Unexpected Selective Gas Adsorption on a ‘Non-Porous’ Metal Organic Framework

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
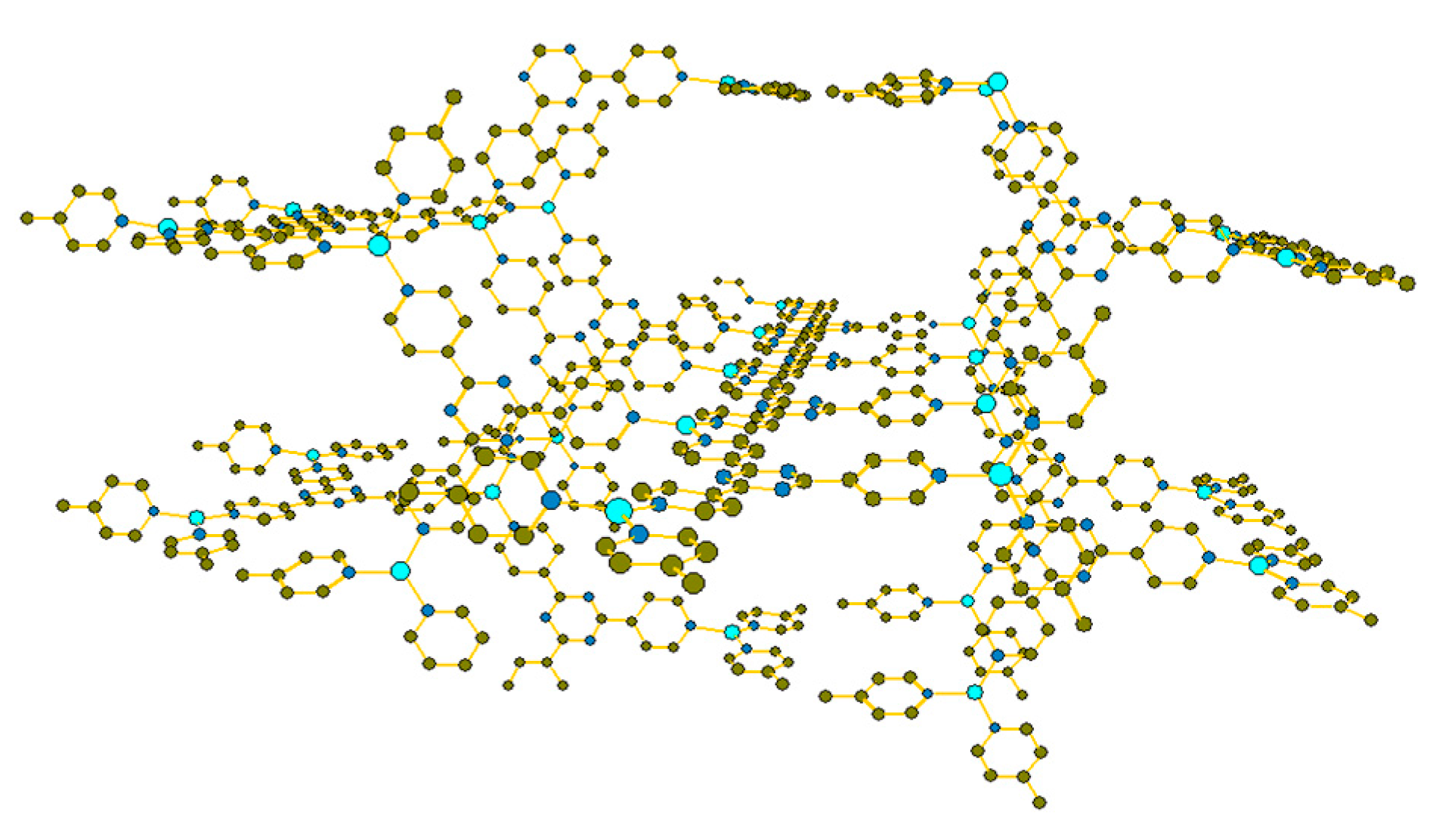
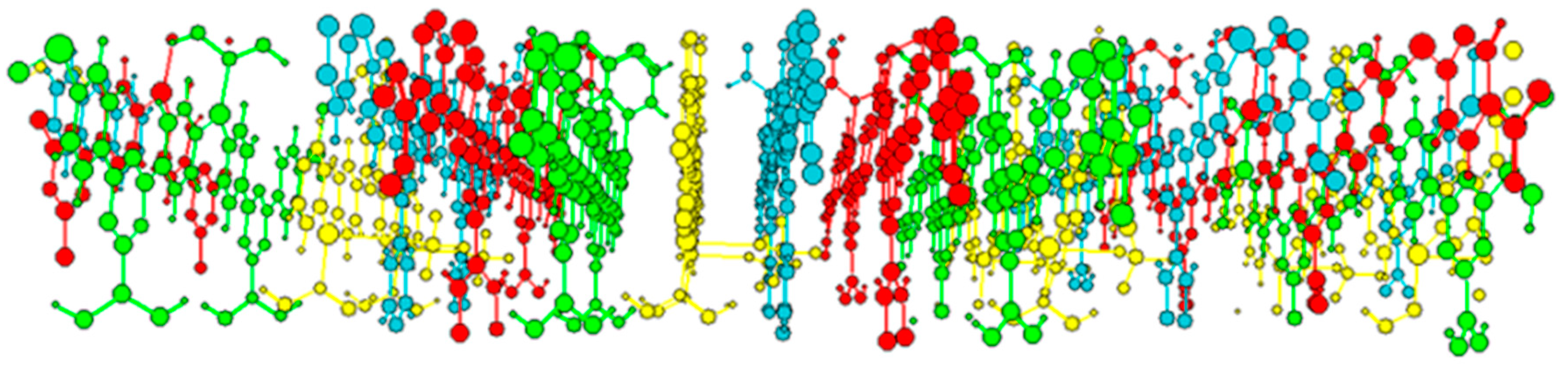
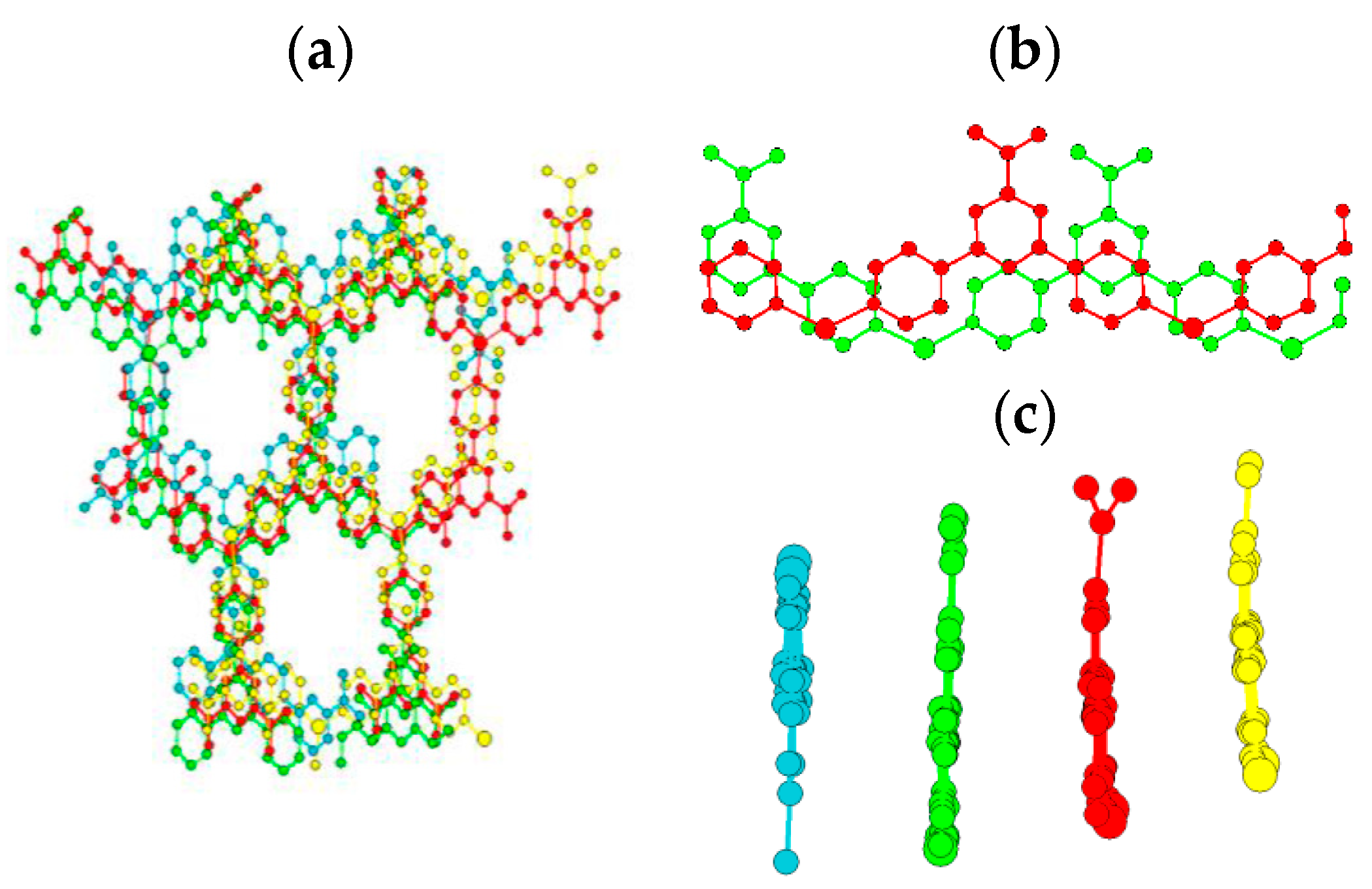
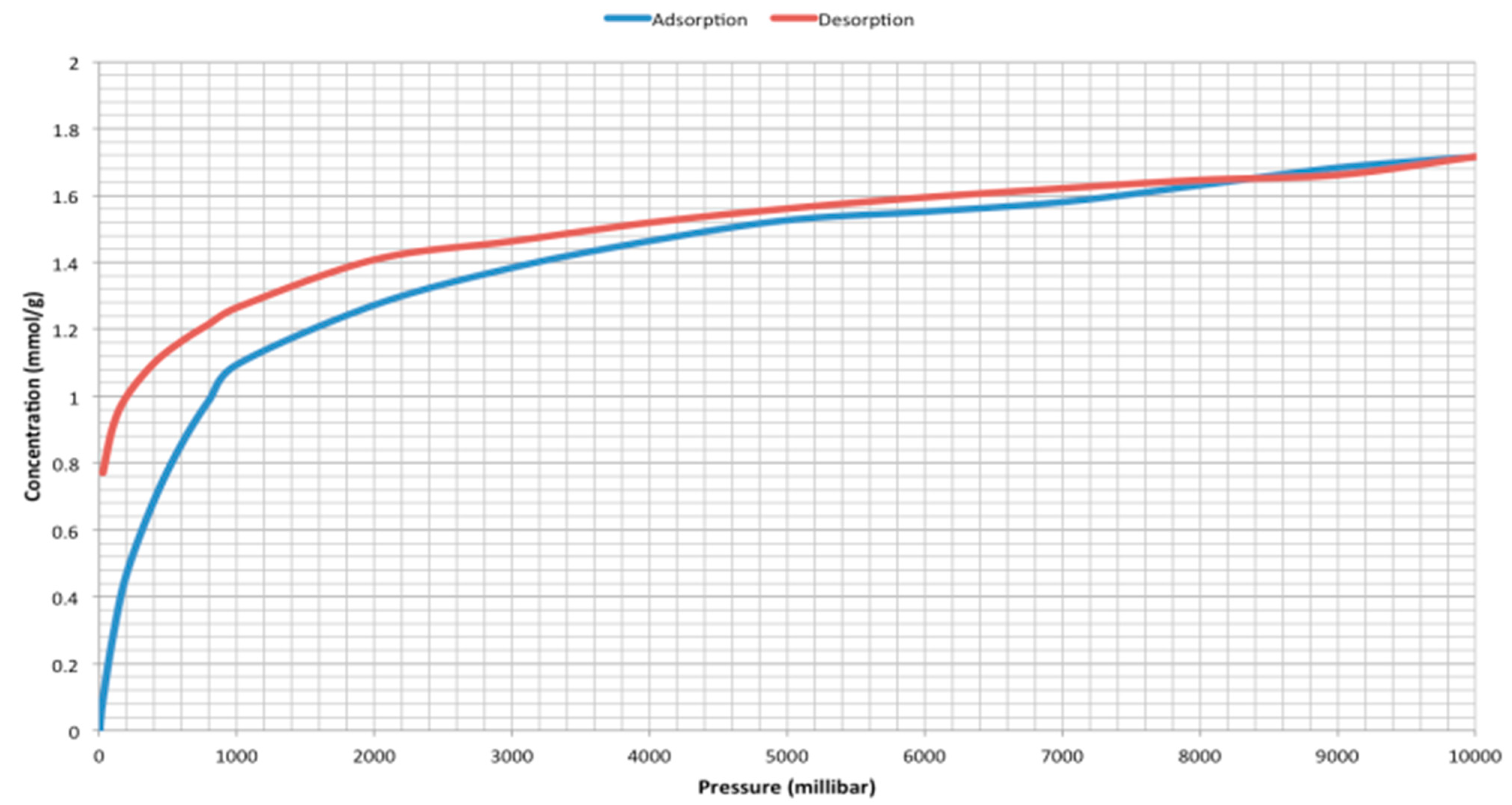
2. Results and Discussion
3. Methodology
3.1. Synthesis of 2,4,6-tri(4-pyridyl)-1,3,5-triazine (tpt)
3.2. Solvothermal Synthesis of Cu(tpt)(BF4)·¾H2O
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Intergovernmental Panel on Climate Change. Intergovernmental Panel on Climate Change (IPCC) Fifth Assessment Report: Climate Change 2014; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2014. [Google Scholar]
- United Nations. Kyoto Protocol to the United Nations Framework Conventions on Climate Change; United Nations: New York, NY, USA, 1998. [Google Scholar]
- Global CCS Institute. CO2 Capture Technologies—Post Combustion Capture (PCC); Global CCS Institute: Melbourne, Australia, 2012. [Google Scholar]
- Pennline, H.W.; Resnik, K.P.; Yeh, J.T. Study of CO2 Absorption and Desorption in a Packed Column. Energy Fuels 2001, 15, 274–278. [Google Scholar]
- Booras, G.S.; Smelser, S.C. An engineering and economic evaluation of CO2 removal from fossil-fuel-fired power plants. Energy Environ. Sci. 1991, 16, 1295–1305. [Google Scholar] [CrossRef]
- Zhang, J.; Webley, P.A.; Xiao, P. Effect of process parameters on power requirements of vacuum swing adsorption technology for CO2 capture from flue gas. Energy Convers. Manag. 2008, 49, 346–356. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.; Mason, J. Carbon dioxide capture in metal-organic frameworks. Chem. Rev. 2011, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Shang, J.; Zhao, Q.; Gu, Q.; Xie, K.; Li, G.; Singh, R.; Xiao, P.; Webley, P.A. A comparative study on conversion of porous and non-porous metal–organic frameworks (MOFs) into carbon-based composites for carbon dioxide capture. Polyhedron 2016, 120, 30–35. [Google Scholar] [CrossRef]
- Dybtsev, D.N.; Chun, H.; Kim, K. Three-dimensional metal–organic framework with (3,4)-connected net, synthesized from an ionic liquid medium. Chem. Commun. 2004, 14, 1594–1595. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. A critical account on π-π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 21, 3885–3896. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Chang, C.-H.; Thanasekaran, P.; Tsai, C.-C.; Tseng, T.-W.; Lee, G.-H.; Peng, S.-M.; Lu, K.-L. Unusual face-to-face pi-pi stacking interactions within an indigo-pillared M3 (tpt)-based triangular metalloprism. Dalton Trans. 2008, 135, 6110–6112. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.F.; Batten, S.R.; Hamit, H.; Hoskins, B.F.; Robson, R. A wellsian ‘three-dimensional’ racemate: Eight interpenetrating, enantiomorphic (10,3)-a nets, four right- and four left-handed. Chem. Commun. 1996, 11, 1313. [Google Scholar] [CrossRef]
- Abrahams, B.F.; Batten, S.R.; Hamit, H.; Hoskins, B.F.; Robson, R. A Cubic (3, 4)-Connected Net with Large Cavities in Solvated [Cu3(tpt)4](ClO4)3 (tpt = 2,4,6-Tri (4-pyridyl)-1,3,5-triazine). Angew. Chem. Int. Ed. Engl. 1996, 35, 1690–1692. [Google Scholar] [CrossRef]
- Batten, S.R.; Hoskins, B.F.; Robson, R. Two Interpenetrating 3D Networks Which Generate Spacious Sealed-Off Compartments Enclosing of the Order of 20 Solvent Molceules in the Structures of Zn (CN)(NO3)(tpt) 2/3. cntdot. solv (tpt = 2,4,6-tri (4-pyridyl)-1,3,5-triazine, solv = apprx. 3/4C2H2Cl4. cntdot. 3/4CH3OH or. apprx. 3/2CHCl3. cntdot. 1/3CH3OH). J. Am. Chem. Soc. 1995, 117, 5385–5386. [Google Scholar]
- Singh, S. Cu (I)/Ag (I)-3-(2-Pyridyl)-5, 6-diphenyl-1, 2, 4-triazine-p, p’-disulfonate Based Coordination Polymers: Synthesis, Structures and Photoluminescent Properties. ChemistrySelect 2018, 3, 6786–6790. [Google Scholar] [CrossRef]
- Jin, C.-M.; Zhu, Z.; Yao, M.-X.; Meng, X.-G. In situ reduction from CuX2 (X = Br, Cl) to Cu (I) halide clusters based on ligand bis (2-methylimidazo-1-yl) methane. CrystEngComm 2010, 12, 358–361. [Google Scholar] [CrossRef]
- Jana, S.; Bhowmik, P.; Chattopadhyay, S. Unique in situ reduction of copper (ii) forming an interesting photoluminescent stair-polymer of copper (i) with a Cu2 S2 core. Dalton Trans. 2012, 41, 10145–10149. [Google Scholar] [CrossRef] [PubMed]
- Banthia, S.; Samanta, A. In situ reduction of copper (II) forming an unusually air stable linear complex of copper (I) with a fluorescent tag. Inorg. Chem. 2004, 43, 6890–6892. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, N.N.; Earnshaw, A. Copper, Silver and Gold Complexes. In Chemistry of the Elements; Butterworth Heinemann: Waltham, MA, USA, 1998. [Google Scholar]
- Pavelka, M.; Burda, J.V. Theoretical description of copper Cu(I)/Cu(II) complexes in mixed ammine-aqua environment. DFT and ab initio quantum chemical study. Chem. Phys. 2005, 312, 193–204. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- American Society for Testing and Materials. ASTM D2892-13 Standard Test Method for Distillation of Crude Petroleum; American Society for Testing and Materials: West Conshohocken, PA, USA, 2013. [Google Scholar]
- Mazari, S.A.; Ali, B.S.; Jan, B.M.; Saeed, I.M.; Nizamuddin, S. An overview of solvent management and emissions of amine-based CO2 capture technology. Int. J. Greenh. Gas Control 2015, 34, 129–140. [Google Scholar] [CrossRef]
- Webster, C.E.; Drago, R.S.; Zerner, M.C. Molecular dimensions for adsorptives. J. Am. Chem. Soc. 1998, 120, 5509–5516. [Google Scholar] [CrossRef]
- Fick, A. Ueber diffusion. Annalen der Physik 1855, 170, 59–86. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beveridge, S.; McAnally, C.A.; Nichol, G.S.; Kennedy, A.R.; Cussen, E.J.; Fletcher, A.J. Unexpected Selective Gas Adsorption on a ‘Non-Porous’ Metal Organic Framework. Crystals 2020, 10, 548. https://doi.org/10.3390/cryst10060548
Beveridge S, McAnally CA, Nichol GS, Kennedy AR, Cussen EJ, Fletcher AJ. Unexpected Selective Gas Adsorption on a ‘Non-Porous’ Metal Organic Framework. Crystals. 2020; 10(6):548. https://doi.org/10.3390/cryst10060548
Chicago/Turabian StyleBeveridge, Stuart, Craig A. McAnally, Gary S. Nichol, Alan R. Kennedy, Edmund J. Cussen, and Ashleigh J. Fletcher. 2020. "Unexpected Selective Gas Adsorption on a ‘Non-Porous’ Metal Organic Framework" Crystals 10, no. 6: 548. https://doi.org/10.3390/cryst10060548
APA StyleBeveridge, S., McAnally, C. A., Nichol, G. S., Kennedy, A. R., Cussen, E. J., & Fletcher, A. J. (2020). Unexpected Selective Gas Adsorption on a ‘Non-Porous’ Metal Organic Framework. Crystals, 10(6), 548. https://doi.org/10.3390/cryst10060548