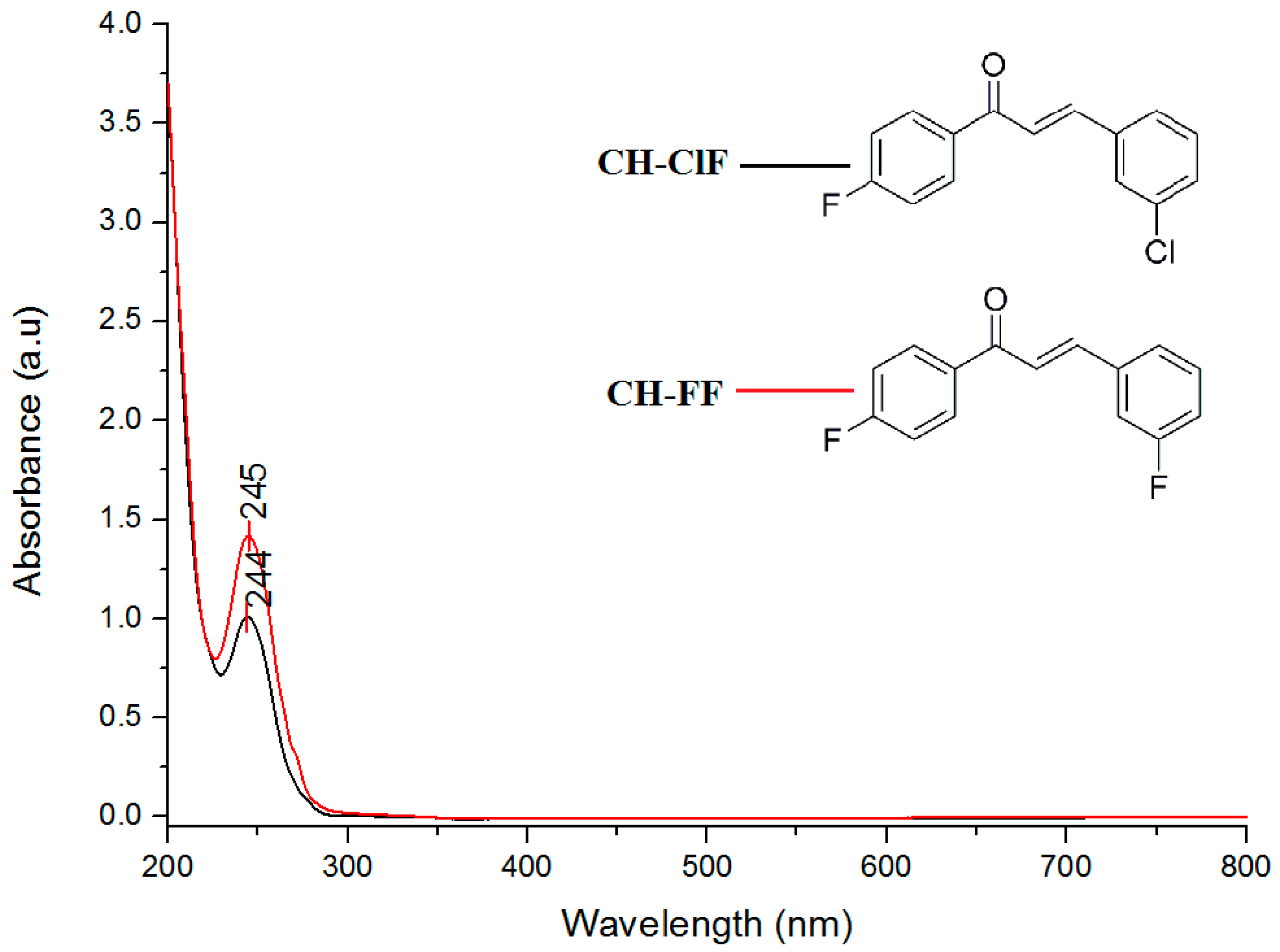
3.5. Ultraviolet–Visible (UV–Vis) and Frontier Molecular Orbital (FMO) Analysis
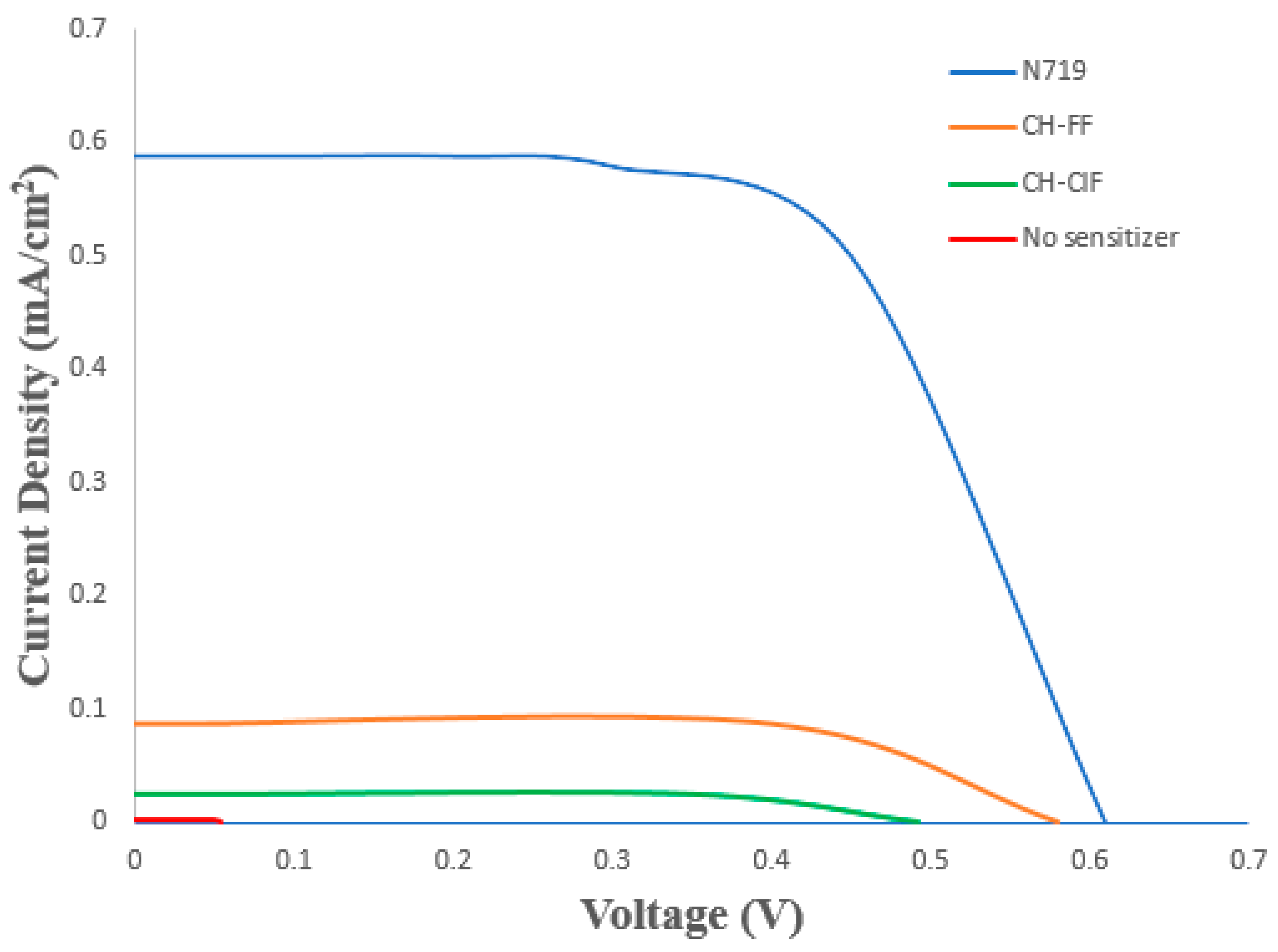
3.6. DSSC Applications and Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prabhu, A.N.; Upadhyaya, V.; Jayarama, A.; Bhat, K.S. Synthesis, growth and characterization of π conjugated organic nonlinear optical chalcone derivative. Mater. Chem. Phys. 2013, 138, 179–185. [Google Scholar] [CrossRef]
- Shehzad, R.A.; Muhammad, S.; Chaudhry, A.R.; Ito, S.; Iqbal, J.; Khalid, M.; Aloui, Z.; Xu, H.-L. Electro-Optical and Charge Transport Properties of Chalcone Derivatives Using a Dual Approach from Molecule to Material Level Simulations. Comput. Theor. Chem. 2021, 1203, 113349. [Google Scholar] [CrossRef]
- Alidmat, M.M.; Khairuddean, M.; Norman, N.M.; Asri, A.N.M.; Suhaimi, M.H.M.; Sharma, G. Synthesis, Characterization, Docking Study and Biological Evaluation of New Chalcone, Pyrazoline, and Pyrimidine Derivatives as Potent Antimalarial Compounds. Arab. J. Chem. 2021, 14, 103304. [Google Scholar] [CrossRef]
- Wu, X.; Shi, Y.; Ju, Y.; Jia, J.; Fang, Y.; Chen, Y.; Wu, Q.; Song, Y. Investigation of the intensity dependent transition from saturable absorption to reverse saturable absorption and ultrafast negative refraction in terthiophene-based chalcone derivatives. Opt. Mater. 2021, 117, 111174. [Google Scholar] [CrossRef]
- Gomes, L.R.; Low, J.N.; Wardell, J.L. Crystal structures, Hirshfeld surface analysis and PIXEL calculations of two chalcone derivatives, containing isopropoxy substituents: Importance of dispersion energy. J. Mol. Struct. 2021, 1237, 130354. [Google Scholar] [CrossRef]
- Kiran, M.S.; Anand, B.; Sankara, S.S.; Rao, G.N. Second- and third-order nonlinear optical properties of bis-chalcone derivatives. J. Photochem. Photobiol. A Chem. 2014, 290, 38–42. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Al Hatahta, A. Spectroscopic evaluation of chalcone derivatives and their zinc metal complexes: A combined experimental and computational approach studying the interactions of the complexes with the serum albumin. J. Mol. Struct. 2021, 1232, 130052. [Google Scholar] [CrossRef]
- Makhlouf, M.M.; Radwan, A.S.; Ghazal, B. Experimental and DFT insights into molecular structure and optical properties of new chalcones as promising photosensitizers towards solar cell applications. Appl. Surf. Sci. 2018, 452, 337–351. [Google Scholar] [CrossRef]
- Anizaim, A.H.; Zainuri, D.A.; Zaini, M.F.; Razak, I.A.; Bakhtiar, H.; Arshad, S. Comparative analyses of new donor-π-acceptor ferrocenyl-chalcones containing fluoro and methoxy-fluoro acceptor units as synthesized dyes for organic solar cell material. PLoS ONE 2020, 15, e0241113. [Google Scholar] [CrossRef]
- Anizaim, A.H.; Zaini, M.F.; Razak, I.A.; Arshad, S. Insight into the impact of the substituent modification on the photovoltaic performance of ferrocenyl chalcones based DSSCs. J. Solid State Chem. 2021, 304, 122551. [Google Scholar] [CrossRef]
- Phan, T.P.; Teo, K.Y.; Liu, Z.Q.; Tsai, J.K.; Tay, M.G. Application of unsymmetrical bis-chalcone compounds in dye sensitized solar cell. Chem. Data Collect. 2019, 22, 100256. [Google Scholar] [CrossRef]
- Rajakumar, P.; Thirunarayanan, A.; Raja, S.; Ganesan, S.; Maruthamuthu, P. Photophysical properties and dye-sensitized solar cell studies on thiadiazole–triazole–chalcone dendrimers. Tetrahedron Lett. 2012, 53, 1139–1143. [Google Scholar] [CrossRef]
- Rajakumar, P.; Kalpana, V.; Ganesan, S.; Maruthamuthu, P. Synthesis and DSSC application of novel dendrimers with benzothiazole and triazole units. Tetrahedron Lett. 2011, 52, 5812–5816. [Google Scholar] [CrossRef]
- Rajakumar, P.; Visalakshi, K.; Ganesan, S.; Maruthamuthu, P.; Suthanthiraraj, S.A. Pyreno-chalcone dendrimers as an additive in the redox couple of dye-sensitized solar cells. J. Mater. Sci. 2012, 47, 1811–1818. [Google Scholar] [CrossRef]
- Chauhan, R.; Yadav, R.; Singh, A.K.; Trivedi, M.; Kociok-Köhn, G.; Kumar, A.; Gosavi, S.; Rane, S. Ferrocenyl chalcones with phenolic and pyridyl anchors as potential sensitizers in dye-sensitized solar cells. RSC Adv. 2016, 6, 97664–97675. [Google Scholar] [CrossRef]
- Omar, A.; Ali, M.S.; Rahim, N.A. Electron transport properties analysis of titanium dioxide dye-sensitized solar cells (TiO2-DSSCs) based natural dyes using electrochemical impedance spectroscopy concept: A review. Sol. Energy 2020, 207, 1088–1121. [Google Scholar] [CrossRef]
- Yahya, M.; Bouziani, A.; Ocak, C.; Seferoğlu, Z.; Sillanpää, M. Organic/metal-organic photosensitizers for dye-sensitized solar cells (DSSC): Recent developments, new trends, and future perceptions. Dye. Pigment. 2021, 192, 109227. [Google Scholar] [CrossRef]
- Patil, S.S.; Mane, R.M.; Khot, K.V.; Mali, S.S.; Hong, C.K.; Bhosale, P.N. Surfactant assisted approach to development of efficient WO3 photoanode for natural dye sensitized solar cell. Sol. Energy 2021, 220, 371–383. [Google Scholar] [CrossRef]
- Dimitrov, V.; Komatsu, T. Correlation among electronegativity, cation polarizability, optical basicity and single bond strength of simple oxides. J. Solid State Chem. 2012, 196, 574–578. [Google Scholar] [CrossRef]
- Mujica-Martínez, C.A.; Arce, J.C. Mini-bandstructure tailoring in pi-conjugated periodic block copolymers using the envelope crystalline-orbital method. Int. J. Quantum Chem. J. 2010, 110, 2532–2540. [Google Scholar]
- Pan, S.; Kong, X.; Zhang, Q.; Xu, Q.; Wang, M.; Wei, C.; Zhao, Y.; Zhang, X. Rational modulating electronegativity of substituents in amorphous metal-organic frameworks for water oxidation catalysis. Int. J. Hydrogen Energy 2020, 45, 9723–9732. [Google Scholar] [CrossRef]
- Zhang, B.; Chou, L.; Bi, Y. Tuning surface electronegativity of BiVO4 photoanodes toward high-performance water splitting. Appl. Catal. B Environ. 2020, 262, 118267. [Google Scholar] [CrossRef]
- Bruker, A. Saint and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; van de Streek, J.; Wood, P. Mercury CSD 2.0– new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Wolff, S.; Grimwood, D.; McKinnon, J.; Turner, M.; Jayatilaka, D.; Spackman, M. Crystal Explorer; The University of Western Australia: Perth, Australia, 2012. [Google Scholar]
- Ternavisk, R.R.; Camargo, A.J.; Machado, F.B.C.; Rocco, J.A.F.F.; Aquino, G.L.B.; Silva, V.H.C.; Napolitano, H.B. Synthesis, characterization, and computational study of a new dimethoxy-chalcone. J. Mol. Model. 2014, 20, 2526. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kumar, V.; Gupta, R.; Bansal, A. Role of chenodeoxycholic acid as co-additive in improving the efficiency of DSSCs. Sol. Energy 2020, 196, 589–596. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to Read and Interpret FTIR Spectroscope of Organic Material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Vinaya, P.; Prabhu, A.; Bhat, K.S.; Upadhyaya, V. Synthesis, growth and characterization of a long-chain π-conjugation based methoxy chalcone derivative single crystal; a third order nonlinear optical material for optical limiting applications. Opt. Mater. 2019, 89, 419–429. [Google Scholar] [CrossRef]
- Thanigaimani, K.; Arshad, S.; Khalib, N.C.; Razak, I.A.; Arunagiri, C.; Subashini, A.; Sulaiman, S.F.; Hashim, N.S.; Ooi, K.L. A new chalcone structure of (E)-1-(4-Bromophenyl)-3-(napthalen-2-yl)prop-2-en-1-one: Synthesis, structural characterizations, quantum chemical investigations and biological evaluations. Spectrochim. Acta Part A. Mol. Biomol. Spectrosc. 2015, 149, 90–102. [Google Scholar] [CrossRef]
- Naik, V.S.; Patil, P.S.; Wong, Q.A.; Quah, C.K.; Gummagol, N.B.; Jayanna, H. Molecular structure, linear optical, second and third-order nonlinear optical properties of two non-centrosymmetric thiophene-chalcone derivatives. J. Mol. Struct. 2020, 1222, 128901. [Google Scholar] [CrossRef]
- Saravanan, R.R.; Seshadri, S.; Gunasekaran, S.; Mendoza-Meroño, R.; Garcia-Granda, S. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy MEP and molecular docking studies on 1- (1- (3-methoxyphenyl) ethylidene) thiosemicarbazide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 139, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Anizaim, A.H.; Arshad, S.; Zaini, M.F.; Abdullah, M.; Zainuri, D.A.; Razak, I.A. Third order nonlinear optical properties of selected fluorinated chalcone derivatives. Opt. Mater. 2019, 98, 109406. [Google Scholar] [CrossRef]
- Arshad, S.; Pilli, R.R.; Zainuri, D.A.; Khalib, N.C.; Razak, I.A.; Armaković, S.; Armaković, S.J.; Renjith, R.; Panicker, Y.; Alsenoye, C.V. Synthesis, XRD crystal structure, spectroscopic characterization, local reactive properties using DFT and molecular dynamics simulations and molecular docking study of (E)-1-(4-bromophenyl)-3-(4-(trifluoromethoxy)phenyl)prop-2-en-1-one. J. Mol. Struct. 2017, 1137, 419–430. [Google Scholar] [CrossRef]
- Zainuri, D.A.; Abdullah, M.; Arshad, S.; Aziz, M.S.A.; Krishnan, G.; Bakhtiar, H.; Razak, I.A. Crystal structure, spectroscopic and third-order nonlinear optical susceptibility of linear fused ring dichloro-substituent chalcone isomers. Opt. Mater. 2018, 86, 32–45. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, A.; Deval, V.; Gupta, A.; Tandon, P.; Patil, P.S.; Watve, J.G. Molecular structure, spectroscopic (FT-IR, FT Raman, UV, NMR and THz) investigation and hyperpolarizability studies of 3-(2-Chloro-6-fluorophenyl)-1-(2-thienyl) prop-2-en-1-one. J. Mol. Struct. 2017, 1129, 292–304. [Google Scholar] [CrossRef]
- Zaini, M.F.; Arshad, S.; Thanigaimani, K.; Khalib, N.C.; Zainuri, D.A.; Abdullah, M.; Razak, I.A. New halogenated chalcones: Synthesis, crystal structure, spectroscopic and theoretical analyses for third-order nonlinear optical properties. J. Mol. Struct. 2019, 1195, 606–619. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Ng, S.-L.; Razak, I.A.; Fun, H.-K.; Patil, P.S.; Dharmaprakash, S.M. 4’-Fluorochalcone. Acta Crystallogr. Sect. E 2006, 62, o2897–o2899. [Google Scholar] [CrossRef]
- Fun, H.-K.; Chia, T.S.; Sapnakumari, M.; Narayana, B.; Sarojini, B. (E)-3-(4-Chlorophenyl)-1-(4-fluorophenyl) prop-2-en-1-one. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, o629. [Google Scholar] [CrossRef]
- Fun, H.-K.; Farhadikoutenaei, A.; Narayana, B.; Nayak, P.S.; Sarojini, B. (2E)-3-(2-Fluorophenyl)-1-(4-fluorophenyl) prop-2-en-1-one. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, o2658. [Google Scholar] [CrossRef] [Green Version]
- Alwani Zainuri, D.; Arshad, S.; Che Khalib, N.; Fikri Zaini, M.; Razak, I.A. Molecular Structure Investigation on Organic Chalcone Derivative of (E)-3-(4-Bromothiophen-2-1-(3-nitrophenyl)prop-2-en-1-one: A Combined Experimental and Theoretical Study. J. Phys. Conf. Ser. 2018, 1083, 1. [Google Scholar] [CrossRef] [Green Version]
- Velasques, J.M.; Gervini, V.C.; Bortoluzzi, A.J.; De Farias, R.L.; De Oliveira, A.B. Crystal structure of (3E)-5-nitro-3-(2-phenylhydrazinylidene)-1H-indol-2(3H)-one. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, 73, 168–172. [Google Scholar] [CrossRef]
- Tripathi, A.; Ganjoo, A.; Chetti, P. Influence of internal acceptor and thiophene based π-spacer in D-A-π-A system on photophysical and charge transport properties for efficient DSSCs: A DFT insight. Sol. Energy 2020, 209, 194–205. [Google Scholar] [CrossRef]
- Reichardt, C.; Eschner, M.; Schäfer, G. Syntheses and UV-visible spectroscopic properties of new ‘fluorophilic’ fluorine- and perfluoroalkyl-substituted solvatochromic pyridiniumN-phenolate betaine dyes. J. Phys. Org. Chem. 2001, 14, 737–751. [Google Scholar] [CrossRef]
- Zaini, M.F.; Arshad, S.; Ibrahim, A.R.; Khalib, N.C.; Zainuri, D.A. Structural Properties and Quantum Chemical Analysis on a New Chalcone Derivative of (E)-3-(4-bromophenyl)-1-(4-fluorophenyl)prop-2-en-1-one. J. Physics Conf. Ser. 2018, 1083, 012047. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.K.; Pervez, M.F.; Mia, N.H.; Mortuza, A.A.; Rahman, M.S.; Rahaman, M.R.; Karim, M.R.; Islam, J.M.M.; Ahmed, F.; Khan, M.A. Effect of dye extracting solvents and sensitization time on photovoltaic performance of natural dye sensitized solar cells. Results Phys. 2017, 7, 1516–1523. [Google Scholar] [CrossRef]
- Borbόn, S.; Lugo, S.; Pourjafari, D.; Pineda Aguilar, N.; Oskam, G.; Lόpez, I. Open-Circuit Voltage (VOC) Enhancement in TiO2-Based DSSCs: Incorporation of ZnO Nanoflowers and Au Nanoparticles. ACS Omega 2020, 5, 10977–10986. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.-D. Comment on “Energy storage via polyvinylidene fluoride dielectric on the counter electrode of dye-sensitized solar cells” by Jiang et al. J. Power Sources 2017, 337, 125–129. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CH-ClF | CH-FF |
|---|---|---|
| CCDC deposition numbers | 1521381 | 1521383 |
| Molecular formula | C15H10ClFO | C15H10F2O |
| Molecular weight | 260.68 | 244.23 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21 | P21/c |
| a/Å | 11.4052 (10) | 11.735 (2) |
| b/Å | 3.9037 (3) | 3.9383 (8) |
| c/Å | 13.8077 (12) | 25.862 (4) |
| β/° | 95.2670 (18) | 100.794 (4) |
| V/Å3 | 1386.42 (9) | 1174.1 (4) |
| Z | 2 | 4 |
| Dcalc (Mg m−3) | 1.414 | 1.382 |
| Crystal Dimensions (mm) | 0.57 × 0.20 × 0.09 | 0.68 × 0.22 × 0.20 |
| μ (mm−1) | 0.31 | 0.11 |
| Radiation λ (Å) | 0.71073 | 0.71073 |
| F(000) | 268 | 504 |
| Tmin/Tmax | 0.8103/0.8835 | 0.8068/0.9393 |
| Reflections measured | 13,236 | 12,518 |
| Ranges/indices (h, k, l) | h = −16→16 k = −5→5 l = −19→19 | h = −16→16 k = −2→5 l = −36→36 |
| θ limit (°) | 1.5–30.2 | 2.2–30.0 |
| Unique reflections | 3616 | 3430 |
| Observed reflections (I > 2σ(I)) | 2664 | 1823 |
| Parameters | 163 | 163 |
| R1 [a], wR2 [b] [I ≥ 2σ(I)] | 0.038/0.110i | 0.048/0.170ii |
| Goodness of fit [c] on F2 | 1.05 | 1.03 |
| Rint | 0.029 | 0.037 |
| Largest diff. peak and hole, e/Å−3 | 0.21 and −0.13 | 0.15 and −0.15 |
| Functional Groups | Wavenumber Frequency (cm−1) | |
|---|---|---|
| CH-ClF | CH-FF | |
| vC = O | 1684.0 | 1683.5 |
| vC = C | 1596.0 | 1683.0 |
| vC—C | 1055.5 | 1055.5 |
| vC—Cl | 691.0 | 506.0 |
| vC—F | 1154.5 | 1155.0 |
| Compound | CH-ClF | CH-FF | ||
|---|---|---|---|---|
| Experimental | DFT | Experimental | DFT | |
| Bond length (Å) | ||||
| C3—F1 | 1.360 (3) | 1.352 | 1.357 (18) | 1.352 |
| C7—O1 | 1.212 (3) | 1.224 | 1.219 (17) | 1.224 |
| C6—C7 | 1.494 (3) | 1.501 | 1.484 (2) | 1.501 |
| C7—C8 | 1.477 (3) | 1.486 | 1.478 (2) | 1.486 |
| C8—C9 | 1.334 (3) | 1.344 | 1.312 (2) | 1.345 |
| C9—C10 | 1.458 (3) | 1.463 | 1.458 (2) | 1.462 |
| C14—Cl1 | 1.742 (2) | 1.759 | - | - |
| C12—F2 | - | - | 1.357 (2) | 1.355 |
| Bond angle (°) | ||||
| O1—C6—C7 | 120.50 (2) | 119.85 | 120.26 (14) | 119.86 |
| O1—C7—C8 | 120.90 (2) | 121.07 | 120.06 (14) | 121.15 |
| C1—C6—C7 | 118.47 (19) | 117.52 | 118.67 (13) | 117.58 |
| C5—C6—C7 | 123.54 (19) | 123.83 | 123.19 (13) | 123.73 |
| C7—C8—C9 | 120.60 (2) | 120.21 | 122.41 (14) | 120.26 |
| C8—C9—C10 | 127.50 (2) | 127.91 | 128.04 (15) | 127.82 |
| C9—C10—C11 | 119.00 (2) | 118.63 | 122.19 (14) | 122.92 |
| C9—C10—C15 | 122.28 (19) | 122.90 | 119.89 (15) | 118.60 |
| Torsion angle (°) | ||||
| C1—C6—C7—O1 | 7.3 (4) | 9.2 | −6.0 (3) | −11.2 |
| C5—C6—C7—O1 | −171.2 (3) | 10.8 | 173.13 (17) | 167.61 |
| O1—C7—C8—C9 | 6.9 (4) | 3.3 | −7.5 (3) | −4.2 |
| C6—C7—C8—C9 | −173.8 (2) | −177.4 | 172.78 (16) | 176.68 |
| C7—C8—C9—C10 | −179.5 (2) | −179.3 | −179.33 (16) | 179.03 |
| C8—C9—C10—C11 | 170.5 (2) | −179.7 | −8.9 (3) | −1.6 |
| C8—C9—C10—C15 | −9.5 (4) | 0.3 | 171.66 (18) | 178.47 |
| Compound | Bond D—H⋯A | Bond Length, (Å) | Angle D—H⋯A (°) | ||
|---|---|---|---|---|---|
| D—H | H⋯A | D⋯A | |||
| CH-FF | C2—H2A⋯O1 (i) | 0.93 | 2.58 | 3.464 (2) | 158 |
| C13—H13A⋯F1 (ii) | 0.93 | 2.55 | 3.422 (2) | 157 | |
| Compound | Current Density, Jsc (mA/cm2) | Open Circuit Voltage, Voc (mV) | Fill Factor, FF (%) | Efficiency, η (%) |
|---|---|---|---|---|
| CH-ClF | 0.024 | 492 | 70.2 | 0.00832 |
| CH-FF | 0.086 | 580 | 72.8 | 0.03632 |
| N719 | 0.589 | 609 | 73.8 | 0.26470 |
| No Sensitizer | 0.002 | 54 | 23.9 | 0.00002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Nizar, S.N.A.; Ab Rahman, S.N.F.; Zaini, M.F.; Anizaim, A.H.; Abdul Razak, I.; Arshad, S. The Photovoltaic Performance of Sensitizers for Organic Solar Cells Containing Fluorinated Chalcones with Different Halogen Substituents. Crystals 2021, 11, 1357. https://doi.org/10.3390/cryst11111357
Mohd Nizar SNA, Ab Rahman SNF, Zaini MF, Anizaim AH, Abdul Razak I, Arshad S. The Photovoltaic Performance of Sensitizers for Organic Solar Cells Containing Fluorinated Chalcones with Different Halogen Substituents. Crystals. 2021; 11(11):1357. https://doi.org/10.3390/cryst11111357
Chicago/Turabian StyleMohd Nizar, Siti Nabilla Aliya, Siti Noor Farhana Ab Rahman, Muhamad Fikri Zaini, Ainizatul Husna Anizaim, Ibrahim Abdul Razak, and Suhana Arshad. 2021. "The Photovoltaic Performance of Sensitizers for Organic Solar Cells Containing Fluorinated Chalcones with Different Halogen Substituents" Crystals 11, no. 11: 1357. https://doi.org/10.3390/cryst11111357
APA StyleMohd Nizar, S. N. A., Ab Rahman, S. N. F., Zaini, M. F., Anizaim, A. H., Abdul Razak, I., & Arshad, S. (2021). The Photovoltaic Performance of Sensitizers for Organic Solar Cells Containing Fluorinated Chalcones with Different Halogen Substituents. Crystals, 11(11), 1357. https://doi.org/10.3390/cryst11111357