Properties of Fourier Syntheses and New Syntheses


 , ,
, , 
Abstract
1. Introduction
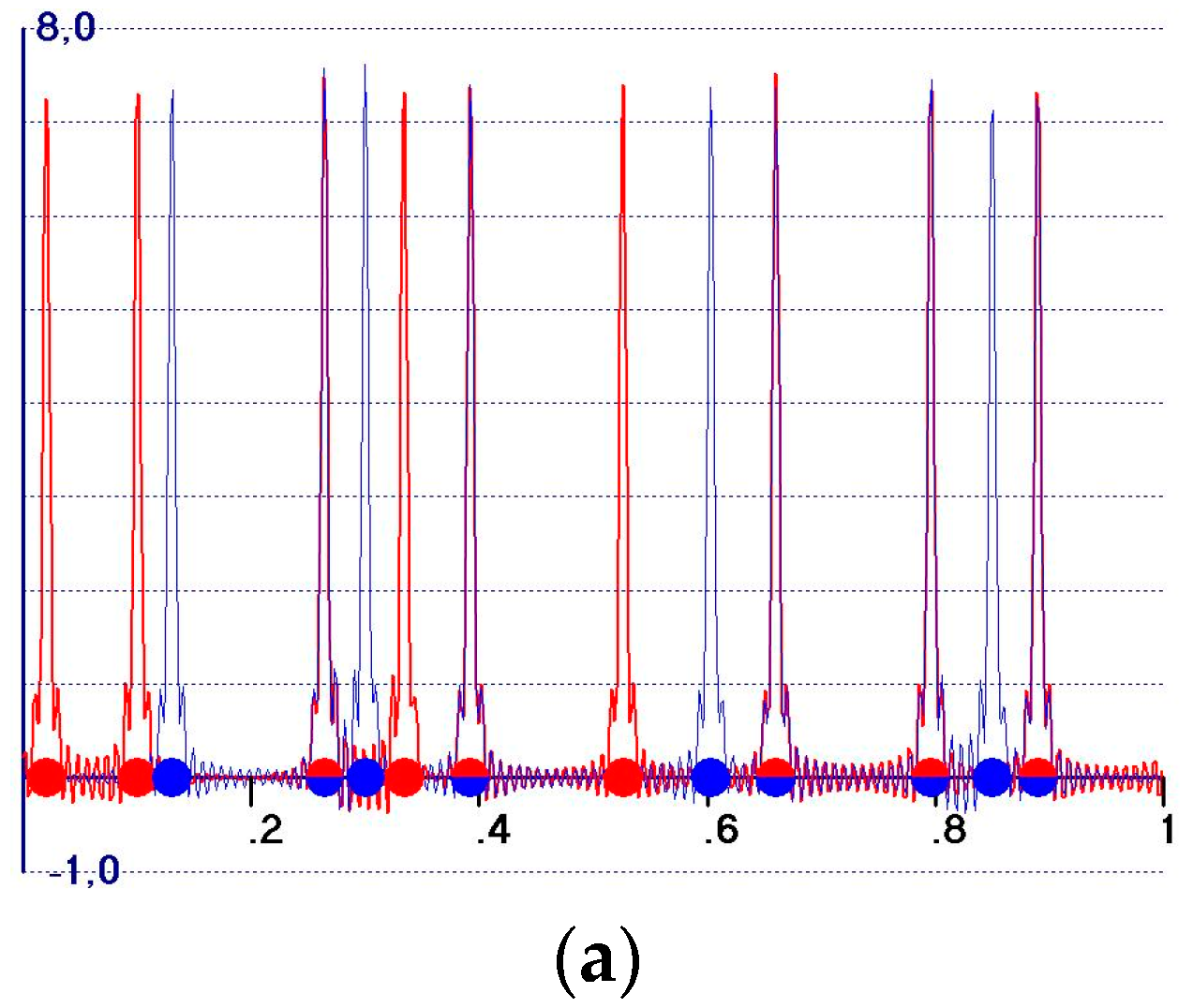
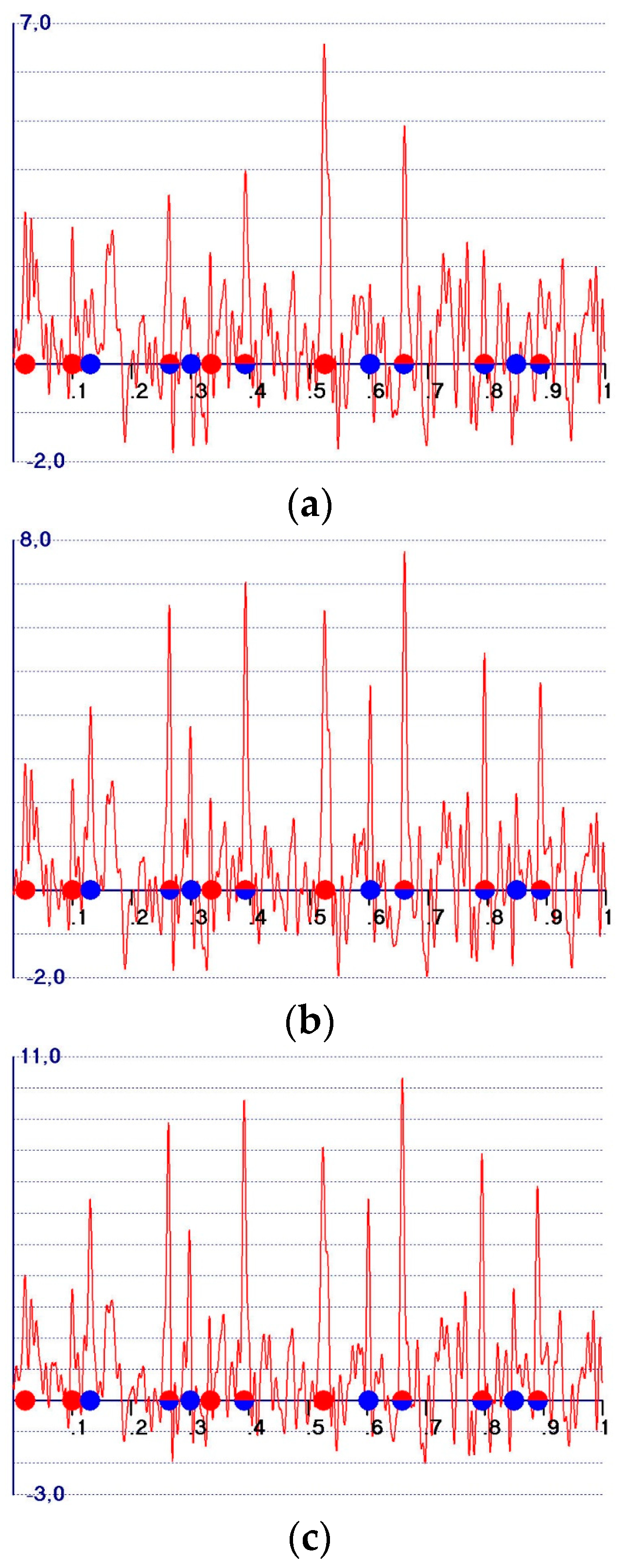
- (a)
- The so-called observed Fourier syntheses (Formula (3))as the Fourier coefficient. They soon became obsolete, and were replaced by the observed weighted syntheses [3,4,5,6,7,8,9,10,11] with (Formula (4)):as Fourier coefficients, where m reflects the probability that the model phase is close to the target phase. m is usually estimated according to Sim [12] or Srinivasan [13]. They play a central role in any modern edm procedure.
- (b)
- (c)
- Main [20] proposed (Formula (6))as the coefficient of difference synthesis. A more recent probabilistic approach [21] generalized Main’s formula for the case in which the target and model have different scattering powers, and used it in the so-called VLD (Vive la Difference) phasing procedure [22,23,24].
- (d)
- Read [25], on the basis of maximum likelihood considerations, suggested, for difference Fourier synthesis, the coefficient (Formula (7))
- (e)
- More recently, the difference Fourier synthesis proposed by Burla et al. [26] used (Formula (8))as the Fourier coefficient amplitude (it is the expected amplitude of the difference ), and the phase if , or if .
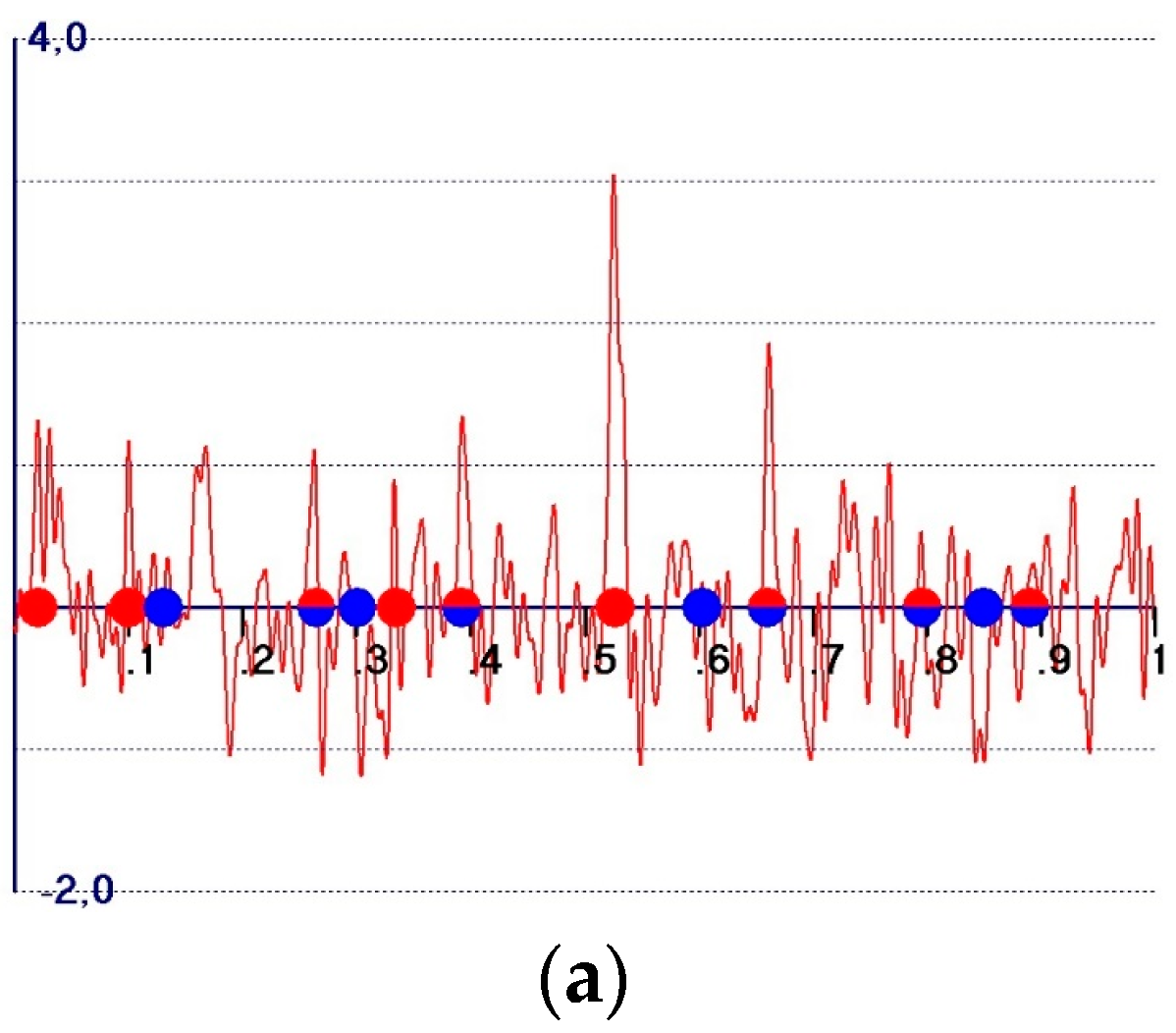
2. The Observed Fourier Synthesis
- -
- The perfect target electron density (Equation (21)); the expected amplitude at the target atomic positions is
- -
- The model electron density (Equation (22)); the expected amplitude at the model atomic positions is
- -
- The observed electron density (Equation (23)); the expected amplitude at the model atomic positions is
- -
- The observed electron density (Equation (24)); the expected amplitude at the target atomic positions is
- (1)
- All of the peaks, Equations (23) and (24) included, are expected to be positive;
- (2)
- A background, practically absent in calculated electron density maps, is present. We call the background the distribution of the electron density outside the model peaks: in the background, it is normal to find peaks corresponding to the target atoms not included int the model, as well as false peaks. The source of the background is quite clear: if and are the sets of amplitudes and phases for a given crystal structure, respectively, the Fourier transform of will provide a vanishing electron density outside the model peaks. Any modification of the amplitudes and/or of the phases will create a background which no longer vanishes outside the model atomic peaks (examples are given in the following sections);
- (3)
- Target and model peaks exhibit quite different behavior, unless the peak is related to both the structures;
- (4)
- The expected amplitudes of the model peaks are given by Equation (23). They are always large and well-distinguishable from the background, and vary in a small range ; the low limit is attained for a random model structure, and the high limit is obtained for a high quality model structure. The amplitude value is a smooth function of ;
- (5)
- The expected amplitudes of the peaks corresponding to the target atoms (not part of the model) are given by Equation (24). They may vary in a large range , according to the model quality. For bad or medium quality models, they are hardly distinguishable from the other background peaks, and only for good models are they comparable with the model amplitudes.
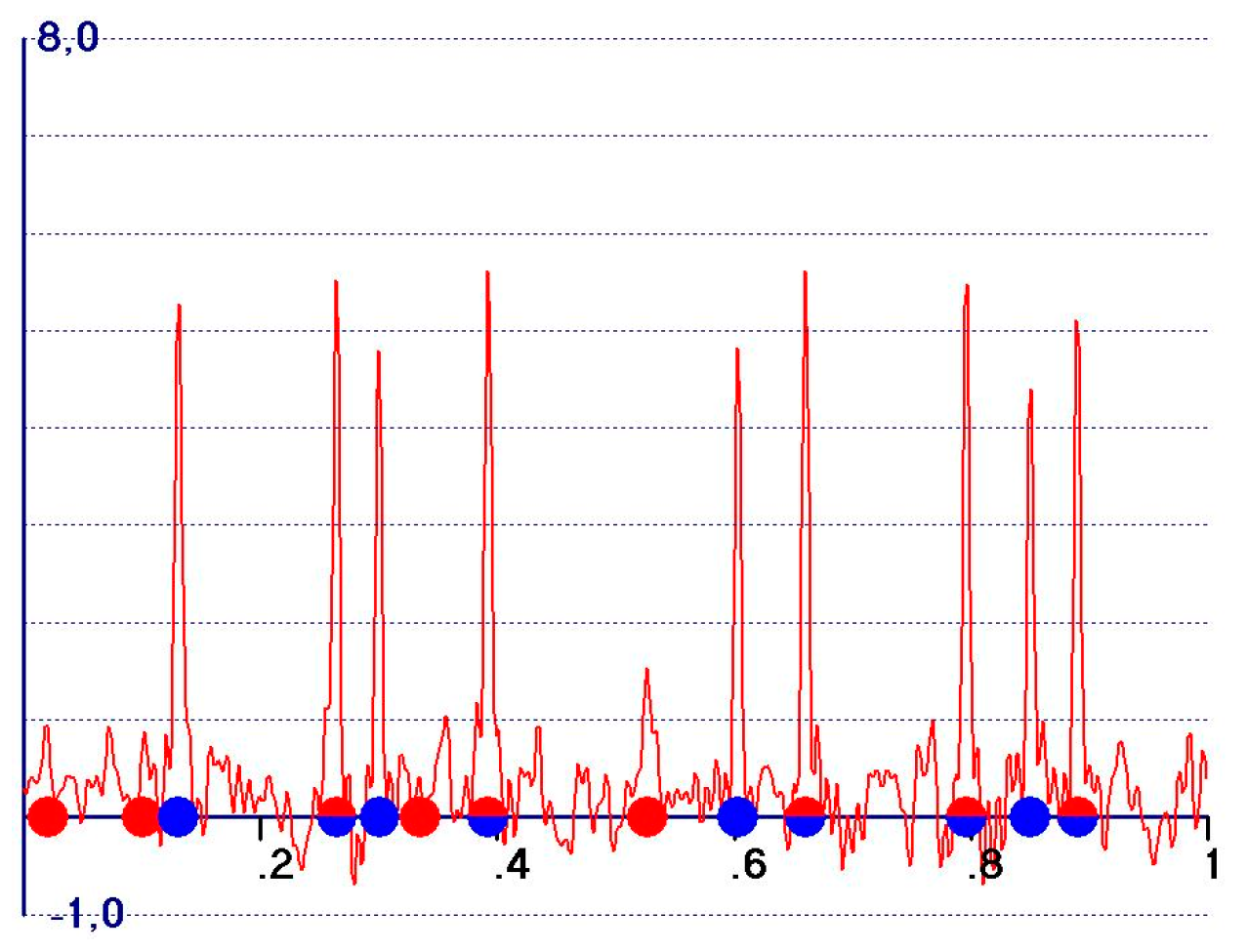
3. The Difference Fourier Synthesis
- (1)
- M.no.T peaks should be negative and strong;
- (2)
- T&M peaks should be weakly negative or, better, weakly positive;
- (3)
- T.no.M peaks should be positive and possibly large;
- (4)
- noT.no.M peaks should be vanishing.
- (i)
- The expected peak amplitude corresponding to a model atom is equal (subtract Equation (22) from Equation (23)) to Equation (29):Since the amplitudes at the model atomic positions are expected to be weakly negative: they asymptotically tend to vanish when the model is of a high quality (then is close to 1);
- (ii)
- The expected peak amplitude corresponding to a target atom (which is not part of the model) coincides with the expected value of the same peak in the observed density (Equation (30)),because the corresponding amplitude in the calculated density is zero;
- (iii)
- In accordance with Section 2, the background present in the difference synthesis essentially coincides with that of the observed density. As a consequence, the missed target atoms remain hidden in the background, with weakly positive peak amplitude values. The basic advantage of the difference synthesis is that model peaks are expected to be weakly negative, so identification of the missed atoms among the positive peaks is easier.
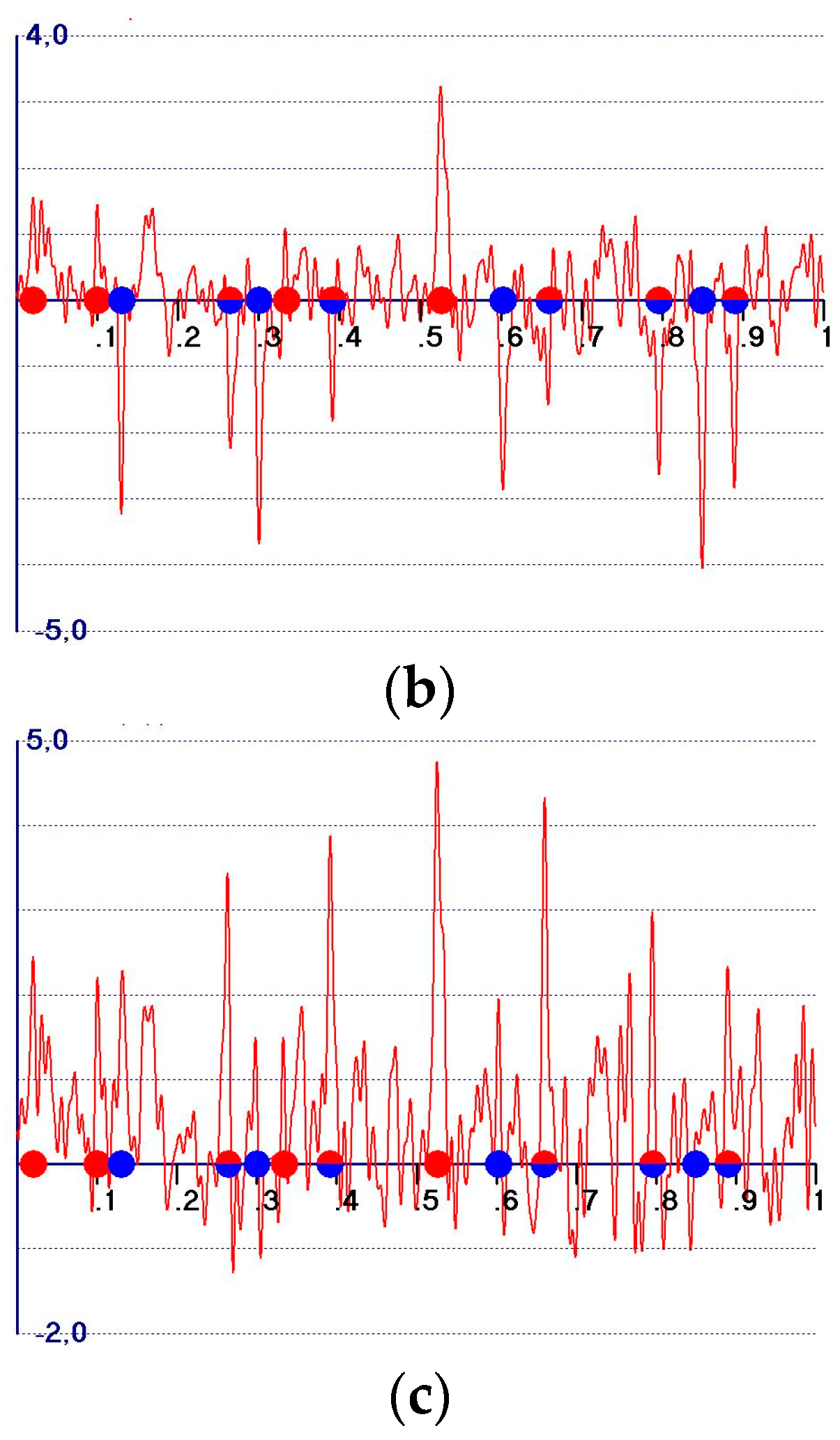
4. General Expression for Hybrid Syntheses
- (i)
- The expected peak amplitude corresponding to a model atom is equal to Equation (33)Since , no matter the quality of the model, if , then , regardless of the quality of the model. The peak amplitude is therefore expected to always be negative, no matter the model quality: the greater the difference the larger the negative amplitudes.
- (ii)
- The expected peak amplitude corresponding to a target atom is given by Equation (34)The presence of the factor suggests that such amplitudes linearly increase with , but the reader should not expect the probability of discovering target atoms not included in the model to be enhanced, because the background is enhanced by the same factor (see Equation (33)).
- (i)
- The expected peak amplitudes at the model atomic positions are positive for high quality models and remain negative for very low quality models. In the first case, a synthesis shares the features of an observed synthesis, and it is more similar to a difference synthesis in the second case;
- (ii)
- The expected peak amplitudes corresponding to a target atom are given by Equation (34): as for the case, the probability of discovering target atoms not included in the model is not enhanced;
- (iii)
- An interesting feature may be found. Equation (33) suggests that model peaks present in are also peaks of the map, but with smaller amplitudes. On the contrary, the amplitudes of target peaks of remain unvaried in (e.g., because does not contribute to the target peaks), so their ratio with the amplitudes of the model peaks increases. Unfortunately, the other background peaks (e.g., those which do not correspond to the target atoms) also remain unvaried, so the difficulty in identifying missed atom peaks among the background false peaks still remains.
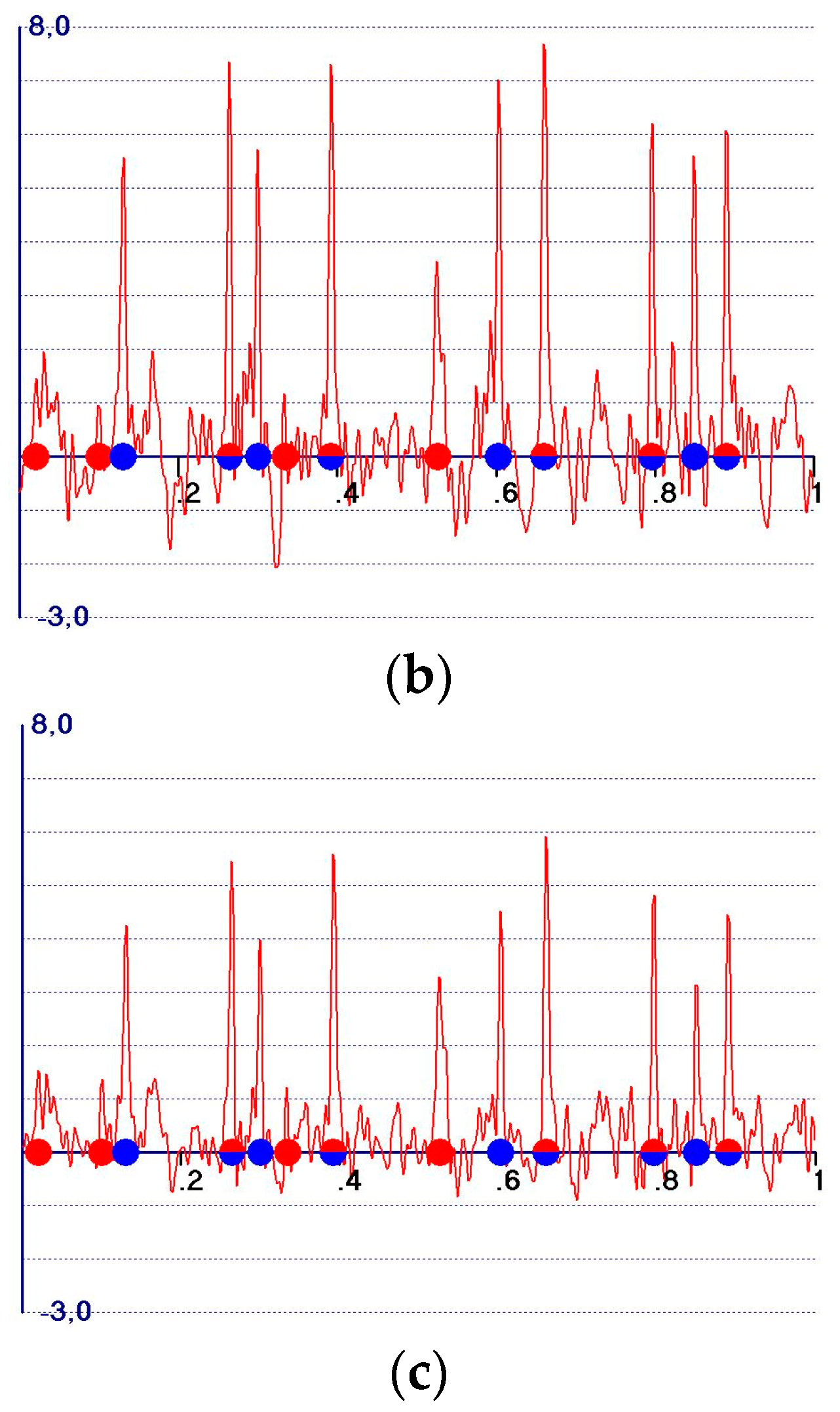
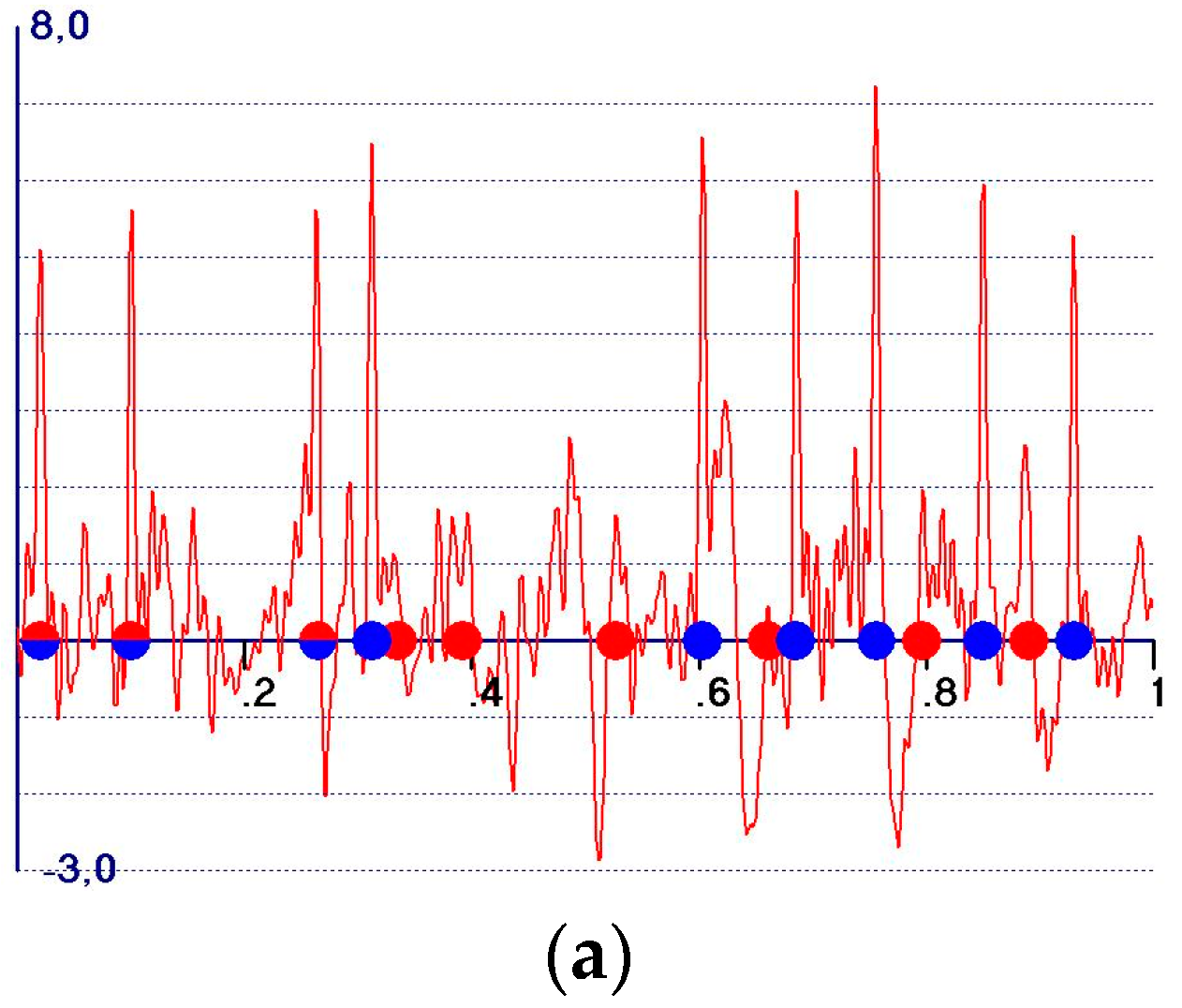
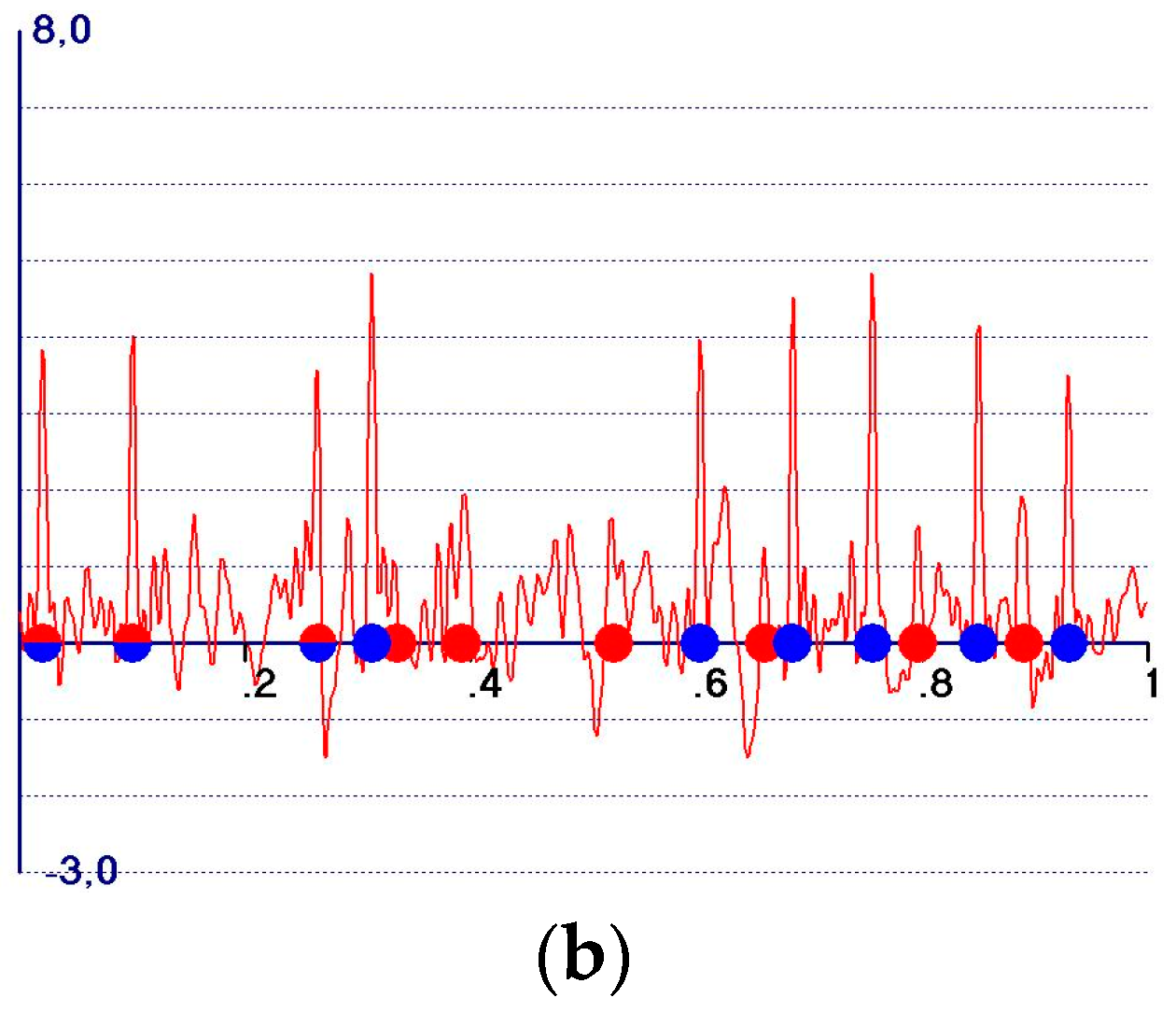
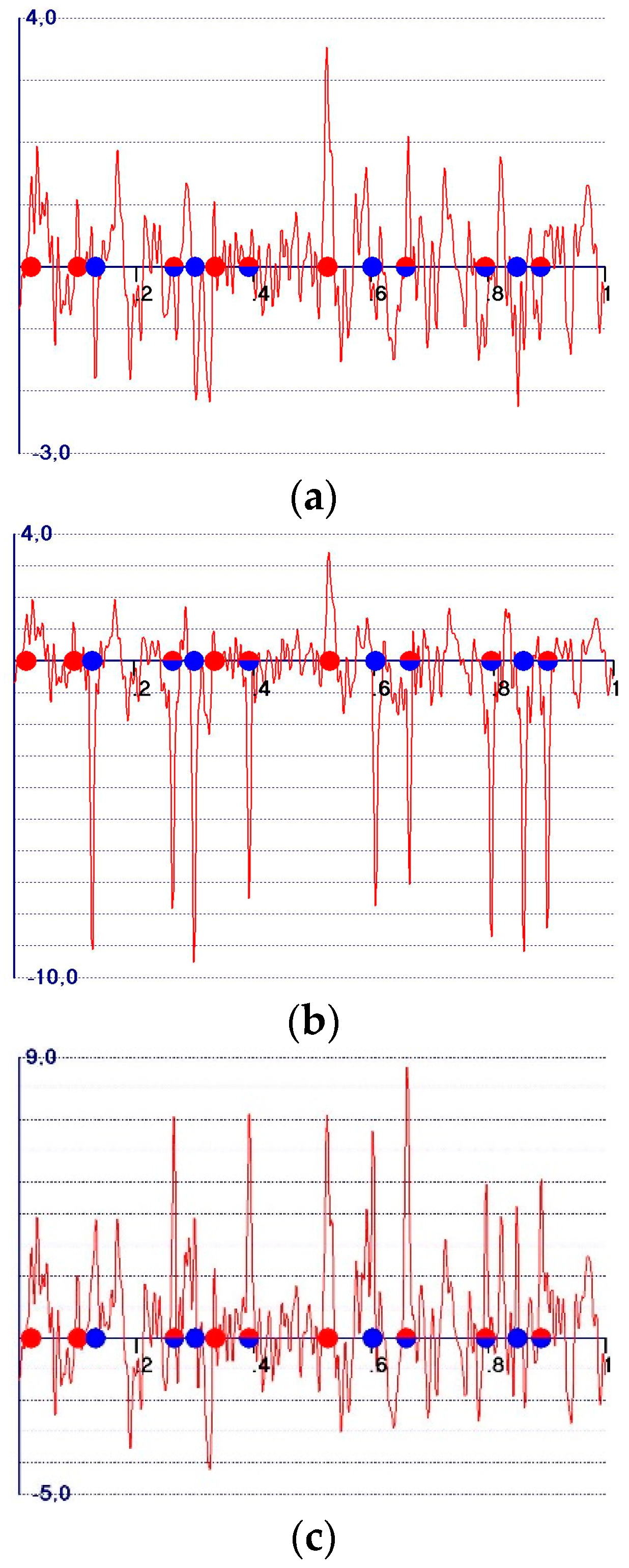
5. The Weighted Observed Fourier Syntheses
- -
- A reduction in the amplitudes of the model and target peaks, as well as the background (see Figure 1b,c). Such peaks remain positive;
- -
- An increase in the ratio between the amplitudes of the target and the model peaks;
- -
- Modification of the background of the unweighted observed synthesis and emphasis of the target peak amplitudes.
6. The Weighted Hybrid Fourier Syntheses
7. New Types of Observed Fourier Synthesis
8. The First Check of the Fourier Syntheses for One-Dimensional Ideal Structures
9. Results
10. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Normalized structure factors of , respectively. | |
| the average is performed per resolution shell. | |
| Modified Bessel function of order i. | |
| where | |
| edm | electron density modification (procedures). |
Appendix A. Useful Relations for Electron Density Maps
References
- Bragg, W.L. The determination of parameters in crystal structures by means of Fourier series. Proc. R. Soc. Lond. Ser. A 1929, 123, 537–559. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Srinivasan, R. Fourier Methods in Crystallography; Wiley-Interscience: New York, NY, USA, 1970. [Google Scholar]
- Cowtan, K.D. ‘Dm’: An automated procedure for phase improvement by density modification. Jt. CCP4 ESF-EACBM Newsl. Protein Crystallogr. 1994, 31, 34–38. [Google Scholar]
- Cowtan, K.D. Error estimation and bias correction in phase-improvement calculations. Acta Cryst. 1999, D55, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P. Bias Reduction in Phase Refinement by Modified Interference Functions: Introducing the γ Correction. Acta Cryst. 1997, D53, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P.; Leslie, A.G.W. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Cryst. 1996, D52, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Refaat, L.S.; Woolfson, M.M. Direct-space methods in phase extension and phase determination. II. Developments of low-density elimination. Acta Cryst. 1993, D49, 367–371. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Siliqi, D. Improving Direct Methods phases by Heavy-Atom information and Solvent Flattening. Acta Cryst. 1997, D53, 789–798. [Google Scholar] [CrossRef]
- Terwilliger, T.C. Reciprocal-space solvent flattening. Acta Cryst. 1999, D55, 1863–1871. [Google Scholar] [CrossRef]
- Terwilliger, T.C. Maximum-likelihood density modification. Acta Cryst. 2000, D56, 965–972. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Macromolecular phasing with SHELXE. Z. Krist. 2002, 217, 644–650. [Google Scholar] [CrossRef]
- Sim, G.A. The distribution of phase angles for structures containing heavy atoms. II. A modification of the normal heavy-atom method for non-centrosymmetrical structures. Acta Cryst. 1959, 12, 813–815. [Google Scholar] [CrossRef]
- Srinivasan, R. Weighting functions for use in the early stage of structure analysis when a part of the structure is known. Acta Cryst. 1966, 20, 143–144. [Google Scholar] [CrossRef]
- Cochran, W. Some properties of the (Fo-Fc)-synthesis. Acta Cryst. 1951, 4, 408–411. [Google Scholar] [CrossRef]
- Henderson, R.; Moffat, J.K. The difference Fourier technique in protein crystallography: Errors and their treatment. Acta Cryst. 1971, B27, 1414–1420. [Google Scholar] [CrossRef]
- Ursby, T.; Bourgeois, D. Improved Estimation of Structure-Factor Difference Amplitudes from Poorly Accurate Data. Acta Cryst. 1997, A53, 564–575. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. The (Fo-Fc) Fourier synthesis: A probabilistic study. Acta Cryst. 2008, A64, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. Advances in the EDM-DEDM procedure. Acta Cryst. 2009, D65, 249–256. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. EDM-DEDM and protein crystal structure solution. Acta Cryst. 2009, D65, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Main, P. A theoretical comparison of the β, γ’ and 2Fo-Fc syntheses. Acta Cryst. 1979, A35, 779–785. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Giacovazzo, C.; Polidori, G. The difference electron density: A probabilistic reformulation. Acta Cryst. 2010, D66, 347–361. [Google Scholar] [CrossRef]
- Burla, M.C.; Giacovazzo, C.; Polidori, G. From a random to the correct structure: The VLD algorithm. J. Appl. Cryst. 2010, 43, 825–836. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. Advances in the VLD algorithm. J. Appl. Cryst. 2011, 44, 1143–1151. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. VLD algorithm and hybrid Fourier syntheses. J. Appl. Cryst. 2012, 45, 1287–1294. [Google Scholar] [CrossRef]
- Read, R.J. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst. 1986, A42, 140–149. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. About difference electron densities and their properties. Acta Cryst. 2017, A73, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M. On the Fourier refinement of protein structures. Acta Cryst. 1980, A36, 295–298. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.D. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997, D53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo., C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D. The revenge of the Patterson methods. II. Substructure applications. J. Appl. Cryst. 2007, 40, 211–217. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D. The revenge of the Patterson methods. III. Ab initio phasing from powder diffraction data. J. Appl. Cryst. 2007, 40, 834–840. [Google Scholar] [CrossRef]
- Loll, P.J.; Bevivino, A.E.; Korty, B.D.; Axelsen, P.H. Simultaneous Recognition of a Carboxylate-Containing Ligand and an Intramolecular Surrogate Ligand in the Crystal Structure of an Asymmetric Vancomycin Dimer. J. Am. Chem. Soc. 1997, 119, 1516–1522. [Google Scholar] [CrossRef]
- Burkhart, B.M.; Gassman, R.M.; Langs, D.A.; Pangborn, W.A.; Duax, W.L. Heterodimer Formation and Crystal Nucleation of Gramicidin, D. Biophysics 1998, 75, 2135–2146. [Google Scholar] [CrossRef][Green Version]
- Prive, G.G.; Anderson, D.H.; Wesson, L.; Cascio, D.; Eisenberg, D. Packed protein bilayers in the 0.90 Å resolution structure of a designed alpha helical bundle. Protein Sci. 1999, 8, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Gonzalez, A.; Rypniewski, W.R.; Wilson, K.S.; Van Beeumen, J.J.; Ciurli, S. Crystal structure of oxidized Bacillus pasteurii cytochrome c553 at 0.97 Å resolution. Biochemistry 2000, 39, 13115–13126. [Google Scholar] [CrossRef] [PubMed]
- Frazao, C.; Soares, C.M.; Carrondo, M.A.; Pohl, E.; Dauter, Z.; Wilson, K.S.; Hervas, M.; Navarro, J.A.; Rosa, M.A.; Sheldrick, G.M. Ab initio determination of the crystal structure of cytochrome c6 and comparison with plastocyanin. Structure 1995, 3, 1159–1169. [Google Scholar] [CrossRef]
- Lehmann, C.; Bunkoczi, G.; Vertesy, L.; Sheldrick, G.M. Structures of Glycopeptide Antibiotics with Peptides that Model Bacterial Cell-Wall Precursors. J. Mol. Biol. 2002, 318, 723–732. [Google Scholar] [CrossRef]
- Dauter, Z.; Adamiak, D.A. Anomalous signal of phosphorus used for phasing DNA oligomer: Importance of data redundancy. Acta Cryst. 2001, D57, 990–995. [Google Scholar] [CrossRef]
- Dauter, Z.; Wilson, K.S.; Sieker, L.C.; Moulis, J.M.; Meyer, J. Zinc-and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: A high-precision model of a ZnS4 coordination unit in a protein. Proc. Natl. Acad. Sci. USA 1996, 93, 8836–8840. [Google Scholar] [CrossRef]
- Lee, D.L.; Ivaninskii, S.; Burkhard, P.; Hodges, R.S. Unique stabilizing interactions identified in the two-stranded alpha-helical coiled-coil: Crystal structure of a cortexillin I/GCN4 hybrid coiled-coil peptide. Protein Sci. 2003, 12, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Schneider, T.R.; Sheldrick, G.M. Crystal Structure of Vancomycin. Structure 1996, 4, 1509–1515. [Google Scholar] [CrossRef]
- Almog, O.; Gonzalez, A.; Klein, D.; Greenblat, H.M.; Braun, S.; Shoham, G. The 0.93 Å Crystal Structure of Sphericase: A Calcium-Loaded Serine Protease from Bacillus Sphaericus. J. Mol. Biol. 2003, 332, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Meijers, R.; Morris, R.J.; Adolph, H.W.; Merli, A.; Lamzin, V.S.; Cedergen-Zeppezauer, E.S. On the Enzymatic Activation of Nadh. J. Biol. Chem. 2001, 276, 9316–9321. [Google Scholar] [CrossRef]
- Rubach, J.K.; Plapp, B.V. Amino Acid Residues in the Nicotinamide Binding Site Contribute to Catalysis by Horse Liver Alcohol Dehydrogenase. Biochemistry 2003, 42, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh Resolution Drug Design I: Details of Interactions in Human Aldose Reductase-Inhibitor Complex at 0.66 Å. Proteins 2004, 55, 792–804. [Google Scholar] [CrossRef]
- Perret, S.; Sabin, C.; Dumon, C.; Pokorna, M.; Gautier, C.; Galanina, O.; Ilia, S.; Bovin, N.; Nicaise, M.; Desmadril, M.; et al. Structural Basis for the Interaction between Human Milk Oligosaccharides and the Bacterial Lectin Pa-Iil of Pseudomonas Aeruginosa. Biochem. J. 2005, 389, 325–332. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Strange, R.W.; Sawers, G.; Eady, R.R.; Hasnain, S.S. Atomic Resolution Structures of Resting-State, Substrate-and Product-Complexed Cu-Nitrite Reductase Provide Insight Into Catalytic Mechanis. Proc. Natl. Acad. Sci. USA 2005, 102, 12041–12046. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. How far are we from automatic crystal structure solution via molecular-replacement techniques? Acta Cryst. 2020, D76, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.H.; Parkinson, G.N.; Reszka, A.P.; Neidle, S. Structural basis of DNA quadruplex recognition by an acridine drug. J. Am. Chem. Soc. 2008, 130, 6722–6724. [Google Scholar] [CrossRef] [PubMed]
- Millonig, H.; Pous, J.; Gouyette, C.; Subirana, J.A.; Campos, J.L. The interaction of manganese ions with DNA. J. Inorg. Biochem. 2009, 103, 876–880. [Google Scholar] [CrossRef]
- Juan, E.C.M.; Shimizu, S.; Ma, X.; Kurose, T.; Haraguchi, T.; Zhang, F.; Tsunoda, M.; Ohkubo, A.; Sekine, M.; Shibata, T.; et al. Insights into the DNA stabilizing contributions of a bicyclic cytosine analogue: Crystal structures of DNA duplexes containing 7,8-dihydropyrido [2,3-d]pyrimidin-2-one. Nucleic Acids Res. 2010, 38, 6737–6745. [Google Scholar] [CrossRef]
- Carter, M.; Ho, P.S. Assaing the energies of biological halogen bonds. Cryst. Growth Des. 2011, 11, 5087–5095. [Google Scholar] [CrossRef]
- Carter, M.; Voth, A.R.; Scholfield, M.R.; Rummel, B.; Sowers, L.C.; Ho, P.S. Enthalpy-entropy compensation in biomolecular halogen bonds measured in DNA junctions. Biochemistry 2013, 52, 4891–4903. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.P. The Effects of Disubstitution on the Binding of Ruthenium Complexes to DNA. Ph.D. Thesis, University of Reading, Reading, UK, 2014. [Google Scholar]
- Rohner, M.; Medina-Molner, A.; Spingler, B. N,N,O and N,O,N meridional cis coordination of two guanines to copper(II) by d(CGCGCG)2. Inorg Chem. 2016, 55, 6130–6140. [Google Scholar] [CrossRef]
- Krauss, I.R.; Ramaswamy, S.; Neidle, S.; Haider, S.; Parkinson, G.N. Structural insights into the quadruplex-duplex 3′ interface formed from a telomeric repeat: A potential molecular target. J. Am. Chem. Soc. 2016, 138, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Sobri, A.F.A.; Brady, R.L. Non-Natural DNA pair Z (6-Amino-5-Nitro-2[1H] Pyridone Heterocycle)-Guanosine. Unpublished work. 2016. [Google Scholar]
- Sbirkova, H.I.; Schivachev, B.L. Crystal structure of a DNA sequence d(CGTGAATTCACG) at 130K. Bulg. Chem. Commun. 2016, 48, 589–593. [Google Scholar]
- Reichenbach, L.F.; Sobri, A.A.; Zaccai, N.R.; Agnew, C.R.J.; Burton, N.; Eperon, L.P.; De Ornellas, S.; Eperon, I.C.; Brady, R.L.; Burley, G.A. Structural basis of the mispairing of an artificially expanded genetic information system. Chemistry 2016, 1, 946–958. [Google Scholar] [CrossRef][Green Version]
- Hardwick, J.S.; Ptchelkine, D.; El-Sagheer, A.H.; Tear, I.; Singleton, D.; Phillips, S.E.V.; Lane, A.N.; Brown, T. 5-Formylcytosine does not change the global structure of DNA. Nat. Struct. Mol. Biol. 2017, 24, 544–552. [Google Scholar] [CrossRef]
- Sbirkova, H.I.; Schivachev, B.L. The effect of berenil and cacodylate on the crystal structure of d(CGTGAATTCACG). Unpublished work. 2017. [Google Scholar]
- Sbirkova-Dimitrova, H.I.; Shivachev, B.L. Crystal structure of the DNA sequence d(CGTGAATTCACG)2 with DAPI. Acta Cryst. 2017, 73, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Cruse, W.; Saludjian, P.; Neuman, A.; Prange, T. Destabilizing effect of a fluorouracil extra base in a hybrid RNA duplex compared with bromo and chloro analogues. Acta Cryst. 2001, D57, 1609–1613. [Google Scholar] [CrossRef]
- Gherghe, C.M.; Krahn, J.M.; Weeks, K.M. Crystal structures, reactivity and inferred acylation transition states for 2′-amine substituted RNA. J. Am. Chem. Soc. 2005, 127, 13622–13628. [Google Scholar] [CrossRef]
- Haeberli, P.; Berger, I.; Pallan, P.S.; Egli, M. Syntheses of 4′-thioribonucleosides and thermodynamic stability and crystal structure of RNA oligomers with incorporated 4′-thiocytosine. Nucleic Acids Res. 2005, 33, 3965–3975. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ennifar, E.; Paillart, J.C.; Bodlenner, A.; Walter, P.; Weibel, J.-M.; Aubertin, A.-M.; Pale, P.; Dumas, P.; Marquet, R. Targeting the dimerization initiation site of HIV-1 RNA with aminoglycosides: From crystal to cell. Nucleic Acids Res. 2006, 34, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Han, Q.; Kissinger, C.R.; Hermann, T.; Thompson, P.A. Structure of hepatitis C virus IRES subdomain IIa. Acta Cryst. 2008, D64, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Thore, S.; Frick, C.; Ban, N. Structural basis of thiamine pyrophosphate analogues binding to the eukaryotic riboswitch. J. Am. Chem. Soc. 2008, 130, 8116–8117. [Google Scholar] [CrossRef]
- Pitt, J.N.; Ferre-D’Amare, A.R. Structure-guided engineering of the regioselectivity of RNA ligase ribozymes. J. Am. Chem. Soc. 2009, 131, 3532–3540. [Google Scholar] [CrossRef]
- Ren, A.; Rajashankar, K.R.; Patel, D.J. Fluoride ion encapsulation by Mg2+ ions and phosphates in a fluoride riboswitch. Nature 2012, 486, 85–89. [Google Scholar] [CrossRef]
- Vorobiev, S.M.; Ma, L.-C.; Montelione, G.T. Crystal structure of the 16-mer double stranded RNA. Unpublished work. 2016. [Google Scholar]
- Monestier, A.; Aleksandrov, A.; Coureux, P.D.; Panvert, M.; Mechulam, Y.; Schmitt, E. The structure of an E. coli tRNAf(Met) A1-U72 variant shows an unusual conformation of the A1-U72 base pair. RNA 2017, 23, 673–682. [Google Scholar] [CrossRef]
- Huang, L.; Wang, J.; Wilson, T.J.; Lilley, D.M.J. Structure of the guanidine III riboswitch. Cell Chem. Biol. 2017, 24, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Z. DNA 8mer containing two 2SeT modifications. Unpublished work. 2016. [Google Scholar]
- Zhang, W.; Tam, C.P.; Zhou, L.; Oh, S.S.; Wang, J.; Szostak, J.W. Structural Rationale for the Enhanced Catalysis of Nonenzymatic RNA Primer Extension by a Downstream Oligonucleotide. J. Am. Chem. Soc. 2018, 140, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.A.; Tykac, M.; Kovalevskiy, O.; Murshudov, G.N. Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM. Acta Cryst. 2018, D74, 492–505. [Google Scholar] [CrossRef]
- Burla, M.C.; Giacovazzo, C.; Mazzone, A.; Polidori, G.; Siliqi, D. About the σA estimate. Acta Cryst. 2011, A67, 276–283. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model With | ||||||
|---|---|---|---|---|---|---|
| 5 Overlapping Atoms | 3 Overlapping Atoms | |||||
| SYNTH. | CRIT1 | CRIT2 | CRIT3 | CRIT1 | CRIT2 | CRIT3 |
| F | 0.71 | 0.29 | 0.18 | 0.45 | 0.21 | 0.19 |
| mF | 0.80 | 0.46 | 0.24 | 0.54 | 0.34 | 0.27 |
| mFp | 0.66 | 0.19 | 0.12 | 0.42 | 0.13 | 0.13 |
| w1F | 0.87 | 0.64 | 0.27 | 0.61 | 0.44 | 0.30 |
| w2F | 0.81 | 0.45 | 0.34 | 0.51 | 0.30 | 0.37 |
| w3F | 0.87 | 0.60 | 0.39 | 0.57 | 0.39 | 0.43 |
| F-(1-m)Fp | 0.92 | 0.77 | 0.29 | 0.66 | 0.52 | 0.31 |
| F-Fp | 0.43 | 0.40 | ||||
| mF-DFp | 0.60 | 0.53 | ||||
| mF-Fp | 0.51 | 0.52 | ||||
| <Fq> | 0.49 | 0.41 | ||||
| wq<Fq> | 0.48 | 0.41 | ||||
| F-mFp | 0.20 | 0.19 | ||||
| mF-(1-m)Fp | 0.33 | 0.39 | ||||
| m(F-Fp) | 0.45 | 0.43 | ||||
| (1-m)(F-Fp) | 0.35 | 0.28 | ||||
| w1|ΔF| | 0.53 | 0.57 | ||||
| F-2Fp | 0.46 | 0.44 | ||||
| mF-2Fp | 0.50 | 0.54 | ||||
| F-2mFp | −0.03 | 0.01 | ||||
| mF-2(1-m)Fp | 0.38 | 0.45 | ||||
| mF-2DFp | 1.13 | 1.15 | ||||
| 2F-Fp | 0.92 | 0.76 | 0.28 | 0.65 | 0.53 | 0.28 |
| 2mF-Fp | 1.69 | 6.65 | 0.39 | 2.31 | 2.48 | 0.46 |
| 2F-mFp | 0.74 | 0.36 | 0.19 | 0.47 | 0.26 | 0.20 |
| 2mF-(1-m)Fp | 0.95 | 0.84 | 0.29 | 0.71 | 0.58 | 0.34 |
| 2mF-DFp | 0.98 | 0.95 | 0.35 | 0.61 | 0.45 | 0.36 |
| PDB | nc1 | nc2 | nc3 | nc4 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1aa5 [32] | 65 | 12 | 18 | 12 | 17 | 8 | 21 | 12 | 18 |
| 1alz [33] | 68 | 16 | 24 | 12 | 23 | 12 | 26 | 16 | 24 |
| 1byz [34] | 73 | 24 | 23 | 16 | 21 | 12 | 22 | 16 | 20 |
| 1c75 [35] | 69 | 21 | 19 | 13 | 18 | 13 | 21 | 13 | 18 |
| 1ctj [36] | 55 | 13 | 44 | 9 | 41 | 9 | 42 | 17 | 43 |
| 1hhy [37] | 74 | 20 | 23 | 14 | 23 | 16 | 25 | 16 | 23 |
| 1ick [38] | 73 | 16 | 18 | 12 | 17 | 12 | 19 | 12 | 17 |
| 1irn [39] | 64 | 9 | 58 | 21 | 32 | 17 | 32 | 25 | 30 |
| 1p9i [40] | 74 | 24 | 51 | 24 | 40 | 24 | 36 | 24 | 36 |
| 1sho [41] | 75 | 11 | 58 | 11 | 55 | 2 | 68 | 11 | 49 |
| 1ea7 [42] | 75 | 25 | 73 | 25 | 66 | 25 | 28 | 25 | 41 |
| 1het [43] | 75 | 25 | 72 | 25 | 71 | 25 | 71 | 25 | 73 |
| 1heu [43] | 64 | 25 | 43 | 25 | 41 | 25 | 42 | 25 | 41 |
| 1n8k [44] | 74 | 25 | 71 | 25 | 68 | 25 | 61 | 25 | 68 |
| 1us0 [45] | 72 | 25 | 24 | 25 | 14 | 25 | 26 | 25 | 24 |
| 1w8f [46] | 73 | 25 | 47 | 25 | 35 | 25 | 34 | 25 | 34 |
| 2bw4 [47] | 75 | 25 | 15 | 25 | 15 | 25 | 18 | 25 | 16 |
| PDB | |||||
|---|---|---|---|---|---|
| 3ce5 [49] | 62 | 49 | 47 | 49 | 47 |
| 3eil [50] | 64 | 46 | 41 | 44 | 41 |
| 3n4o [51] | 35 | 34 | 30 | 32 | 31 |
| 3tok [52] | 61 | 49 | 44 | 47 | 44 |
| 4gsg [53] | 53 | 53 | 50 | 52 | 51 |
| 4ms5 [54] | 73 | 57 | 59 | 60 | 58 |
| 4xqz [55] | 52 | 49 | 46 | 45 | 45 |
| 5dwx [56] | 77 | 67 | 65 | 65 | 65 |
| 5i4s [57] | 50 | 36 | 33 | 36 | 33 |
| 5ihd [55] | 70 | 39 | 39 | 38 | 39 |
| 5ju4 [58] | 51 | 26 | 22 | 25 | 23 |
| 5lj4 [59] | 45 | 30 | 27 | 28 | 27 |
| 5mvt [60] | 64 | 28 | 25 | 28 | 26 |
| 5nt5 [61] | 24 | 25 | 20 | 23 | 20 |
| 5t4w [62] | 46 | 25 | 20 | 23 | 20 |
| 1iha [63] | 38 | 41 | 38 | 41 | 38 |
| 1z7f [64] | 38 | 34 | 31 | 33 | 31 |
| 2a0p [65] | 33 | 31 | 29 | 30 | 28 |
| 2fd0 [66] | 49 | 33 | 30 | 33 | 30 |
| 2pn4 [67] | 46 | 38 | 35 | 36 | 35 |
| 3d2v [68] | 78 | 58 | 58 | 57 | 57 |
| 3fs0 [69] | 68 | 32 | 29 | 41 | 28 |
| 4enc [70] | 29 | 28 | 26 | 26 | 26 |
| 5kvj [71] | 60 | 49 | 49 | 50 | 50 |
| 5l4o [72] | 67 | 35 | 38 | 34 | 37 |
| 5nz6 [73] | 40 | 26 | 24 | 25 | 24 |
| 5tgp [74] | 70 | 34 | 32 | 37 | 32 |
| 5uz6 [75] | 75 | 34 | 32 | 34 | 32 |
| 6az4 [75] | 55 | 42 | 41 | 42 | 40 |
| Overall | 54.2 | 38.9 | 36.6 | 38.4 | 36.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. Properties of Fourier Syntheses and New Syntheses. Crystals 2020, 10, 538. https://doi.org/10.3390/cryst10060538
Burla MC, Carrozzini B, Cascarano GL, Giacovazzo C, Polidori G. Properties of Fourier Syntheses and New Syntheses. Crystals. 2020; 10(6):538. https://doi.org/10.3390/cryst10060538
Chicago/Turabian StyleBurla, Maria Cristina, Benedetta Carrozzini, Giovanni Luca Cascarano, Carmelo Giacovazzo, and Giampiero Polidori. 2020. "Properties of Fourier Syntheses and New Syntheses" Crystals 10, no. 6: 538. https://doi.org/10.3390/cryst10060538
APA StyleBurla, M. C., Carrozzini, B., Cascarano, G. L., Giacovazzo, C., & Polidori, G. (2020). Properties of Fourier Syntheses and New Syntheses. Crystals, 10(6), 538. https://doi.org/10.3390/cryst10060538