Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature

 ,
, 

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Experimental
3. Results and Discussion
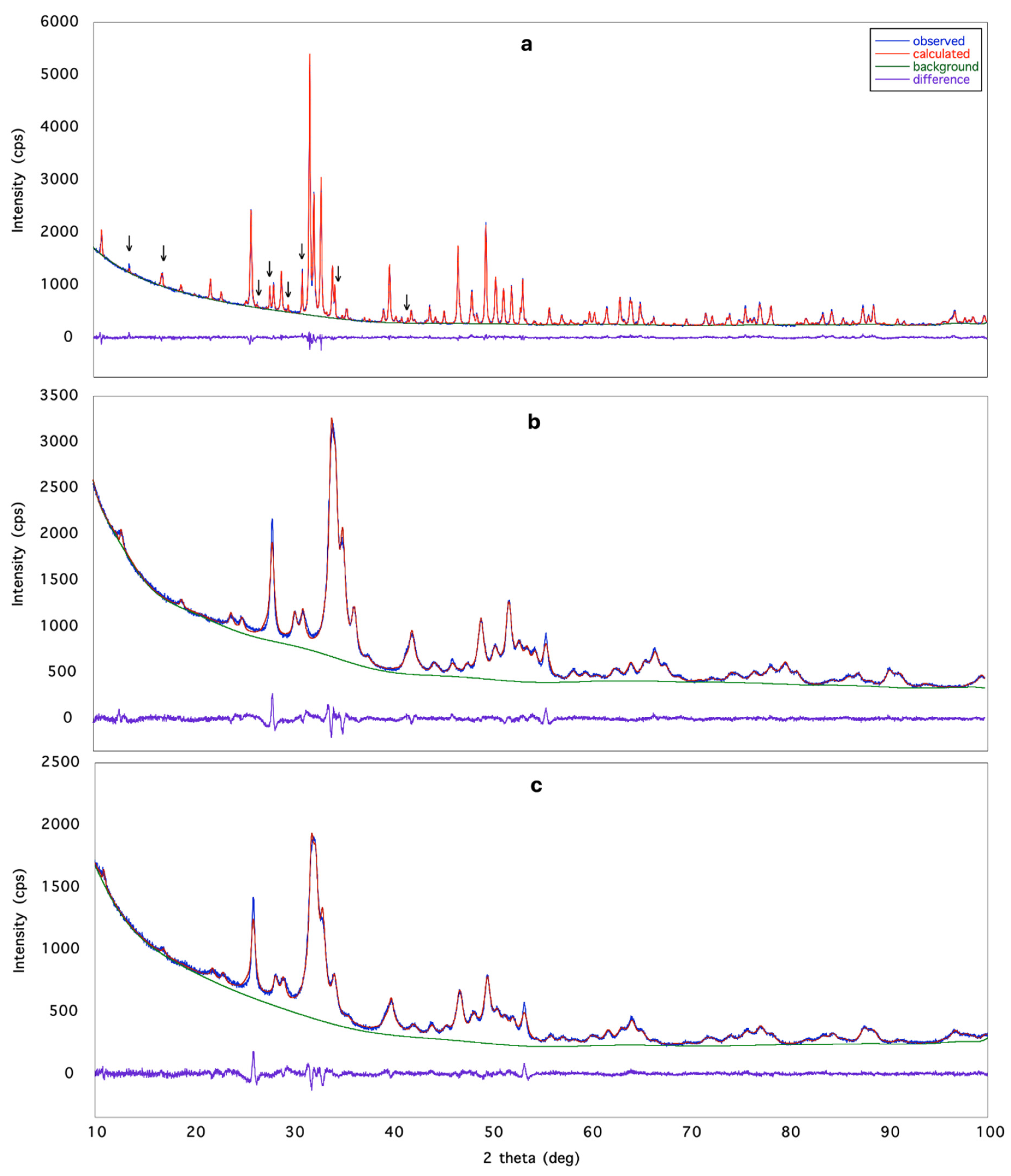
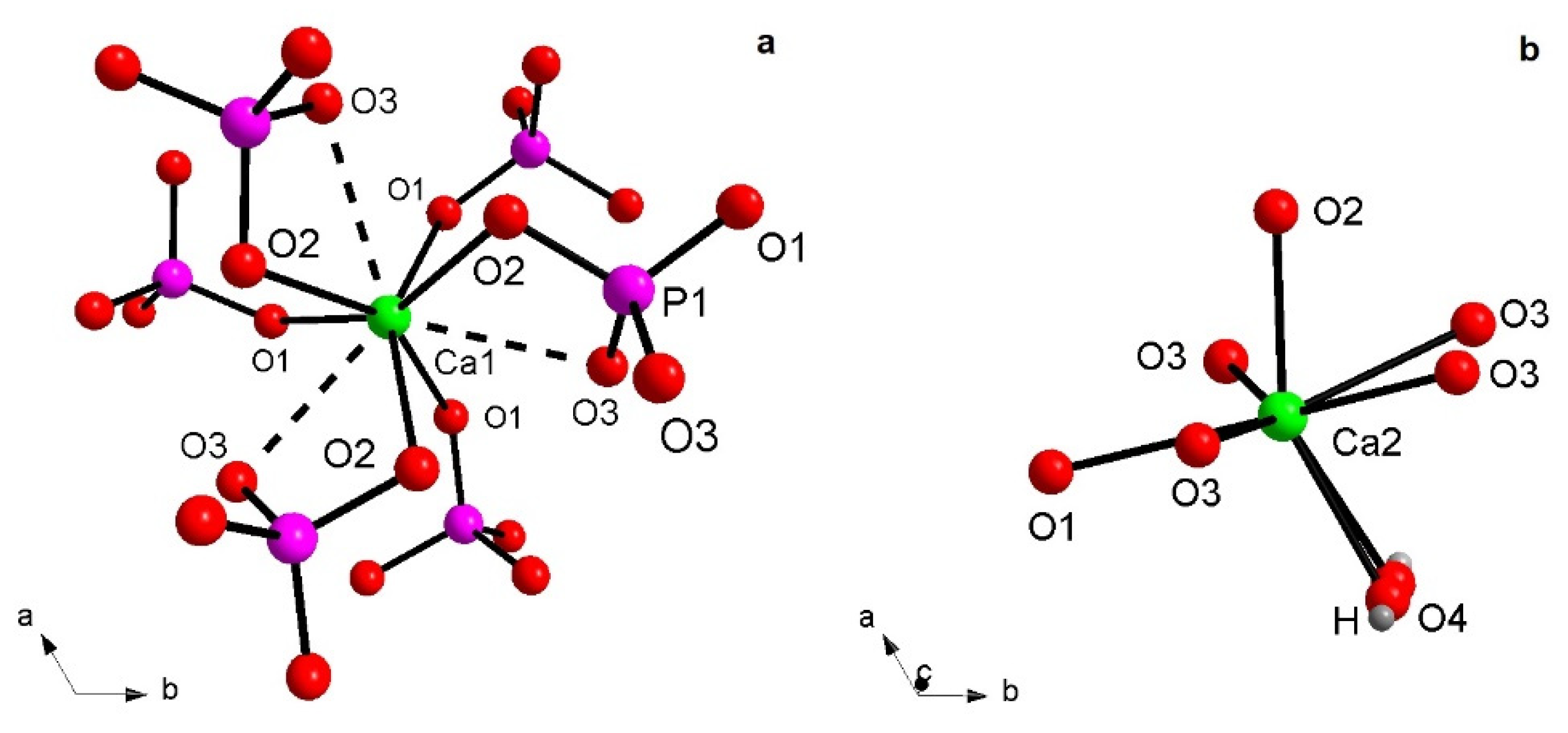
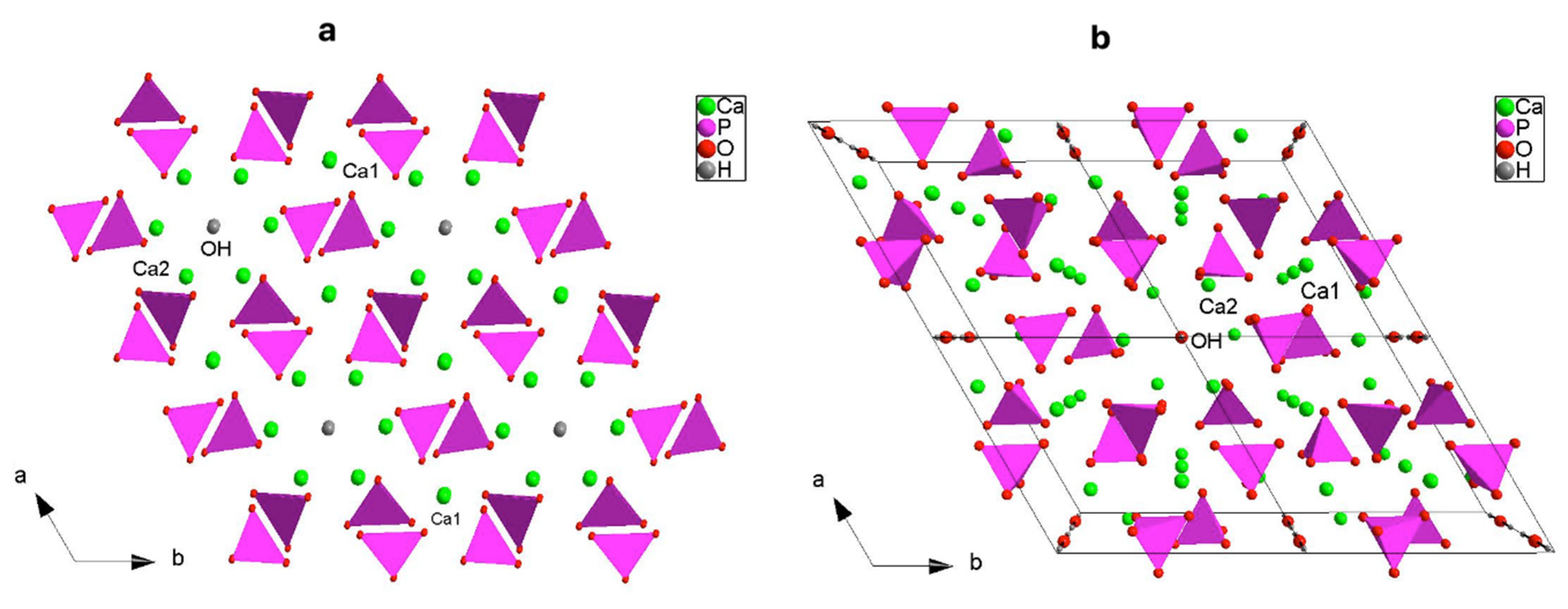
3.1. X-ray Structural Study
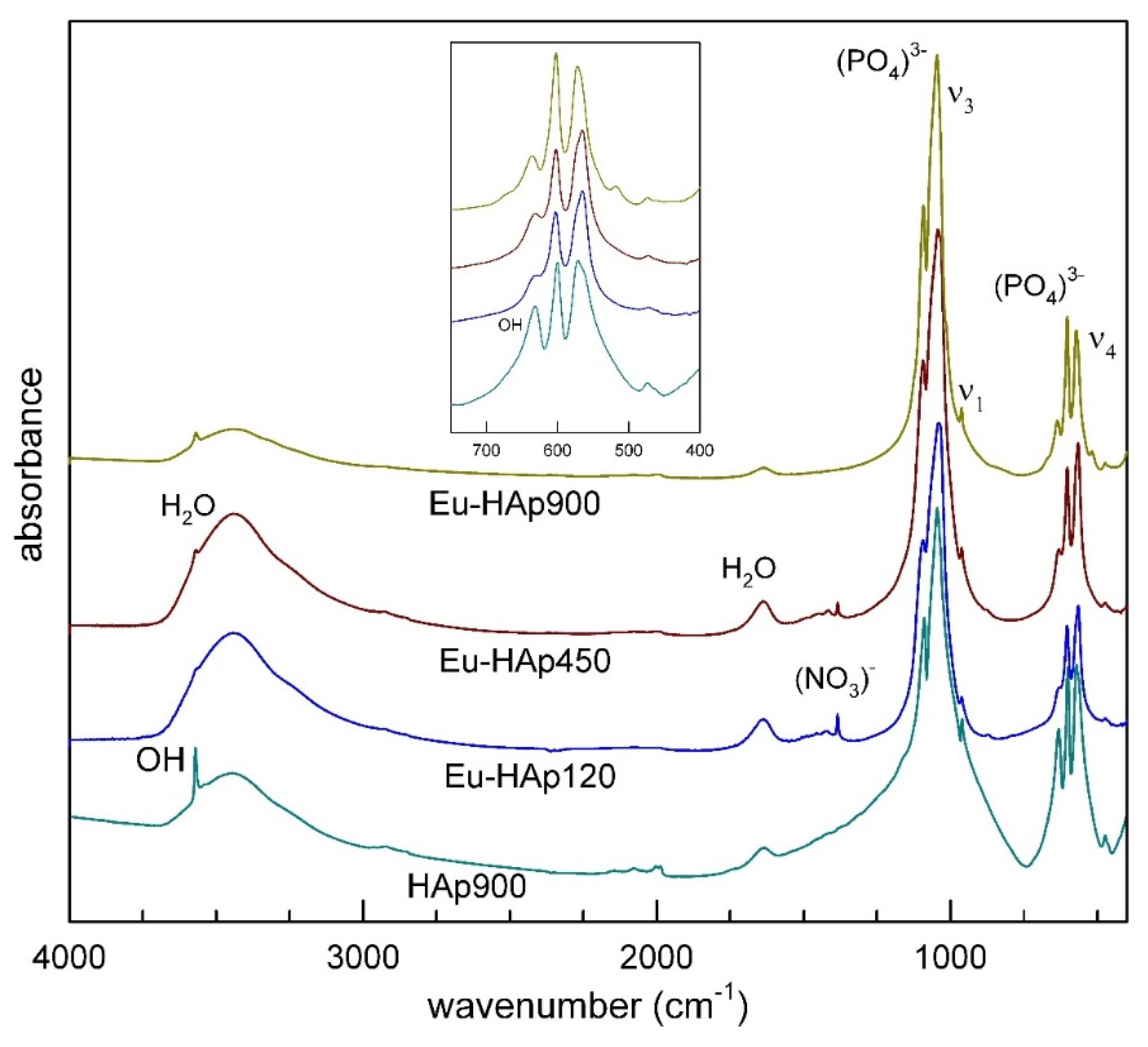
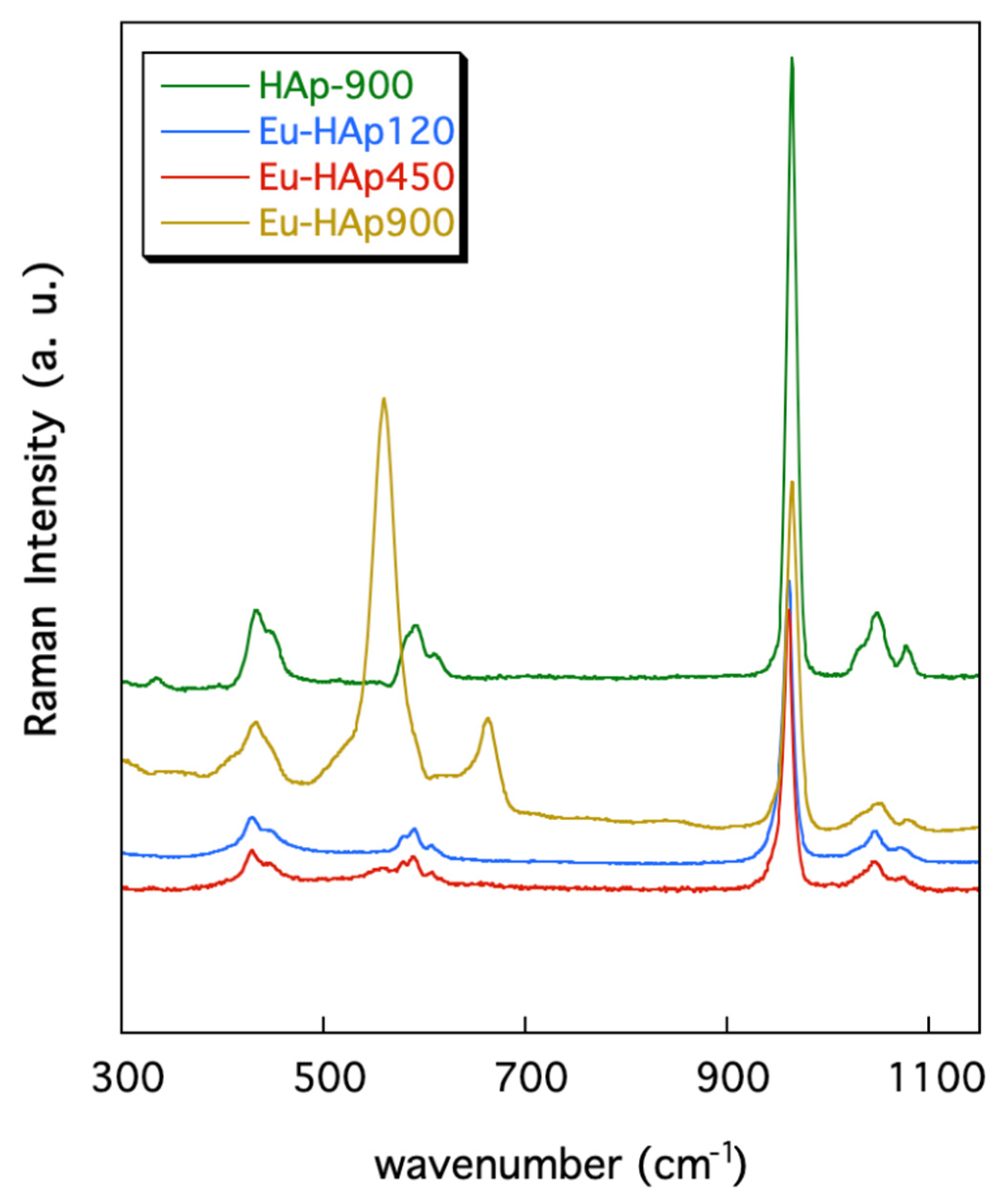
3.2. FTIR and Raman Spectroscopy
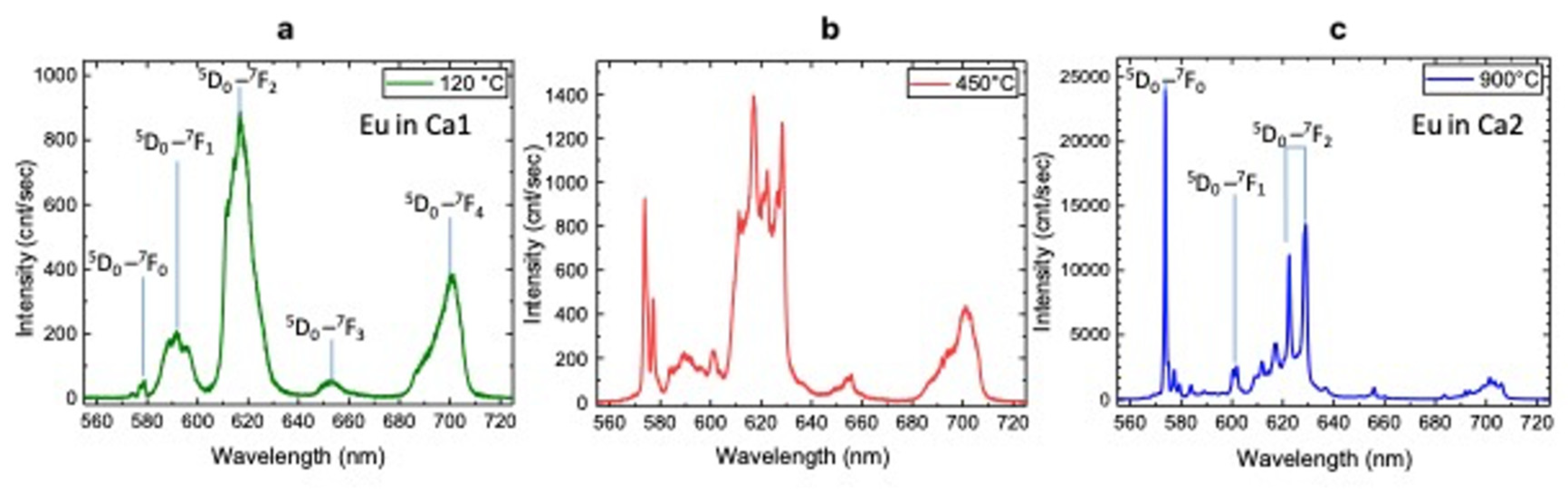
3.3. Luminescence Spectroscopy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorozhkin, S.V. Calcium Orthophosphates: Occurrence, Properties and Major Applications. Bioceram. Dev. Appl. 2014, 4, 2. [Google Scholar] [CrossRef]
- Prichodko, A.; Enrichi, F.; Stankeviciute, Z.; Benedetti, A.; Grigoraviciute-Puroniene, I.; Kareiva, A. Study of Eu3+ and Tm3+ substitution effects in sol–gel fabricated calcium hydroxyapatite. J. Sol-Gel Sci. Technol. 2017, 81, 261–267. [Google Scholar] [CrossRef]
- Hughes, J.M.; Rakovan, J. The Crystal Structure of Apatite, Ca5 (PO4)3 (F, OH, Cl). Rev. Mineral. Geochem. 2002, 48, 1–12. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphate bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Ignjatović, N.L.; Mančić, L.; Vuković, M.; Stojanović, Z.; Nikolić, M.G.; Škapin, S.; Jovanović, S.; Veselinović, L.; Uskoković, V.; Lazić, S.; et al. Rare-earth (Gd3+,Yb3+/Tm3+, Eu3+) co-doped hydroxyapatite as magnetic, up-conversion and down-conversion materials for multimodal imaging. Sci. Rep. 2019, 9, 16305. [Google Scholar] [CrossRef] [PubMed]
- Suchanek, W.; Yoshimura, M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 1998, 13, 94–117. [Google Scholar] [CrossRef]
- El Khouri, A.; Elaatmani, M.; Della Ventura, G.; Sodo, A.; Rizzi, R.; Rossi, M.; Capitelli, F. Synthesis, structure refinement and vibrational spectroscopy of new rare-earth tricalcium phosphates Ca9RE(PO4)7 (RE = La, Pr, Nd, Eu, Gd, Dy, Tm, Yb). Ceram. Int. 2017, 43, 15645–15653. [Google Scholar] [CrossRef]
- Capitelli, F.; Rossi, M.; ElKhouri, A.; Elaatmani, M.; Corriero, N.; Sodo, A.; Della Ventura, G. Synthesis, structural model and vibrational spectroscopy of lutetium tricalcium phosphate Ca9Lu(PO4)7. J. Rare Earths 2018, 36, 1162–1168. [Google Scholar] [CrossRef]
- Neacsu, I.A.; Stoica, A.E.; Vasile, B.S.; Andronescu, E. Luminescent Hydroxyapatite Doped with Rare Earth Elements for Biomedical Applications. Nanomaterials 2019, 9, 239. [Google Scholar] [CrossRef]
- Albin, M.; Horrocks, W.D. Europium (III) luminescence excitation spectroscopy. Quantitive correlation between the total charge on the ligands and the 7F0 → 5D0 transition frequency in europium (III) complexes. Inorg. Chem. 1985, 24, 895–900. [Google Scholar] [CrossRef]
- Wiglusz, R.J.; Bednarkiewicz, A.; Strek, W. Synthesis and optical properties of Eu3+ ion doped nanocrystalline hydroxya patites embedded in PMMA matrix. J. Rare Earths 2011, 29, 1111–1116. [Google Scholar] [CrossRef]
- Zavala-Sanchez, L.A.; Hirata, G.A.; Novitskaya, E.; Karandikar, K.; Herrera, M.; Graeve, O.A. Distribution of Eu2+ and Eu3+ Ions in Hydroxyapatite: A Cathodoluminescence and Raman Study. ACS Biomater. Sci. Eng. 2015, 1, 1306–1313. [Google Scholar] [CrossRef]
- Garcia Dominguez, G.; Garrido Hernandez, A.; Ceron Montes, O.; Morales Ramirez, A.J.; Diaz De La Torre, S. Structural and luminescent properties of europium-doped and undoped hydroxyapatite powders sintered by spark plasma. Ceram. Silik. 2019, 100–109. [Google Scholar] [CrossRef]
- Chen, F.; Zhu, Y.-J.; Zhang, K.-H.; Wu, J.; Wang, K.-W.; Tang, Q.-L.; Mo, X.-M. Europium-doped amorphous calcium phosphate porous nanospheres: Preparation and application as luminescent drug carriers. Nanoscale Res. Lett. 2011, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Dembski, S.; Rupp, S.; Milde, M.; Gellermann, C.; Dyrba, M.; Schweizer, S.; Batentschuk, M.; Osvet, A.; Winnacker, A. Synthesis and optical properties of luminescent core–shell structured silicate and phosphate nanoparticles. Opt. Mater. 2011, 33, 1106–1110. [Google Scholar] [CrossRef]
- Mobasherpour, I.; Heshajin, M.S.; Kazemzadeh, A.; Zakeri, M. Synthesis of nanocrystalline hydroxyapatite by using precipitation method. J. Alloys Compd. 2007, 430, 330–333. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.; Gong, H.; Jiang, X.; Wang, H.; Li, K. Effects of synthesis conditions on the morphology of hydroxyapatite nanoparticles produced by wet chemical process. Powder Technol. 2010, 203, 315–321. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.-T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Ciobanu, C.S.; Iconaru, S.L.; Massuyeau, F.; Constantin, L.V.; Costescu, A.; Predoi, D. Synthesis, Structure, and Luminescent Properties of Europium-Doped Hydroxyapatite Nanocrystalline Powders. J. Nanomater. 2012, 2012, 942801. [Google Scholar] [CrossRef]
- Silva, F.R.O.; Lima, N.B.; Guilhen, S.N.; Courrol, L.C.; Bressiani, A.H.A. Evaluation of europium-doped HA/β-TCP ratio fluorescence in biphasic calcium phosphate nanocomposites controlled by the pH value during the synthesis. J. Lumin. 2016, 180, 177–182. [Google Scholar] [CrossRef]
- Nikolaev, A.; Kolesnikov, I.; Frank-Kamenetskaya, O.; Kuz’mina, M. Europium concentration effect on characteristics and luminescent properties of hydroxyapatite nanocrystalline powders. J. Mol. Struct. 2017, 1149, 323–331. [Google Scholar] [CrossRef]
- Jarcho, M.; Bolen, C.H.; Thomas, M.B.; Bobick, J.; Kay, J.F.; Doremus, R.H. Hydroxylapatite synthesis and characterization in dense polycrystalline form. J. Mater. Sci. 1976, 11, 2027–2035. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Altomare, A.; Campi, G.; Cuocci, C.; Eriksson, L.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Werner, P.-E. Advances in powder diffraction pattern indexing: N-TREOR09. J. Appl. Crystallogr. 2009, 42, 768–775. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Celotti, G.; Sprio, S. Densification behaviour and mechanisms of synthetic hydroxyapatites. J. Eur. Ceram. Soc. 2000, 20, 2377–2387. [Google Scholar] [CrossRef]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.P. Rietveld refinement of the crystallographic structure of human dental enamel apatites. Am. Mineral. 1999, 84, 1406–1414. [Google Scholar] [CrossRef]
- Rossi, M.; Ghiara, M.R.; Chita, G.; Capitelli, F. Crystal-chemical and structural characterization of fluorapatites in ejecta from Somma-Vesuvius volcanic complex. Am. Mineral. 2011, 96, 1828–1837. [Google Scholar] [CrossRef]
- Pogosova, M.A.; Eliseev, A.A.; Kazin, P.E.; Azarmi, F. Synthesis, structure, luminescence, and color features of the Eu- and Cu-doped calcium apatite. Dye. Pigment. 2017, 141, 209–216. [Google Scholar] [CrossRef]
- Aquilano, D.; Bruno, M.; Rubbo, M.; Massaro, F.R.; Pastero, L. Low Symmetry Polymorph of Hydroxyapatite. Theoretical Equilibrium Morphology of the Monoclinic Ca5(OH)(PO4)3. Cryst. Growth Des. 2014, 14, 2846–2852. [Google Scholar] [CrossRef]
- Yashima, M.; Sakai, A.; Kamiyama, T.; Hoshikawa, A. Crystal structure analysis of β-tricalcium phosphate Ca3 (PO4)2 by neutron powder diffraction. J. Solid State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]
- Kong, L.B.; Ma, J.; Boey, F. Nanosized hydroxyapatite powders derived from coprecipitation process. J. Mater. Sci. 2002, 37, 1131–1134. [Google Scholar] [CrossRef]
- Skorokhod, V.V.; Solonin, S.M.; Dubok, V.A.; Kolomiets, L.L.; Permyakova, T.V.; Shinkaruk, A.V.; Solonin, S.M.; Dubok, V.A.; Kolomiets, L.L.; Permyakova, T.V.; et al. Decomposition activation of hydroxyapatite in contact with β-tricalcium phosphate. Powder Metall. Met. Ceram. 2010, 49, 324–329. [Google Scholar] [CrossRef]
- Altomare, A.; Rizzi, R.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Paterlini, V.; Della Ventura, G.; Capitelli, F. New Ca2.90 (Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties. Crystals 2019, 9, 288. [Google Scholar] [CrossRef]
- Chiari, G.; Ferraris, G. The water molecule in crystalline hydrates studied by neutron diffraction. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 2331–2341. [Google Scholar] [CrossRef]
- Get’man, E.I.; Loboda, S.N.; Tkachenko, T.V.; Yablochkova, N.V.; Chebyshev, K.A. Isomorphous substitution of samarium and gadolinium for calcium in hydroxyapatite structure. Russ. J. Inorg. Chem. 2010, 55, 333–338. [Google Scholar] [CrossRef]
- Kazin, P.E.; Pogosova, M.A.; Trusov, L.A.; Kolesnik, I.V.; Magdysyuk, O.V.; Dinnebier, R.E. Crystal structure details of La- and Bi-substituted hydroxyapatites: Evidence for LaO+ and BiO+ with a very short metal–oxygen bond. J. Solid State Chem. 2016, 237, 349–357. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD), Version 2018-2; Fachinformationszentrum: Karlsruhe, Germany, 2018.
- Fleet, M.E.; Liu, X.; Pan, Y. Site Preference of Rare Earth Elements in Hydroxyapatite [Ca10(PO4)6(OH)2]. J. Solid State Chem. 2000, 149, 391–398. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. B Struct. Sci. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Graeve, O.A.; Kanakala, R.; Madadi, A.; Williams, B.C.; Glass, K.C. Luminescence variations in hydroxyapatites doped with Eu2+ and Eu3+ ions. Biomaterials 2010, 31, 4259–4267. [Google Scholar] [CrossRef]
- Low, H.R.; Phonthammachai, N.; Maignan, A.; Stewart, G.A.; Bastow, T.J.; Ma, L.L.; White, T.J. The Crystal Chemistry of Ferric Oxyhydroxyapatite. Inorg. Chem. 2008, 47, 11774–11782. [Google Scholar] [CrossRef]
- de Aza, P.N.; Guitián, F.; Santos, C.; de Aza, S.; Cuscó, R.; Artús, L. Vibrational Properties of Calcium Phosphate Compounds. 2. Comparison between Hydroxyapatite and β-Tricalcium Phosphate. Chem. Mater. 1997, 9, 916–922. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- El Khouri, A.; Zegzouti, A.; Elaatmani, M.; Capitelli, F. Bismuth-substituted hydroxyapatite ceramics synthesis: Morphological, structural, vibrational and dielectric properties. Inorg. Chem. Commun. 2019, 110, 107568. [Google Scholar] [CrossRef]
- Jastrzębski, W.; Sitarz, M.; Rokita, M.; Bułat, K. Infrared spectroscopy of different phosphates structures. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Iconaru, S.-L.; Motelica-Heino, M.; Predoi, D. Study on Europium-Doped Hydroxyapatite Nanoparticles by Fourier Transform Infrared Spectroscopy and Their Antimicrobial Properties. J. Spectrosc. 2013, 2013, 284285. [Google Scholar] [CrossRef]
- Markovic, M.; Fowler, B.O.; Tung, M.S. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 553. [Google Scholar] [CrossRef]
- Predoi, D.; Barsan, M.; Andronescu, E.; Vatasescu-Baltan, R.A.; Costache, M. Hydroxyapatite-iron oxide bioceramic prepared using nano-size powders. J. Optoelectron. Adv. Mater. 2007, 9, 3609–3613. [Google Scholar]
- Fowler, B.O. Infrared studies of apatites. I. Vibrational assignments for calcium, strontium, and barium hydroxyapatites utilizing isotopic substitution. Inorg. Chem. 1974, 13, 194–207. [Google Scholar] [CrossRef]
- Andronescu, E.; Predoi, D.; Neacsu, I.A.; Paduraru, A.V.; Musuc, A.M.; Trusca, R.; Oprea, O.; Tanasa, E.; Vasile, O.R.; Nicoara, A.I.; et al. Photoluminescent Hydroxylapatite: Eu3+ Doping Effect on Biological Behaviour. Nanomaterials 2019, 9, 1187. [Google Scholar] [CrossRef]
- Silva, F.R.O.; de Lima, N.B.; Bressiani, A.H.A.; Courrol, L.C.; Gomes, L. Synthesis, characterization and luminescence properties of Eu3+-doped hydroxyapatite nanocrystal and the thermal treatment effects. Opt. Mater. 2015, 47, 135–142. [Google Scholar] [CrossRef]
- Jagannathan, R.; Kottaisamy, M. Eu3+ luminescence: A spectral probe in M5 (PO4)3 X apatites (M = Ca or Sr; X = F−, Cl−, Br−or OH−). J. Phys. Condens. Matter 1995, 7, 8453–8466. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ca (wt%) | P (wt%) | Eu (wt%) | (Ca + Eu)/P | Eu/(Ca + Eu) |
|---|---|---|---|---|---|
| HAp900 | 37.85 | 17.84 | 0.00 | 1.66 | 0.00 |
| Eu-HAp120 | 32.84 | 15.74 | 3.70 | 1.66 | 0.03 |
| Eu-HAp450 | 33.84 | 17.15 | 3.68 | 1.57 | 0.03 |
| Eu-HAp900 | 36.22 | 18.75 | 4.03 | 1.54 | 0.03 |
| Crystal Formula | HAp900 | Eu-HAp120 | Eu-HAp450 | Eu-HAp900 |
|---|---|---|---|---|
| Refined formula Formula weight | Ca10(PO4)6(OH)2 1004.61 | Ca9.76Eu0.16(PO4)6(OH)2 1023.40 | Ca9.82Eu0.08(PO4)6(OH)2 1020.48 | Ca9.92Eu0.08(PO4)6(OH)2 1019.71 |
| Color | colorless | colorless | colorless | colorless |
| Temperature (K) | 293 | 293 | 293 | 293 |
| Wavelength (Å) | 1.54056 | 1.54056 | 1.54056 | 1.54056 |
| 2θ range; step (°) | 8–100, 0.02 | 8-100, 0.02 | 8–97, 0.02 | 8–100, 0.02 |
| System, space group | Hex., P63/m | Hex., P63/m | Hex., P63/m | Hex., P63/m |
| a = b (Å) | 9.4145(4) | 9.4108(13) | 9.3952(11) | 9.4147(4) |
| c (Å) | 6.8758(5) | 6.868(2) | 6.8652(17) | 6.8760(5) |
| V (Å3) | 527.77(5) | 526.75(19) | 524.80(16) | 527.81(5) |
| Z; Densitycalc. (Mg·m−3) | 1, 3.161 | 1, 3.226 | 1, 3.229 | 1, 3.208 |
| Refinement method | FMLS | FMLS | FMLS | FMLS |
| Bragg refl., parameters | 615, 57 | 205, 57 | 191, 58 | 615, 59 |
| Rp; Rwp; Rexp (%) | 2.55, 3.61; 4.33 | 2.03, 2.78 4.30 | 1.97, 2.71, 3.57 | 2.17, 3.00, 4.33 |
| Parameter | HAp900 | Eu-HAp120 | Eu-HAp450 | Eu-HAp900 |
|---|---|---|---|---|
| Crystallite Size (nm) | 56 | 25 | 26 | 148 |
| Crystallinity (%) | 81 | 3 | 7 | 87 |
| Atom | Site | x | y | z |
|---|---|---|---|---|
| Ca1 | 4f | 2/3 | 1/3 | z |
| Ca2 | 6h | x | y | ¼ |
| P1 | 6h | x | y | ¼ |
| O1 | 6h | x | y | ¼ |
| O2 | 6h | x | y | z |
| O3 | 12i | x | y | ¼ |
| OH | 4e | 0 | 0 | z |
| F | 2a | 0 | 0 | ¼ |
| Cl | 4e | 0 | 0 | z |
| Bond | HAp900 | bvp | Eu-HAp120 | bvp | Eu-HAp450 | bvp | Eu-HAp900 | bvp |
|---|---|---|---|---|---|---|---|---|
| 3x Ca1-O1 | 2.403(4) | 0.31 | 2.444(5) | 0.28 | 2.444(5) | 0.28 | 2.405(4) | 0.31 |
| 3x Ca1-O2 | 2.432(5) | 0.29 | 2.399(7) | 0.31 | 2.401(6) | 0.31 | 2.460(4) | 0.26 |
| 3x Ca1…O3 | 2.812(4) | 0.10 | 2.835(6) | 0.10 | 2.851(6) | 0.09 | 2.817(3) | 0.10 |
| 2.08 | 2.03 | 2.03 | 2.01 | |||||
| Ca2-O2 | 2.384(5) | 0.32 | 2.385(8) | 0.32 | 2.381(7) | 0.33 | 2.364(4) | 0.34 |
| 2x Ca2-O3 | 2.317(3) | 0.39 | 2.379(4) | 0.33 | 2.368(4) | 0.34 | 2.321(3) | 0.39 |
| 2x Ca2-O3 | 2.517(5) | 0.23 | 2.519(7) | 0.23 | 2.515(7) | 0.23 | 2.517(4) | 0.23 |
| Ca2-O4H | 2.358(2) | 0.35 | 2.365(3) | 0.34 | 2.347(3) | 0.36 | 2.343(2) | 0.36 |
| Ca2-O1 | 2.684(7) | 0.14 | 2.576(10) | 0.19 | 2.576(9) | 0.19 | 2.701(5) | 0.14 |
| 2.25 | 2.08 | 2.18 | 2.28 | |||||
| P1-O1 | 1.564(5) | 1.512 | 1.519(7) | 1.292 | 1.495(7) | 1.39 | 1.537(4) | 1.24 |
| P1-O2 | 1.558(8) | 1.170 | 1.575(12) | 1.121 | 1.587(12) | 1.09 | 1.549(6) | 1.20 |
| 2x P1-O3 | 1.557(3) | 1.176 | 1.511(5) | 1.332 | 1.527(5) | 1.27 | 1.556(3) | 1.18 |
| 4.67 | 5.02 | 5.02 | 4.80 | |||||
| O4-H1 | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 |
| HAp | Eu-HAp120 | Eu-HAp450 | Eu-HAp900 | Assignment |
|---|---|---|---|---|
| 3571 | 3570 vw | 3570 w | 3570 w | νs(OH) |
| 1637 | 1637 | 1637 | 1637 | δ(H2O) |
| 1089 | 1093 | 1093 | 1091 | |
| 1046 | 1040 | 1040 | 1046 | ν3(PO4)3− |
| - | - | - | 1015 sh | |
| 962 | 960 | 960 | 962 | ν1(PO4)3− |
| 632 | 631 | 631 | 634 | δ(OH) |
| 601 | 602 | 602 | 602 | |
| 570 | 565 | 565 | 572 | ν4(PO4)3− |
| - | - | - | 518 | Eu3+- O2− |
| 473 | 473 | 473 | 473 | ν2(PO4)3− |
| HAp | Eu-HAp120 | Eu-HAp450 | Eu-HAp900 | Assignment HAp Assignment TCP | |
|---|---|---|---|---|---|
| - | - | - | 409 | - | ν 2(PO4)3− |
| 434 | 430 | 430 | 433 | ν 2(PO4)3− | - |
| 447 | 448 | 447 | 447 | ||
| - | - | 559 | 560 | - | ν 4(PO4)3− |
| 580 | 579 | 578 | 592 | ν 4(PO4)3− | - |
| 591 | 590 | 588 | |||
| 610 | 608 | 607 | |||
| - | - | - | 664 | Eu3+-O2− | - |
| 964 | 961 | 961 | 964 | ν 1(PO4)3− | - |
| 1032 | 1030 | 1034 | 1032 | ν 3(PO4)3− | - |
| 1049 | 1047 | 1047 | 1051 | ||
| 1079 | 1074 | 1074 | - | ||
| 1082 | - | - | 1080 | ||
| 1086 | - | - | - | ||
| Luminescence Center | λlum (nm) | Electron Transition | T (°C) | Characteristics of Peaks |
|---|---|---|---|---|
| Eu3+ Ca1 site | 579 | (5D0→7F0) | 120 | weak |
| 592 | (5D0→7F1) | strong, broad | ||
| 618 | (5D0→7F2) | strong, broad | ||
| 653 | (5D0→7F3) | broad | ||
| 700 | (5D0→7F4) | strong, broad | ||
| Eu3+ Ca1 site | 579 | (5D0→7F0) | 450 | strong, narrow |
| 592 | (5D0→7F1) | strong, broad | ||
| 618 | (5D0→7F2) | strong, narrow | ||
| 653 | (5D0→7F3) | weak, broad | ||
| 700 | (5D0→7F4) | strong, broad | ||
| Eu3+ Ca2 site | 574 | (5D0→7F0) | 450 | strong, narrow |
| 602 | (5D0→7F1) | weak, narrow | ||
| 622 | (5D0→7F2) | weak, narrow | ||
| 629 | (5D0→7F3) | strong, narrow | ||
| Eu3+ Ca2 site | 574 | (5D0→7F0) | 900 | very strong, narrow |
| 602 | (5D0→7F1) | weak, narrow | ||
| 622, 629 | (5D0→7F2) | couple: strong, narrow |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldassarre, F.; Altomare, A.; Corriero, N.; Mesto, E.; Lacalamita, M.; Bruno, G.; Sacchetti, A.; Dida, B.; Karaj, D.; Ventura, G.D.; et al. Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature. Crystals 2020, 10, 250. https://doi.org/10.3390/cryst10040250
Baldassarre F, Altomare A, Corriero N, Mesto E, Lacalamita M, Bruno G, Sacchetti A, Dida B, Karaj D, Ventura GD, et al. Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature. Crystals. 2020; 10(4):250. https://doi.org/10.3390/cryst10040250
Chicago/Turabian StyleBaldassarre, Francesco, Angela Altomare, Nicola Corriero, Ernesto Mesto, Maria Lacalamita, Giovanni Bruno, Alberto Sacchetti, Bujar Dida, Dafina Karaj, Giancarlo Della Ventura, and et al. 2020. "Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature" Crystals 10, no. 4: 250. https://doi.org/10.3390/cryst10040250
APA StyleBaldassarre, F., Altomare, A., Corriero, N., Mesto, E., Lacalamita, M., Bruno, G., Sacchetti, A., Dida, B., Karaj, D., Ventura, G. D., Capitelli, F., & Siliqi, D. (2020). Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature. Crystals, 10(4), 250. https://doi.org/10.3390/cryst10040250