First-Principles Study of Structure and Magnetism in Copper(II)-Containing Hybrid Perovskites



Abstract

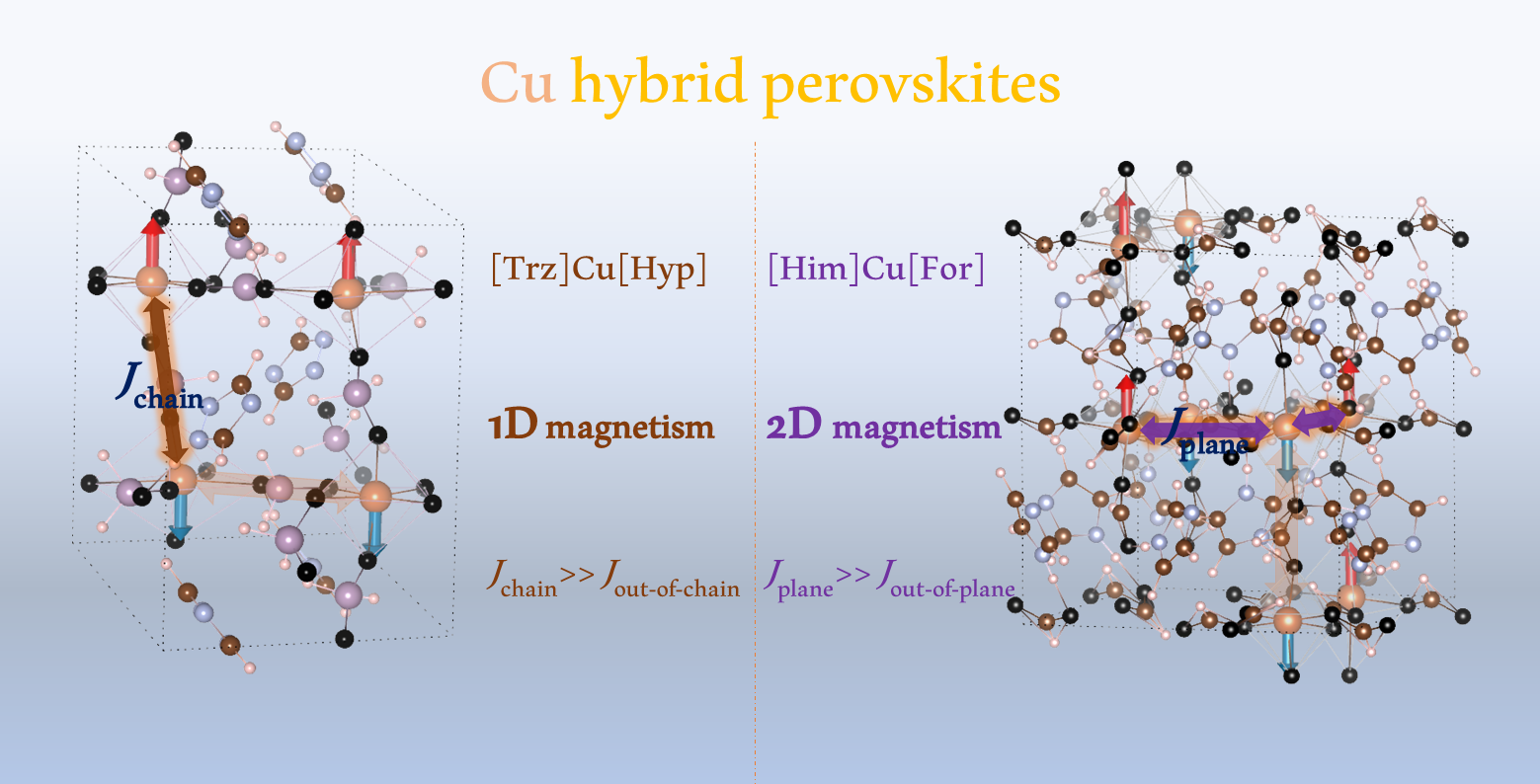
1. Introduction
2. Methods
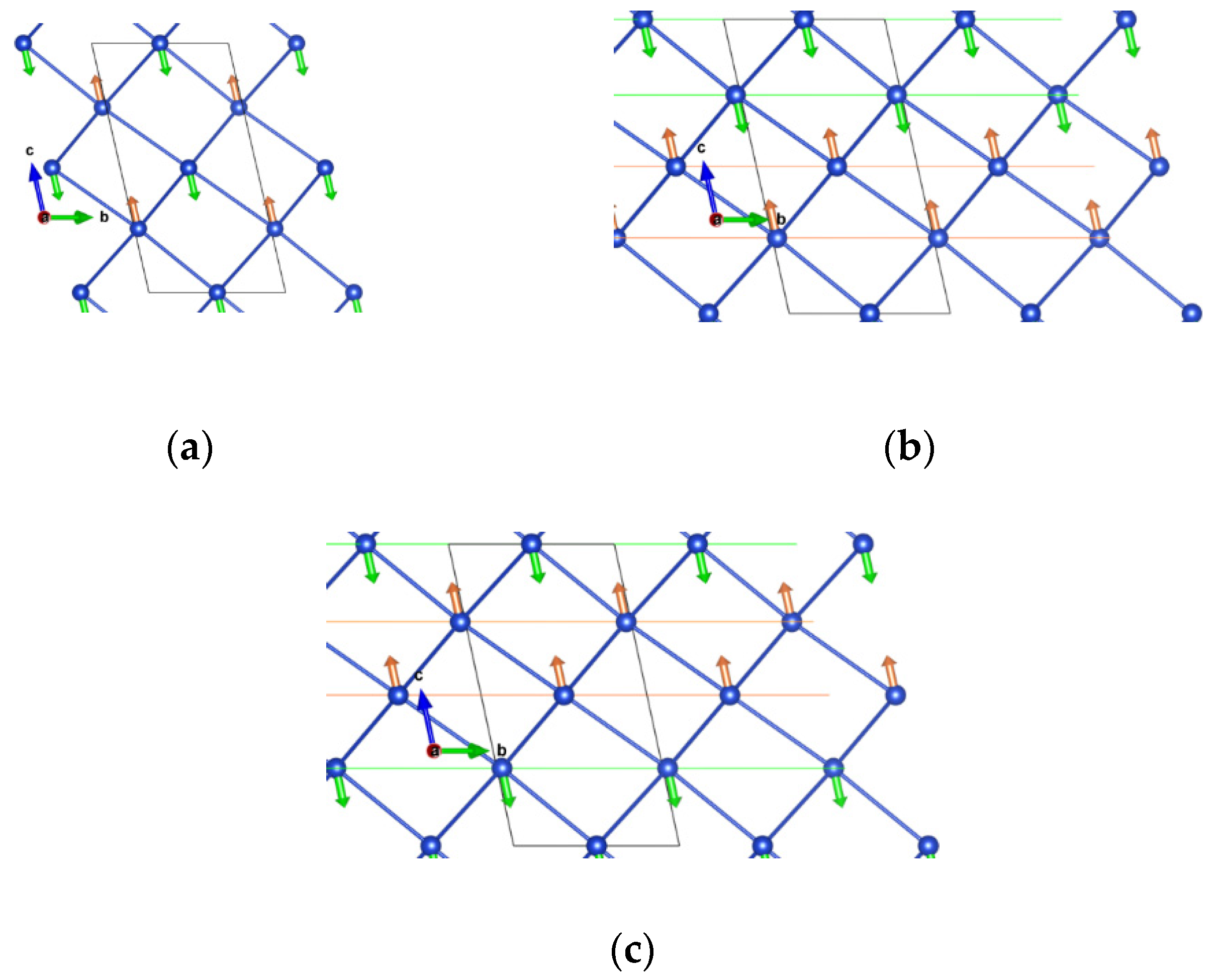
3. Triazolium Cu Hypophosphite
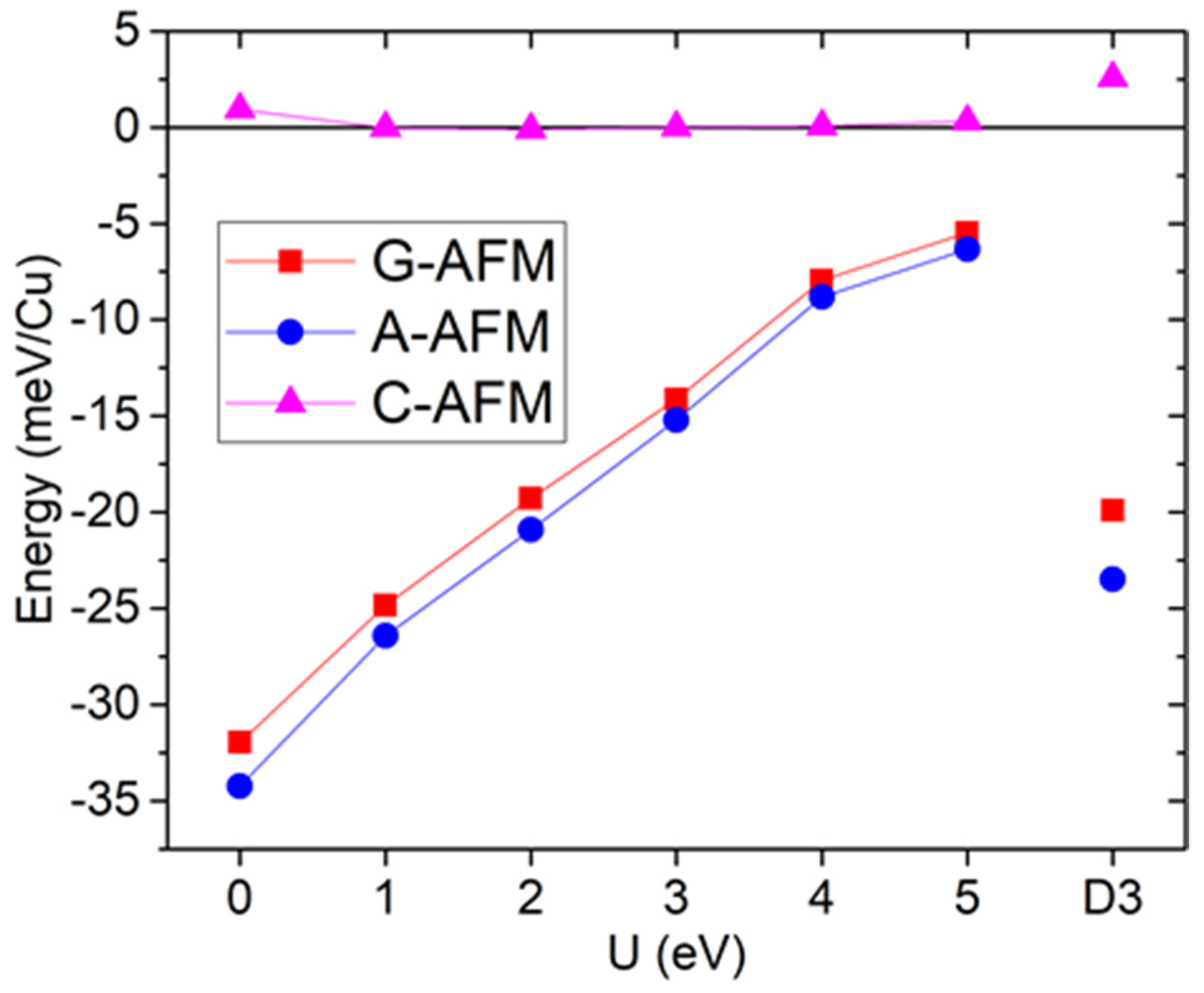
3.1. Magnetism
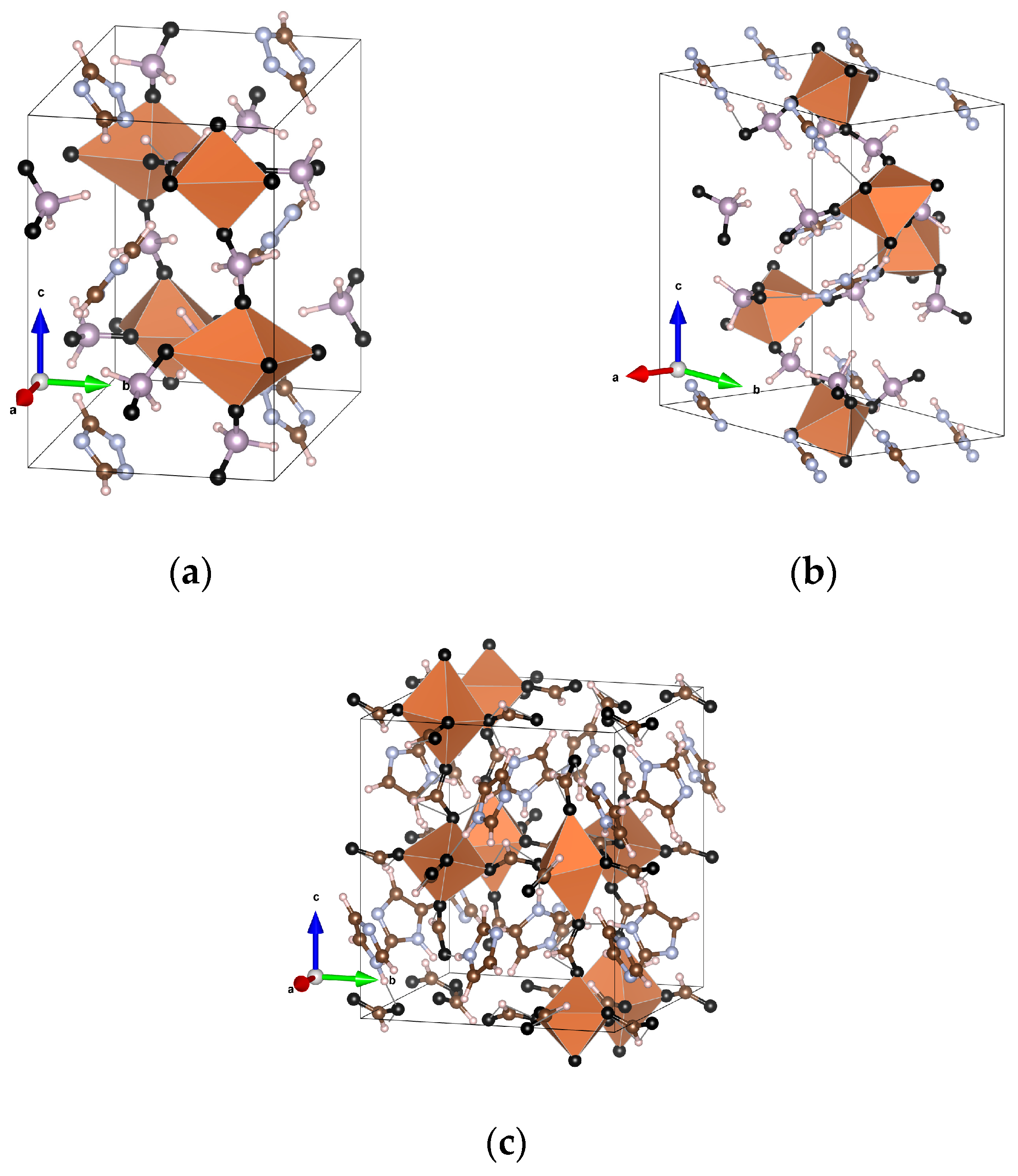
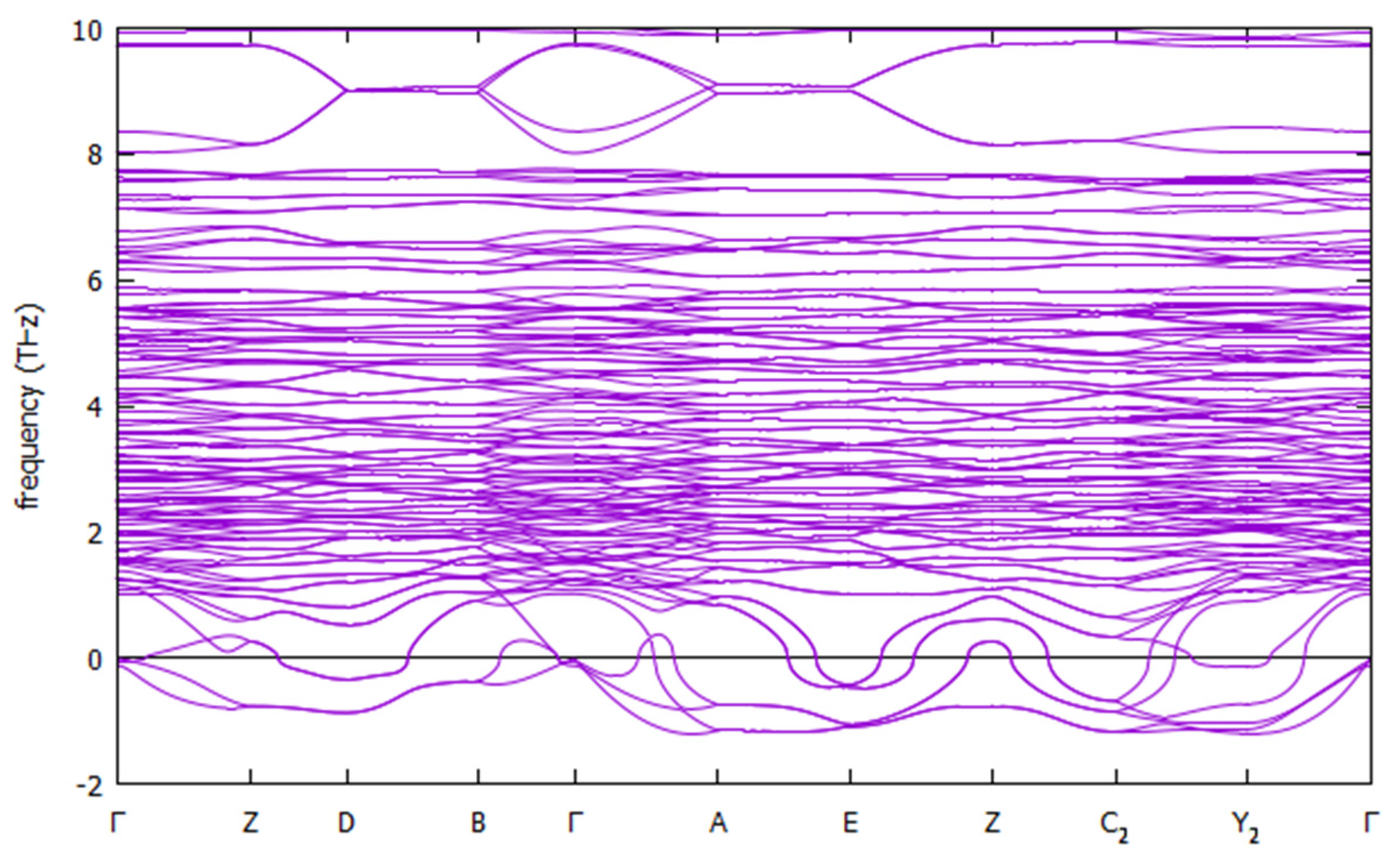
3.2. Structure
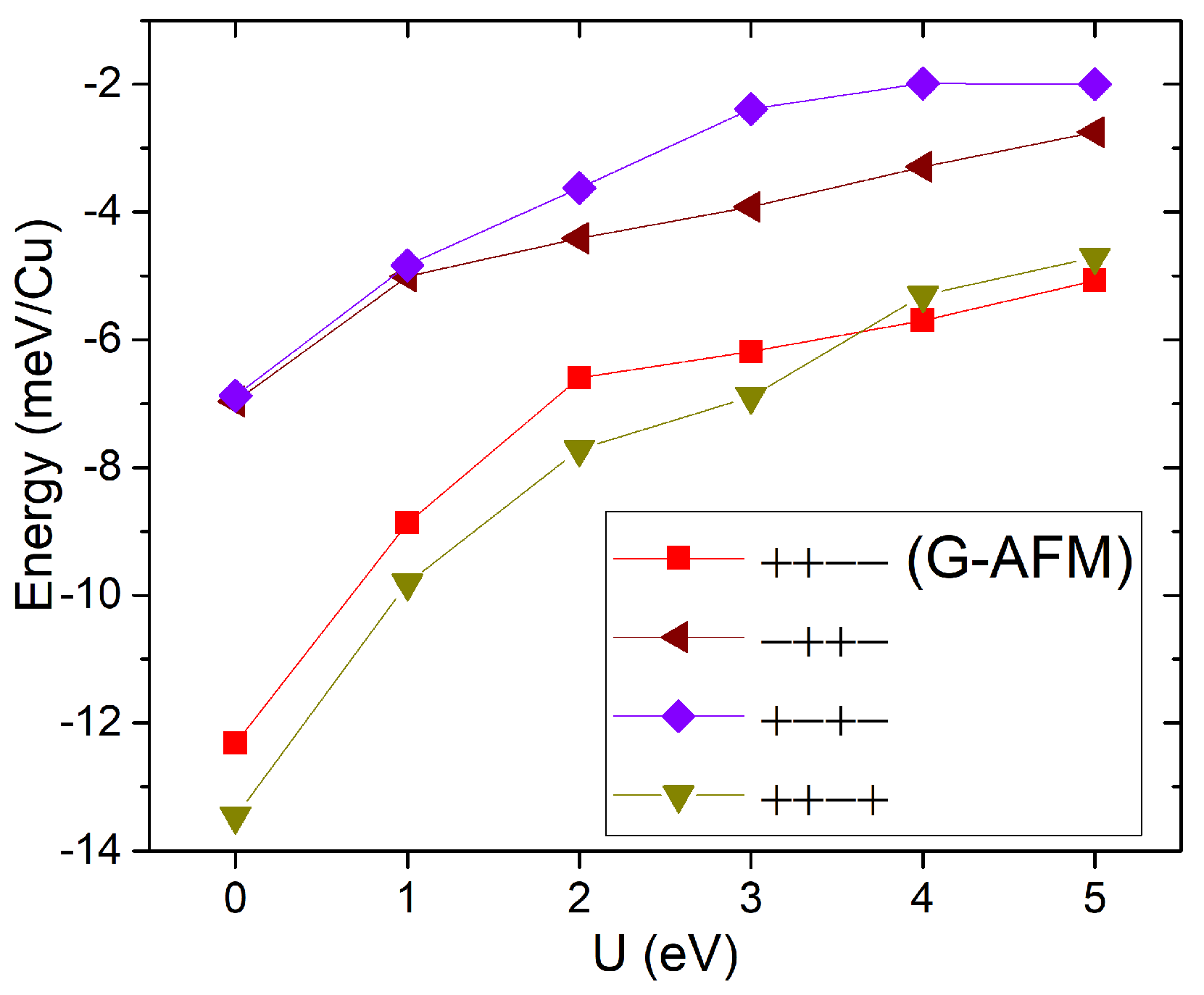
4. Guanidinium Cu Hypophosphite
5. Imidazolium Cu Formate
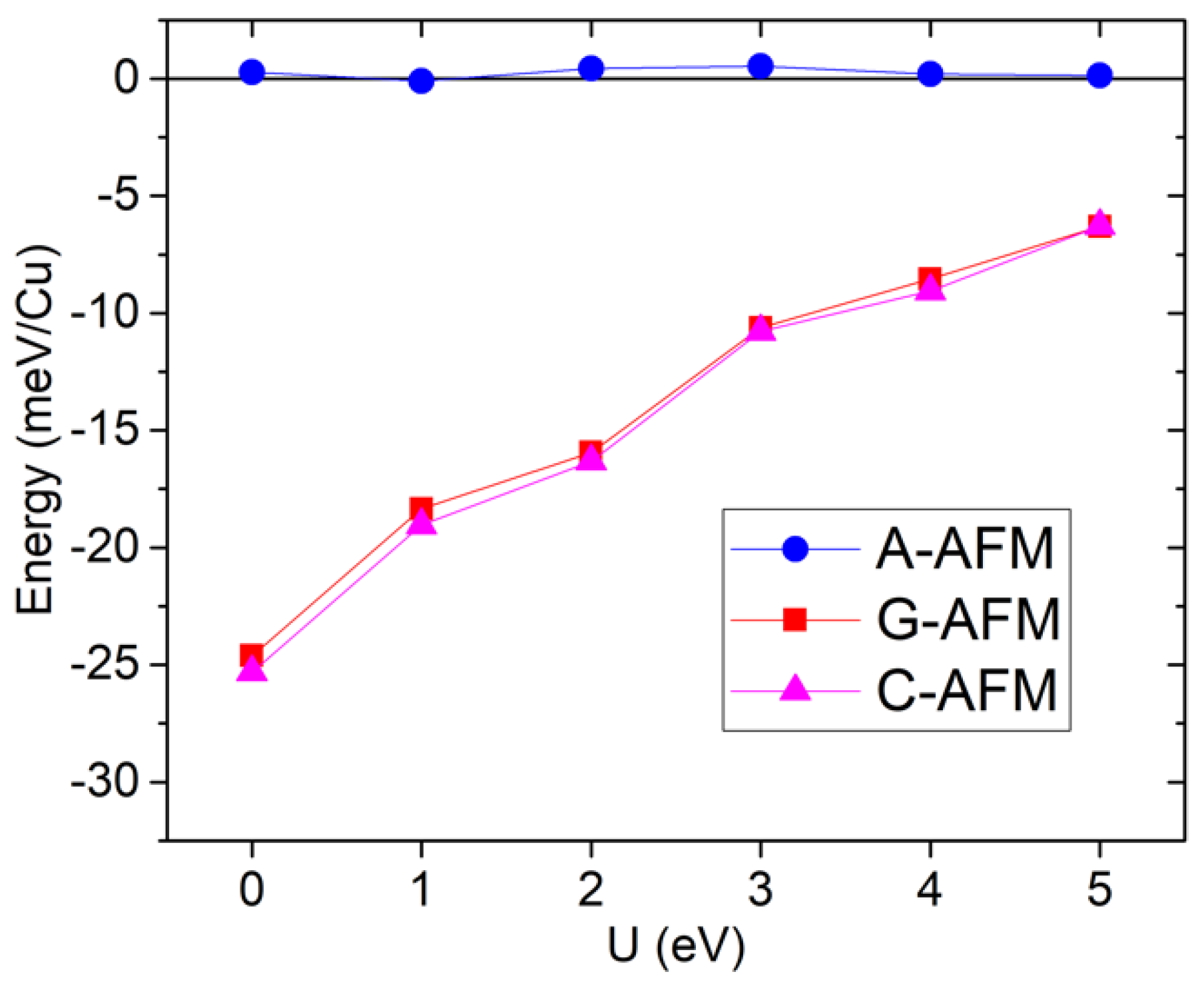
5.1. Magnetism
5.2. Structure
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tian, Y.; Stroppa, A.; Chai, Y.S.; Yan, L.Q.; Wang, S.; Barone, P.; Picozzi, S.; Sun, Y. Cross coupling between electric and magnetic orders in a multiferroic metal–organic framework. Sci. Rep. 2015, 4, 6062. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Aguirre, L.C.; Pato-Doldán, B.; Mira, J.; Castro-García, S.; Señarís-Rodríguez, M.A.; Sánchez-Andújar, M.; Singleton, J.; Zapf, V.S. Magnetic Ordering-Induced Multiferroic Behavior in [CH3NH3][Co(HCOO)3] Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Z.; Deschler, F.; Gao, S.; Friend, R.H.; Cheetham, A.K. Chemically diverse and multifunctional hybrid organic–inorganic perovskites. Nat. Rev. Mater. 2017, 2, 16099. [Google Scholar] [CrossRef]
- Kimura, T.; Goto, T.; Shintani, H.; Ishizaka, K.; Arima, T.; Tokura, Y. Magnetic control of ferroelectric polarization. Nature 2003, 426, 55–58. [Google Scholar] [CrossRef]
- Wang, J.; Neaton, B.; Zheng, H.; Nagarajan, V.; Ogale, S.B.; Liu, B.; Viehland, D.; Vaithyanathan, V.; Schlom, D.G.; Waghmare, U.V.; et al. Epitaxial BiFeO3 multi-ferroic thin film heterostructures. Science 2003, 299, 1719–1722. [Google Scholar] [CrossRef]
- Tian, Y.; Stroppa, A.; Chai, Y.S.; Barone, P.; Perez-Mato, M.; Picozzi, S.; Sun, Y. High-temperature ferro-electricity and strong magnetoelectric effects in a hybrid organic-inorganic perovskite framework, physica status solidi. Rapid Res. Lett. 2015, 9, 62–67. [Google Scholar]
- Stroppa, A.; Jain, P.; Barone, P.; Marsman, M.; Perez-Mato, J.M.; Cheetham, A.K.; Kroto, H.W.; Picozzi, S. Electric Control of Magnetization and Interplay between Orbital Ordering and Ferroelectricity in a Multiferroic Metal-Organic Framework. Angew. Chem. Int. Ed. 2011, 50, 5847–5850. [Google Scholar] [CrossRef]
- Stroppa, A.; Barone, P.; Jain, P.K.; Perez-Mato, J.M.; Picozzi, S. Hybrid Improper Ferroelectricity in a Multiferroic and Magnetoelectric Metal-Organic Framework. Adv. Mater. 2013, 25, 2284–2290. [Google Scholar] [CrossRef]
- Wu, Y.; Shaker, S.; Brivio, F.; Murugavel, R.; Bristowe, P.D.; Cheetham, A.K. [Am]Mn(H2POO)3: A New Family of Hybrid Perovskites Based on the Hypophosphite Ligand. J. Am. Chem. Soc. 2017, 139, 16999–17002. [Google Scholar] [CrossRef]
- Boström, H.L.B.; Senn, M.S.; Goodwin, A.L. Recipes for improper ferroelectricity in molecular perovskites. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Wu, Y.; Binford, T.; Hill, J.A.; Shaker, S.; Wang, J.; Cheetham, A.K. Hypophosphite hybrid perovskites: A platform for unconventional tilts and shifts. Chem. Commun. 2018, 54, 3751–3754. [Google Scholar] [CrossRef]
- Hill, J.A.; Thompson, A.L.; Goodwin, A.L. Di-cyanometallates as model extended frameworks. J. Am. Chem. Soc. 2016, 138, 5886–5896. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, W.; Butler, K.T.; Feng, G.; Howard, C.J.; Carpenter, M.P.; Lu, P.; Walsh, A.; Cheetham, A.K. An Unusual Phase Transition Driven by Vibrational Entropy Changes in a Hybrid Organic-Inorganic Perovskite. Angew. Chem. Int. Ed. 2018, 57, 8932–8936. [Google Scholar] [CrossRef] [PubMed]
- Boström, H.L.B.; Hill, J.A.; Goodwin, A.L. Columnar shifts as symmetry-breaking degrees of freedom in molecular perovskites. Phys. Chem. Chem. Phys. 2016, 18, 31881–31894. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.L.; Thygesen, P.M.M.; Boström, H.L.B.; Reynolds, E.M.; Collings, I.E.; Phillips, A.E.; Goodwin, A.L. Control of Multipolar and Orbital Order in Perovskite-like [C(NH2)3]CuxCd1–x(HCOO)3 Metal–Organic Frameworks. J. Am. Chem. Soc. 2016, 138, 9393–9396. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Q.; Yan, H.-B.; Huang, Z.Q.; Zhang, Z. Reversible high-temperature phase transition of a manganese(II) formate framework with imidazolium cations. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2013, 69, 616–619. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. 2006, 73, 195107. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Stokes, H.T.; Hatch, D.M.; Campbell, B.J. FINDSYM, version 7.0, ISOTROPY Software Suite. 2019. Available online: https://iso.byu.edu/iso/isotropy.php (accessed on 9 December 2020).
- Stokes, H.T.; Hatch, D.M. FINDSYM: Program for identifying the space-group symmetry of a crystal. J. Appl. Crystallogr. 2005, 38, 237–238. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Wollan, E.O.; Koehler, W.C. Neutron Diffraction Study of the Magnetic Properties of the Series of Perovskite-Type Compounds[(1−x)La, xCa]MnO3. Phys. Rev. 1955, 100, 545–563. [Google Scholar] [CrossRef]
- Wang, Z.; Jain, P.; Choi, K.-Y.; Van Tol, J.; Cheetham, A.K.; Kroto, H.W.; Koo, H.-J.; Zhou, H.D.; Hwang, J.; Choi, E.S.; et al. Dimethylammonium copper formate [(CH3)2NH2]Cu(HCOO)3: A metal-organic framework with quasi-one-dimensional antiferromagnetism and magnetostriction. Phys. Rev. B 2013, 87, 224406. [Google Scholar] [CrossRef]
- Hu, K.-L.; Kurmoo, M.; Wang, Z.; Gao, S. Metal-Organic Perovskites: Synthesis, Structures, and Magnetic Properties of [C(NH2)3][MII(HCOO)3] (M = Mn, Fe, Co, Ni, Cu, and Zn; C(NH2)3 = Guanidinium). Chem.-A Eur. J. 2009, 15, 12050–12064. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Benedek, N.A.; Fennie, C.J. Hybrid Improper Ferroelectricity: A Mechanism for Controllable Polarization-Magnetization Coupling. Phys. Rev. Lett. 2011, 106, 107204. [Google Scholar] [CrossRef]
- Baroni, S.; De Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U = 0 eV | D3 | U = 5 eV | [DMA]Cu(HCO2)3 1 | |
|---|---|---|---|---|
| J1 | 780 | 533 | 141 | 214 |
| J2 | −18.6 | −36 | −6.8 | −8.8 |
| J3 | 7.8 | 5.9 | 3.1 | 2.7 |
| Structural Parameter | GGA | D3 | U = 5 eV |
|---|---|---|---|
| a | 9.88 | 9.58 | 9.86 |
| b | 9.18 | 8.54 | 9.04 |
| c | 12.68 | 12.66 | 12.73 |
| β | 99.47 | 99.14 | 99.30 |
| V | 285.91 | 255.45 | 279.29 |
| Cu-O eq. | 1.95–1.99 | 1.95–1.99 | 1.96–1.99 |
| Cu-O ax. | 2.56 & 2.95 | 2.39 & 2.63 | 2.49 & 2.76 |
| U = 0 eV | U = 5 eV | |
|---|---|---|
| J1 (out-of-plane) | −11.7 | −1.5 |
| J2 (in-plane) | +307.3 | +77.9 |
| J3 (out-of-plane) | 2.5 | −1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, J.N.; Phillips, A.E.; Li, W.; Stroppa, A. First-Principles Study of Structure and Magnetism in Copper(II)-Containing Hybrid Perovskites. Crystals 2020, 10, 1129. https://doi.org/10.3390/cryst10121129
Gonçalves JN, Phillips AE, Li W, Stroppa A. First-Principles Study of Structure and Magnetism in Copper(II)-Containing Hybrid Perovskites. Crystals. 2020; 10(12):1129. https://doi.org/10.3390/cryst10121129
Chicago/Turabian StyleGonçalves, João N., Anthony E. Phillips, Wei Li, and Alessandro Stroppa. 2020. "First-Principles Study of Structure and Magnetism in Copper(II)-Containing Hybrid Perovskites" Crystals 10, no. 12: 1129. https://doi.org/10.3390/cryst10121129
APA StyleGonçalves, J. N., Phillips, A. E., Li, W., & Stroppa, A. (2020). First-Principles Study of Structure and Magnetism in Copper(II)-Containing Hybrid Perovskites. Crystals, 10(12), 1129. https://doi.org/10.3390/cryst10121129