Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands


Abstract
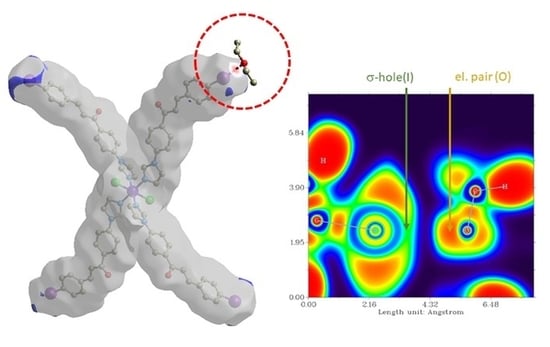
{kind=link}
{kind=link}
{kind=link}
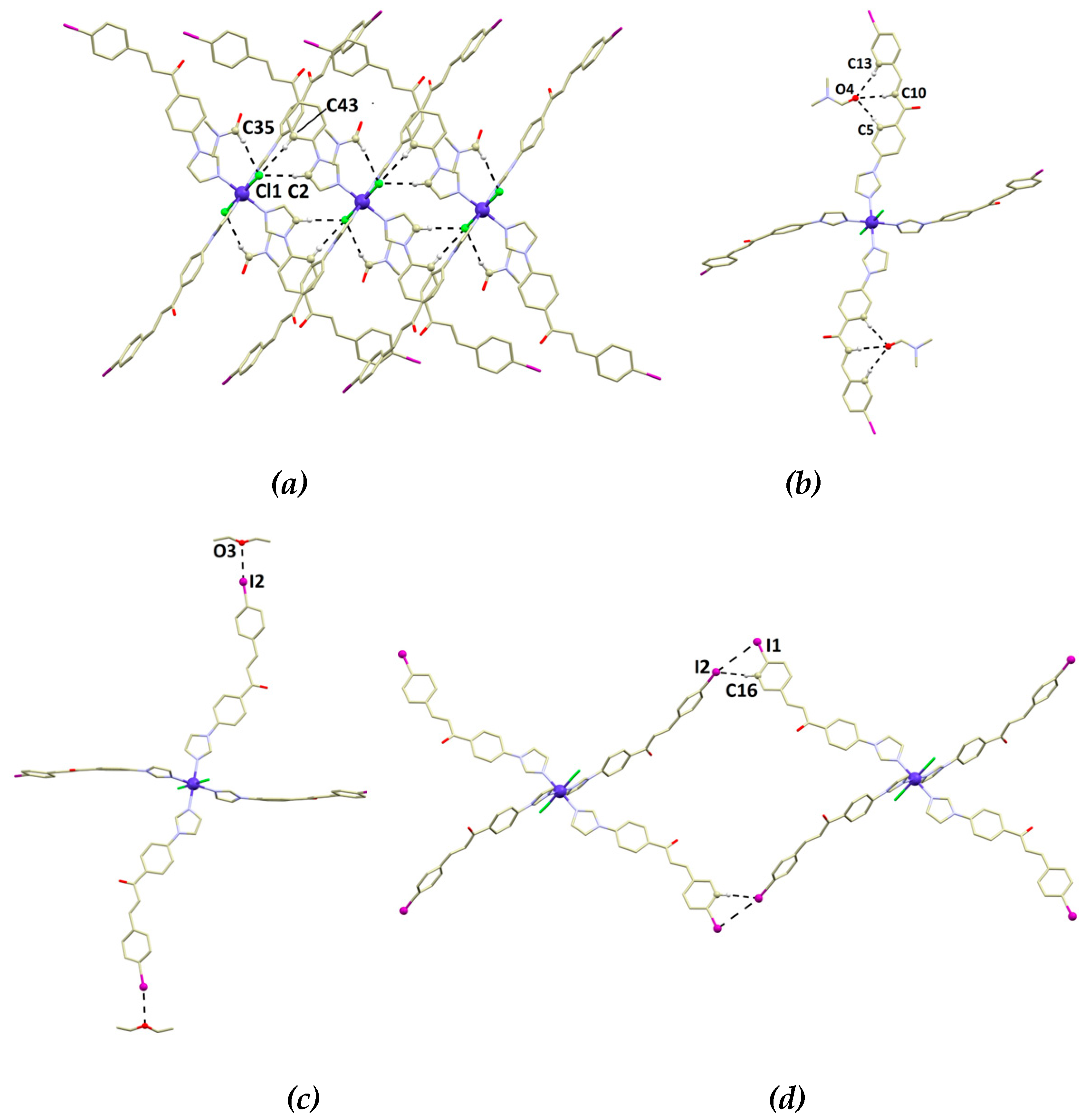{kind=link}
{kind=link}
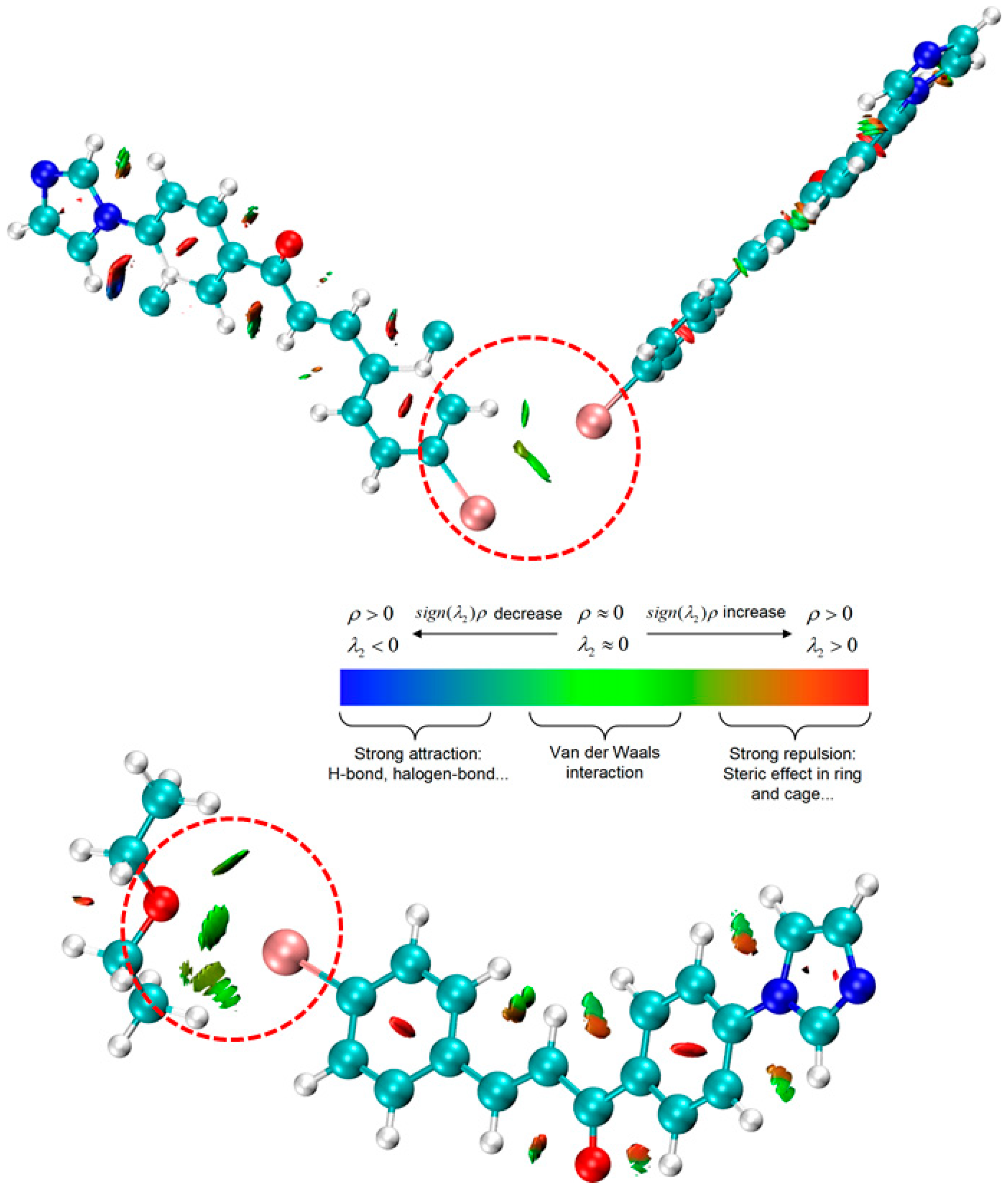{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials
2.2. General Methods
2.3. Crystal Structure Determination
2.4. Theoretical Calculations
2.5. Synthesis of (2E)-1-[4-(1H-imidazol-1-yl)phenyl]-3-(4-iodophenyl)prop-2-en-1-one (4I-L)
2.6. Complex [Co(4I-L)2(Cl)2] (1a)
3. Results and Discussion
3.1. Synthesis and Basic Characterizations
3.2. Crystal Structure
3.3. Theoretical Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jeffrey, G.A. Hydrogen-Bonding: An update. Crystallogr. Rev. 2003, 9, 135–176. [Google Scholar] [CrossRef]
- Steiner, T. C–H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Tiekink, E.R.T.; Zukerman-Schpector, J. Gold···π aryl interactions as supramolecular synthons. CrystEngComm 2009, 11, 1176. [Google Scholar] [CrossRef]
- Chen, T.; Li, M.; Liu, J. π–π Stacking Interaction: A Nondestructive and Facile Means in Material Engineering for Bioapplications. Cryst. Growth Des. 2018, 18, 2765–2783. [Google Scholar] [CrossRef]
- Watt, M.M.; Collins, M.S.; Johnson, D.W. Ion−π Interactions in Ligand Design for Anions and Main Group Cations. Acc. Chem. Res. 2012, 46, 955–966. [Google Scholar] [CrossRef]
- Bauzá, A.; Seth, S.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From Noncovalent Chalcogen–Chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Carreras, L.; Benet-Buchholz, J.; Franconetti, A.; Frontera, A.; Van Leeuwen, P.W.N.M.; Vidal-Ferran, A. Halogen bonding effects on the outcome of reactions at metal centres. Chem. Commun. 2019, 55, 2380–2383. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cotelle, Y.; Sakai, N.; Matile, S. Unorthodox Interactions at Work. J. Am. Chem. Soc. 2016, 138, 4270–4277. [Google Scholar] [CrossRef] [PubMed]
- Amabilino, D.B.; Smith, D.K.; Steed, J.W. Supramolecular materials. Chem. Soc. Rev. 2017, 46, 2404–2420. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011, 111, 6810–6918. [Google Scholar] [CrossRef]
- El-Mellouhi, F.; Bentria, E.T.; Marzouk, A.; Rashkeev, S.N.; Kais, S.; Alharbi, F.H. Hydrogen bonding: A mechanism for tuning electronic and optical properties of hybrid organic–inorganic frameworks. npj Comput. Mater. 2016, 2, 16035. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; Van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Sivaguru, J. Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition. Chem. Rev. 2016, 116, 9914–9993. [Google Scholar] [CrossRef]
- Mako, T.; Racicot, J.M.; Levine, M. Supramolecular Luminescent Sensors. Chem. Rev. 2018, 119, 322–477. [Google Scholar] [CrossRef]
- Kahn, O. Magnetism: A Supramolecular Function; Springer: Dordrecht, The Netherlands, 1996; ISBN 978-94-015-8707-5. [Google Scholar]
- Nemec, I.; Herchel, R.; Šilha, T.; Trávníček, Z. Towards a better understanding of magnetic exchange mediated by hydrogen bonds in Mn(III)/Fe(III) salen-type supramolecular dimers. Dalton Trans. 2014, 43, 15602–15616. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Šalitroš, I.; Travnicek, Z.; Moncol, J.; Fuess, H.; Ruben, M.; Linert, W. Anion driven modulation of magnetic intermolecular interactions and spin crossover properties in an isomorphous series of mononuclear iron(iii) complexes with a hexadentate Schiff base ligand. CrystEngComm 2012, 14, 7015. [Google Scholar] [CrossRef]
- Herchel, R.; Nemec, I.; Machata, M.; Travnicek, Z. Experimental and Theoretical Investigations of Magnetic Exchange Pathways in Structurally Diverse Iron(III) Schiff-Base Complexes. Inorg. Chem. 2015, 54, 8625–8638. [Google Scholar] [CrossRef] [PubMed]
- Nemec, I.; Herchel, R.; Travnicek, Z. Ferromagnetic coupling mediated by Co···π non-covalent contacts in a pentacoordinate Co(ii) compound showing field-induced slow relaxation of magnetization. Dalton Trans. 2016, 45, 12479–12482. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Travnicek, Z. The relationship between the strength of hydrogen bonding and spin crossover behaviour in a series of iron(iii) Schiff base complexes. Dalton Trans. 2015, 44, 4474–4484. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.F.; Taher, D.; Haddad, S.F.; Turnbull, M.M. Competition between Hydrogen and Halogen Bonding Interactions: Theoretical and Crystallographic Studies. Cryst. Growth Des. 2014, 14, 1961–1971. [Google Scholar] [CrossRef]
- Gómez-Coca, S.; Aravena, D.; Morales, R.; Ruiz, E. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289, 379–392. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Kern, M.; Neugebauer, P.; Van Slageren, J.; Travnicek, Z. Magnetic Anisotropy and Field-induced Slow Relaxation of Magnetization in Tetracoordinate CoII Compound [Co(CH3-im)2Cl2]. Materials 2017, 10, 249. [Google Scholar] [CrossRef]
- Alvarez, S. Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Sobczyk, L.; Chudoba, D.; Tolstoy, P.M.; Filarowski, A. Some Brief Notes on Theoretical and Experimental Investigations of Intramolecular Hydrogen Bonding. Molecules 2016, 21, 1657. [Google Scholar] [CrossRef]
- Sen, S.; Kaseman, D.C.; Colas, B.; Jacob, D.; Clark, S. Hydrogen bonding induced distortion of CO 3 units and kinetic stabilization of amorphous calcium carbonate: Results from 2D 13 C NMR spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 20330–20337. [Google Scholar] [CrossRef]
- Fielden, J.; Long, D.L.; Speldrich, M.; Kögerler, P.; Cronin, L. [CoxCu1−x(DDOP)(OH2)(NO3)](NO3): Hydrogen bond-driven distortion of cobalt(ii) by solid solution ‘network mismatch’. Dalton Trans. 2012, 41, 4927. [Google Scholar] [CrossRef] [PubMed]
- Dahl, E.W.; Szymczak, N.K. Hydrogen Bonds Dictate the Coordination Geometry of Copper: Characterization of a Square-Planar Copper(I) Complex. Angew. Chem. 2016, 128, 3153–3157. [Google Scholar] [CrossRef]
- Hussain, T.; Siddiqui, H.L.; Zia-Ur-Rehman, M.; Yasinzai, M.M.; Parvez, M. Anti-oxidant, anti-fungal and anti-leishmanial activities of novel 3-[4-(1H-imidazol-1-yl) phenyl] prop-2-en-1-ones. Eur. J. Med. Chem. 2009, 44, 4654–4660. [Google Scholar] [CrossRef] [PubMed]
- Bauman, V.T.; Shults, E.E.; Kononchuk, V.V.; Bagryanskaya, I.Y.; Shakirov, M.M.; Tolstikov, G.A. Synthetic Transformations of Isoquinoline Alkaloids. 1-Alkynyl-3,6-dimethoxy-N-methyl-4,5α-epoxy-6,18-endoethenobenzo[i]isomorphinans and Their Transformations. Russ. J. Org. Chem. 2013, 49, 1502–1513. [Google Scholar] [CrossRef]
- Cinčić, D.; Friščić, T.; Jones, W. Isostructural Materials Achieved by Using Structurally Equivalent Donors and Acceptors in Halogen-Bonded Cocrystals. Chem. A Eur. J. 2008, 14, 747–753. [Google Scholar] [CrossRef]
- Turunen, L.; Beyeh, N.K.; Pan, F.; Valkonen, A.; Rissanen, K. Tetraiodoethynyl resorcinarene cavitands as multivalent halogen bond donors. Chem. Commun. 2014, 50, 15920–15923. [Google Scholar] [CrossRef]
- Bock, H.; Holl, S. Wechselwirkungen in Molekülkristallen, 167 [1,2]. Kristallzüchtung und Strukturbestimmung von σ-Donator/Akzeptor-Komplexen zwischen 1,4-Dioxan und den Polyiod-Molekülen I2,I2C=CI2, (IC)4S sowie (IC)4NR (R = H, CH3)/Interaction in Molecular Crystals, 167 [1,2]. C rystallization and Structure Determination of σ-Donor/Acceptor Complexes between 1,4-Dioxane and the Polyiodine Molecules I2,I2C=CI2, (IC)4S and (IC)4NR (R = H, CH3). Z. Für Naturforsch. B 2001, 56, 111–121. [Google Scholar] [CrossRef]
- Turunen, L.; Pan, F.; Beyeh, N.K.; Trant, J.F.; Ras, R.H.A.; Rissanen, K. Bamboo-like Chained Cavities and Other Halogen-Bonded Complexes from Tetrahaloethynyl Cavitands with Simple Ditopic Halogen Bond Acceptors. Cryst. Growth Des. 2017, 18, 513–520. [Google Scholar] [CrossRef]
- Chu, Q.; Wang, Z.; Huang, Q.; Yan, C.; Zhu, S. Fluorine-containing donor-acceptor complexes: Crystallographic study of the interactions between electronegative atoms (N, O, S) and halogen atoms (I, Br). New J. Chem. 2003, 27, 1522. [Google Scholar] [CrossRef]
- Roper, L.C.; Präsang, C.; Kozhevnikov, V.N.; Whitwood, A.C.; Karadakov, P.B.; Bruce, D. Experimental and Theoretical Study of Halogen-Bonded Complexes of DMAP with Di- and Triiodofluorobenzenes. A Complex with a Very Short N···I Halogen Bond. Cryst. Growth Des. 2010, 10, 3710–3720. [Google Scholar] [CrossRef]
- Pearson, R.J.; Evans, K.M.; Slawin, A.M.Z.; Philp, D.; Westwood, N.J. Controlling the Outcome of an N-Alkylation Reaction by UsingN-Oxide Functional Groups. J. Org. Chem. 2005, 70, 5055–5061. [Google Scholar] [CrossRef] [PubMed]
- Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A.D. Tri-s-triazine derivatives. Part I. From trichloro-tri-s-triazine to graphitic C3N4 structures. New J. Chem. 2002, 26, 508–512. [Google Scholar] [CrossRef]
- Sun, W.; Zhu, G.; Wu, C.; Li, G.; Hong, L.; Wang, R. Organocatalytic Diastereo- and Enantioselective 1,3-Dipolar Cycloaddition of Azlactones and Methyleneindolinones. Angew. Chemie Int. Ed. 2013, 52, 8633–8637. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, A.; Reglinski, J.; Kennedy, A.R. The use of hydrogenated Schiff base ligands in the synthesis of multi-metallic compounds. Inorg. Chim. Acta 2009, 362, 1267–1274. [Google Scholar] [CrossRef]
- Groom, C.; Bruno, I.J.; Lightfoot, M.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment Olex2 dissected. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Koziskova, J.; Hahn, F.; Richter, J.; Kozisek, J. Comparison of different absorption corrections on the model structure of tetrakis (μ2-acetato)-diaquadicopper(II). Acta Chim. Slovaca. 2016, 9, 136–140. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Najibi, A.; Goerigk, L. The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches. J. Chem. Theory Comput. 2018, 14, 5725–5738. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Sekiya, M.; Koga, T. Segmented contracted basis sets for atoms H through Xe: Sapporo-(DK)-nZP sets (n = D, T, Q). Theor. Chem. Acc. 2012, 131, 131. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Mitra, M.; Choudhury, S.R.; Bauzá, A.; Frontera, A.; Mukhopadhyay, S. Experimental and Computational Study of Counterintuitive ClO4−…ClO4− Interactions and the Interplay between and Anion + Interactions. Cryst. Growth Des. 2014, 14, 5812–5821. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 3.1; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- Osborne, S.; Wellens, S.; Felton, S.; Gunaratne, H.Q.N.; Nockemann, P.; Ward, C.; Bowman, R.; Binnemans, K.; Swadzba-Kwasny, M. Thermochromism and switchable paramagnetism of cobalt(ii) in thiocyanate ionic liquids. Dalton Trans. 2015, 44, 11286–11289. [Google Scholar] [CrossRef] [PubMed]
- Lomjanský, D.; Varga, F.; Rajnák, C.; Moncol, J.; Boča, R.; Titis, J. Redetermination of Zero-Field Splitting in [Co(qu)2Br2] and [Ni(PPh3)2Cl2] Complexes. Nova Biotechnol. Chim. 2016, 15, 200–211. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Mitra, M.; Das, A.; Singh, N.J.; Choudhury, S.R.; Kar, T.; Mukhopadhyay, S. A successive layer-by-layer assembly of supramolecular frameworks driven by a novel type of face-to-face π+–π+ interactions. CrystEngComm 2013, 15, 7879–7886. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Seth, S.; Zubkov, F.I.; Torres, E.S.L.; Bacchi, A.; Stilinović, V.; Frontera, A. Supramolecular Assemblies in Pb(II) Complexes with Hydrazido-Based Ligands. Crystals 2019, 9, 323. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-Garcia, J.; Cohen, A.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J.; Becke, A. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Meaning and Functional Form of the Electron Localization Function. Acta Phys. Chim. Sin. 2011, 27, 2786–2792. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masaryk, L.; Moncol, J.; Herchel, R.; Nemec, I. Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands. Crystals 2020, 10, 354. https://doi.org/10.3390/cryst10050354
Masaryk L, Moncol J, Herchel R, Nemec I. Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands. Crystals. 2020; 10(5):354. https://doi.org/10.3390/cryst10050354
Chicago/Turabian StyleMasaryk, Lukáš, Ján Moncol, Radovan Herchel, and Ivan Nemec. 2020. "Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands" Crystals 10, no. 5: 354. https://doi.org/10.3390/cryst10050354
APA StyleMasaryk, L., Moncol, J., Herchel, R., & Nemec, I. (2020). Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands. Crystals, 10(5), 354. https://doi.org/10.3390/cryst10050354