Synthesis of Mono- and Bis-Pyrazoles Bearing Flexible p-Tolyl Ether and Rigid Xanthene Backbones, and Their Potential as Ligands in the Pd-Catalysed Suzuki–Miyaura Cross-Coupling Reaction

 , ,
, ,
Abstract
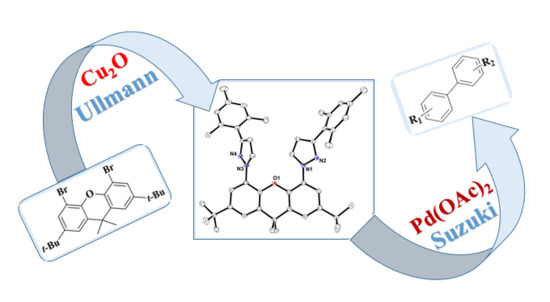
:
1. Introduction
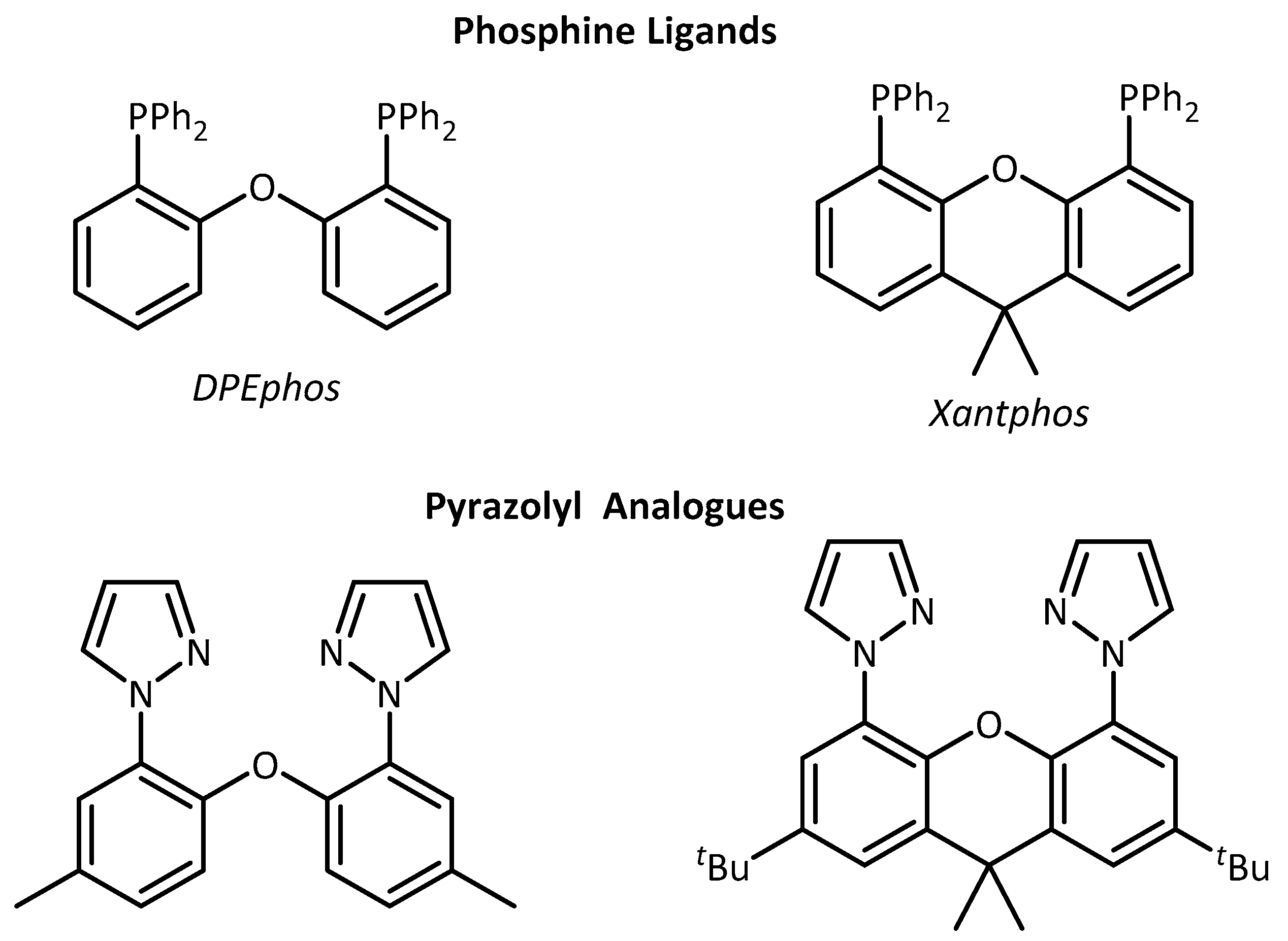
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Pyrazolyl Analogues of DPEphos and tBu-Xantphos
3.2.1. Dibromination of p-Tolyl Ether (Preparation of 1)
3.2.2. Preparation of 3-Mesityl-1H-pyrazole
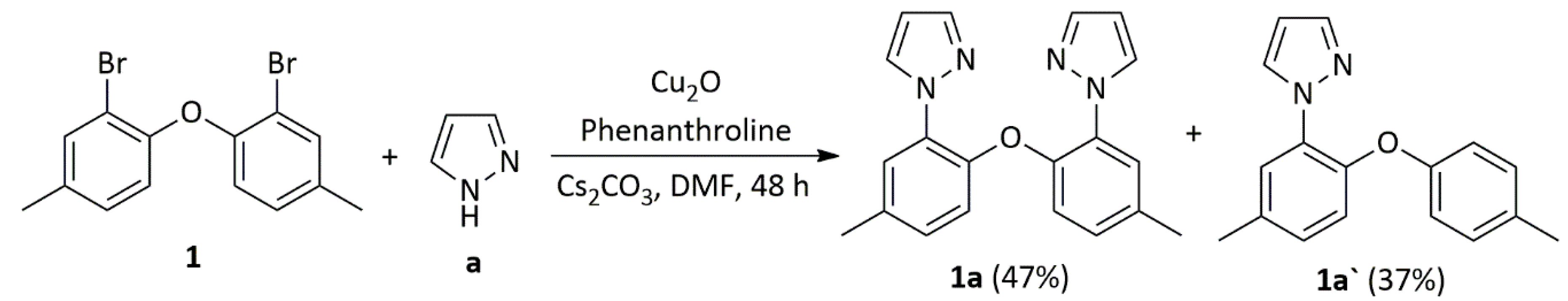
3.2.3. Synthesis of Ligands 1a and 1a`
3.2.4. Synthesis of Ligands 2a and 2a`
3.2.5. Synthesis of Ligands 2b and 2b`
3.2.6. Synthesis of hybrid Ligand 2b`c
3.2.7. Synthesis of Hybrid Ligand 2a`d
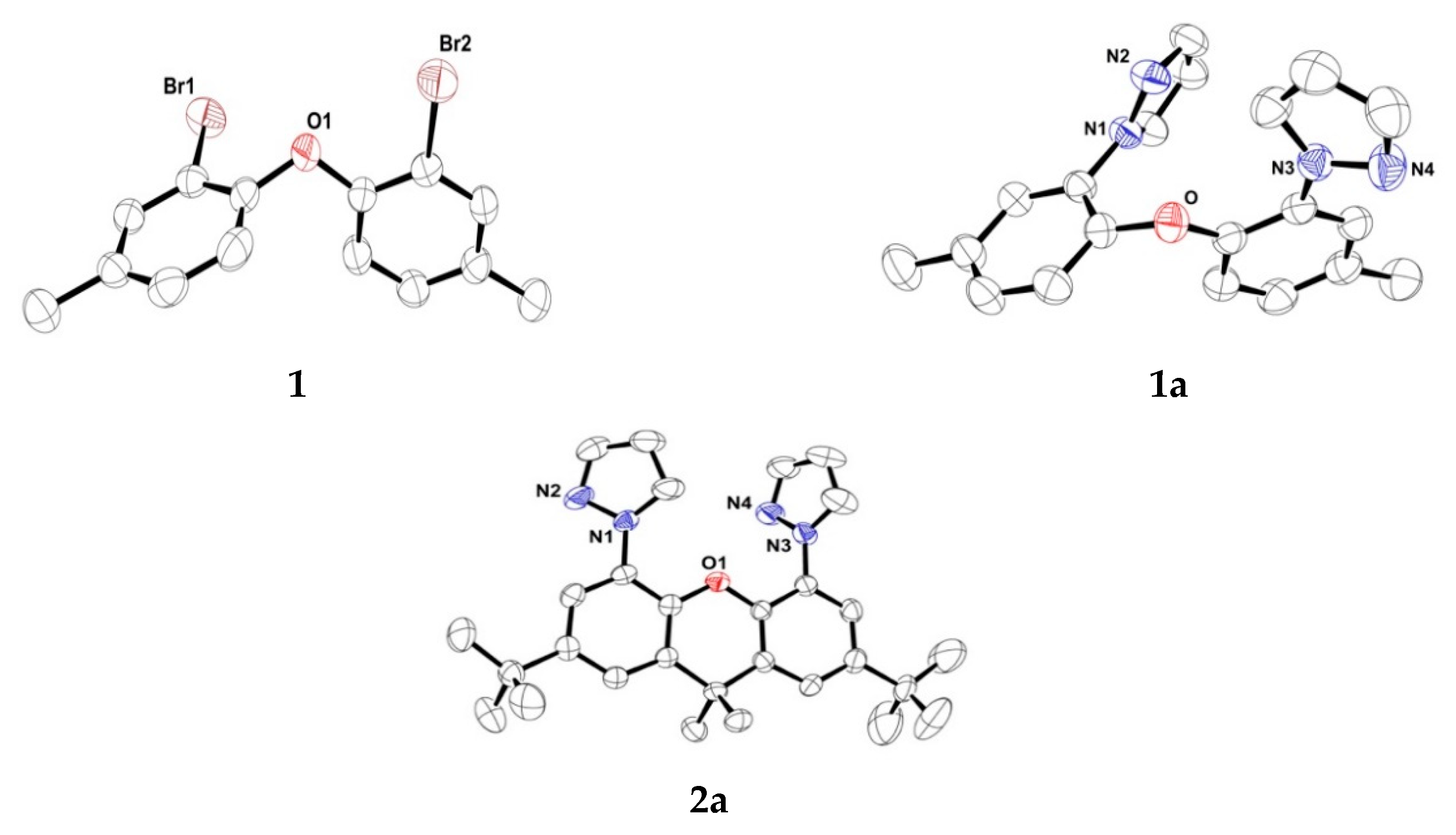
3.3. X-ray Crystallography of 1, 1a, 2a, 2b and 2b`
3.4. General Procedure for SM Cross-Coupling Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fairlamb, I. Metal-Catalyzed Cross-Coupling Reactions and More. 3 Volume Set. Edited by Armin de Meijere, Stefan Brse and Martin Oestreich. Angew. Chem. Int. Ed. 2014, 53. [Google Scholar] [CrossRef]
- Shinde, V.V.; Jeong, D.; Jung, S. An Amino-Chain Modified β-cyclodextrin: A Supramolecular Ligand for Pd(OAc)2 Acceleration in Suzuki–Miyaura Coupling Reactions in Water. Catalysts 2019, 9, 111. [Google Scholar] [CrossRef]
- Zuidema, E.; Goudriaan, P.E.; Swennenhuis, B.H.G.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Lutz, M.; Spek, A.L. Phenoxaphosphine-Based Diphosphine Ligands. Synthesis and Application in the Hydroformylation Reaction. Organometallics 2010, 29, 1210–1221. [Google Scholar] [CrossRef]
- Li, C.; Xiao, G.; Zhao, Q.; Liu, H.; Wang, T.; Tang, W. Sterically demanding aryl–alkyl Suzuki–Miyaura coupling. Org. Chem. Front. 2014, 1, 225–229. [Google Scholar] [CrossRef]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Reek, J.N.H. Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.M.; Weller, A.S. POP-type ligands: Variable coordination and hemilabile behaviour. Coord. Chem. Rev. 2018, 355, 150–172. [Google Scholar] [CrossRef]
- Birkholz, M.N.; Freixa, Z.; van Leeuwen, P.W.N.M. Bite angle effects of diphosphines in C–C and C–X bond forming cross coupling reactions. Chem. Soc. Rev. 2009, 38, 1099–1118. [Google Scholar] [CrossRef]
- Andreychuk, N.R.; Emslie, D.J.H. Potassium–Alkane Interactions within a Rigid Hydrophobic Pocket. Angew. Chem. Int. Ed. 2013, 52, 1696–1699. [Google Scholar] [CrossRef]
- Takano, S.; Takeuchi, D.; Osakada, K. Olefin Polymerization Catalyzed by Double-Decker Dipalladium Complexes: Low Branched Poly(α-Olefin)s by Selective Insertion of the Monomer Molecule. Chem. A Eur. J. 2015, 21, 16209–16218. [Google Scholar] [CrossRef]
- Komuro, T.; Kitano, T.; Yamahira, N.; Ohta, K.; Okawara, S.; Mager, N.; Okazaki, M.; Tobita, H. Directed ortho-C–H Silylation Coupled with trans-Selective Hydrogenation of Arylalkynes Catalyzed by Ruthenium Complexes of a Xanthene-Based Si,O,Si-Chelate Ligand, “Xantsil”. Organometallics 2016, 35, 1209–1217. [Google Scholar] [CrossRef]
- Diéguez, M.; Claver, C.; Masdeu-Bultó, A.M.; Ruiz, A.; van Leeuwen, P.W.N.M.; Schoemaker, G.C. High-Pressure Infrared Studies of Rhodium Complexes Containing Thiolate Bridge Ligands under Hydroformylation Conditions. Organometallics 1999, 18, 2107–2115. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J. Featuring Xantphos. Catal. Sci. Technol. 2018, 8, 26–113. [Google Scholar] [CrossRef]
- Yoo, K.S.; Yoon, C.H.; Jung, K.W. Oxidative Palladium(II) Catalysis: A Highly Efficient and Chemoselective Cross-Coupling Method for Carbon−Carbon Bond Formation under Base-Free and Nitrogenous-Ligand Conditions. J. Am. Chem. Soc. 2006, 128, 16384–16393. [Google Scholar] [CrossRef]
- Punji, B.; Ganesamoorthy, C.; Balakrishna, M.S. Suzuki cross-coupling reactions catalyzed by palladium complex of an inexpensive phosphinite, 2-diphenylphosphinoxynaphthyl. J. Mol. Catal. A Chem. 2006, 259, 78–83. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Kashinath, D. Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar] [CrossRef]
- Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- Biajoli, A.F.P.; Schwalm, C.S.; Limberger, J.; Claudino, T.S.; Monteiro, A.L. Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds. J. Braz. Chem. Soc. 2014, 25, 2186–2214. [Google Scholar] [CrossRef]
- Kumbhar, A. Functionalized nitrogen ligands for palladium catalyzed cross-coupling reactions (part I). J. Organomet. Chem. 2017, 848, 22–88. [Google Scholar] [CrossRef]
- Kumbhar, A. Functionalized nitrogen ligands (CN) for palladium catalyzed cross-coupling reactions (part II). J. Organomet. Chem. 2019, 881, 79–129. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, W.J. Dabco as an Inexpensive and Highly Efficient Ligand for Palladium-Catalyzed Suzuki−Miyaura Cross-Coupling Reaction. Org. Lett. 2004, 6, 2809–2811. [Google Scholar] [CrossRef]
- Li, J.H.; Zhu, Q.M.; Xie, Y.X. Pd(OAc)2/DABCO-catalyzed Suzuki–Miyaura cross-coupling reaction in DMF. Tetrahedron 2006, 62, 10888–10895. [Google Scholar] [CrossRef]
- Doucet, H. Suzuki–Miyaura Cross-Coupling Reactions of Alkylboronic Acid Derivatives or Alkyltrifluoroborates with Aryl, Alkenyl or Alkyl Halides and Triflates. Eur. J. Org. Chem. 2008, 2008, 2013–2030. [Google Scholar] [CrossRef]
- Weng, Z.; Teo, S.; Hor, T.S.A. Metal Unsaturation and Ligand Hemilability in Suzuki Coupling. Acc. Chem. Res. 2007, 40, 676–684. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, J.; Trudell, M.L.; Nolan, S.P. Palladium−Imidazol-2-ylidene Complexes as Catalysts for Facile and Efficient Suzuki Cross-Coupling Reactions of Aryl Chlorides with Arylboronic Acids. J. Org. Chem. 1999, 64, 3804–3805. [Google Scholar] [CrossRef]
- Ibrahim, M.B.; Shakil Hussain, S.M.; Fazal, A.; Fettouhi, M.; El Ali, B. Effective palladium(II)-bis(oxazoline) catalysts: Synthesis, crystal structure, and catalytic coupling reactions. J. Coord. Chem. 2015, 68, 432–448. [Google Scholar] [CrossRef]
- van Koten, G. The Monoanionic ECE-Pincer Ligand: A Versatile Privileged Ligand Platform—General Considerations. In Organometallic Pincer Chemistry; van Koten, G., Milstein, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Peral, D.; Gómez-Villarraga, F.; Sala, X.; Pons, J.; Carles Bayón, J.; Ros, J.; Guerrero, M.; Vendier, L.; Lecante, P.; García-Antón, J.; et al. Palladium catalytic systems with hybrid pyrazole ligands in C–C coupling reactions. Nanoparticles versus molecular complexes. Catal. Sci. Technol. 2013, 3, 475–489. [Google Scholar] [CrossRef]
- Ojwach, S.O.; Darkwa, J. Pyrazole and (pyrazol-1-yl)metal complexes as carbon–carbon coupling catalysts. Inorg. Chim. Acta 2010, 363, 1947–1964. [Google Scholar] [CrossRef]
- Li, F.; Hor, T.S.A. Benzimidazolium-Pyrazole-Palladium(II) Complexes: New and Efficient Catalysts for Suzuki, Heck and Sonogashira Reactions. Adv. Synth. Catal. 2008, 350, 2391–2400. [Google Scholar] [CrossRef]
- Montoya, V.; Pons, J.; Branchadell, V.; Garcia-Antón, J.; Solans, X.; Font-Bardía, M.; Ros, J. Highly Efficient Pyridylpyrazole Ligands for the Heck Reaction. A Combined Experimental and Computational Study. Organometallics 2008, 27, 1084–1091. [Google Scholar] [CrossRef]
- Ocansey, E.; Darkwa, J.; Makhubela, B.C.E. Synthesis, characterization and evaluation of bulky bis(pyrazolyl)palladium complexes in Suzuki–Miyaura cross-coupling reactions. RSC Adv. 2018, 8, 13826–13834. [Google Scholar] [CrossRef]
- Li, K.; Darkwa, J.; Guzei, I.A.; Mapolie, S.F. Synthesis and evaluation of substituted pyrazoles palladium(II) complexes as ethylene polymerization catalysts. J. Organomet. Chem. 2002, 660, 108–115. [Google Scholar] [CrossRef]
- Obuah, C.; Omondi, B.; Nozaki, K.; Darkwa, J. Solvent and co-catalyst dependent pyrazolylpyridinamine and pyrazolylpyrroleamine nickel(II) catalyzed oligomerization and polymerization of ethylene. J. Mol. Catal. A Chem. 2014, 382, 31–40. [Google Scholar] [CrossRef]
- Matos, K.; de Oliveira, L.L.; Favero, C.; Monteiro, A.L.; Hörner, M.; Carpentier, J.-F.; Gil, M.P.; Casagrande, O.L. Palladium complexes based on tridentate pyrazolyl-ligands: Synthesis, structures and use in Suzuki cross-coupling reactions. Inorg. Chim. Acta 2009, 362, 4396–4402. [Google Scholar] [CrossRef]
- Das, B.; Venkateswarlu, K.; Majhi, A.; Siddaiah, V.; Reddy, K.R. A facile nuclear bromination of phenols and anilines using NBS in the presence of ammonium acetate as a catalyst. J. Mol. Catal. A Chem. 2007, 267, 30–33. [Google Scholar] [CrossRef]
- Kreisel, G.; Rudolph, G.; Schulze, D.K.W.; Poppitz, W. Bromierte Diphenylether—Eine entscheidende Zwischenstufe auf dem Weg zu synthetischen Schmierstoffen. Mon. Für Chem. Chem. Mon. 1992, 123, 1153–1161. [Google Scholar] [CrossRef]
- Mudge, M.; Patel, A.R.; Bingül, M.; Bhadbhade, M.; Colbran, S.B. An aryl-bridged dixanthene scaffold for building multinucleating ligands and supramolecular assemblies: Syntheses and structures. Tetrahedron 2017, 73, 6401–6409. [Google Scholar] [CrossRef]
- Nishino, K.; Tsukahara, S.; Ogiwara, Y.; Sakai, N. Palladium(II)/Copper(II)-Catalyzed C–H Sulfidation or Selenation of Arenes Leading to Unsymmetrical Sulfides and Selenides. Eur. J. Org. Chem. 2019, 2019, 1588–1593. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Unusual Influence of the Structures of Transition Metal Complexes on Catalytic C–S and C–Se Bond Formation Under Homogeneous and Heterogeneous Conditions. Eur. J. Org. Chem. 2007, 2007, 3431–3444. [Google Scholar] [CrossRef]
- Zim, D.; Nobre, S.M.; Monteiro, A.L. Suzuki cross-coupling reaction catalyzed by sulfur-containing palladacycles: Formation of palladium active species. J. Mol. Catal. A Chem. 2008, 287, 16–22. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Wu, Q.; Ma, C.; Wang, Z.; Peng, W.; Song, H. Synthesis and antibacterial evaluation of novel 4-alkyl substituted phenyl β-aldehyde ketone derivatives. Eur. J. Med. Chem. 2009, 44, 1737–1744. [Google Scholar] [CrossRef]
- Rheingold, A.L.; White, C.B.; Trofimenko, S. Hydrotris(3-mesitylpyrazol-1-yl)borate and hydrobis(3-mesitylpyrazol-1-yl)(5-mesitylpyrazol-1-yl)borate: Symmetric and asymmetric ligands with rotationally restricted aryl substituents. Inorg. Chem. 1993, 32, 3471–3477. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [Pd] (1 mol%) | Base | Solvent | Conversion (%) b | CC Yield (%) b | HC Yield (%) c |
|---|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | KOH | MeOH | 74 | 66 | 1 |
| 2 | Pd(OAc)2 | K3PO4 | Dioxane | 48 | 7 | 1 |
| 3 | Pd(OAc)2 | Cs2CO3 | DMF | 87 | 61 | 1 |
| 4 | Pd(OAc)2 | K2CO3 | DMF | 99 | 98 | 1 |
| 5 | Pd(OAc)2 | K2CO3 | MeOH | 75 | 44 | 1 |
| 6 | Pd(OAc)2 | K2CO3 | THF:MeOH d | 65 | 4 | 2 |
| 7 | Pd(OAc)2 | K2CO3 | THF d | 49 | 1 | 2 |
| 8 | Pd(OAc)2 | K2CO3 | Dioxane | 48 | 2 | 1 |
| 9 | PdCl2 | K2CO3 | DMF | 97 | 88 | 4 |
| 10 | PdCl2(COD) e | K2CO3 | DMF | 72 | 55 | 3 |
| 11 | PdCl2(PhCN)2 | K2CO3 | DMF | 93 | 92 | 2 |
| Entry | Temp. (°C) | Time (h) | Conversion (%) b | CC Yield (%) b | HC Yield (%) c |
|---|---|---|---|---|---|
| 1 | 25 | 24 | 73 | 59 | 1 |
| 2 | 50 | 12 | 92 | 70 | 1 |
| 3 | 80 | 6 | 99 | 98 | 1 |
| 4 | 110 | 3 | 90 | 75 | 1 |
| 5 | 80 | 12 | 87 | 82 d | 1 |
| Entry | Ligand | Conversion (%) b | CC Yield (%) b | HC Yield (%) c |
|---|---|---|---|---|
| 1 | 2a | 99 | 98 | 1 |
| 2 | 2b | >99 | 98 | 1 |
| 3 | 2b`c | 99 | 98 | 1 |
| 4 | 2a` | 87 | 86 | 1 |
| 5 | 2a`d | 46 | 5 | 1 |
| 6 | 1a | 83 | 47 | 1 |
| 7 | 1a` | 62 | 29 | 1 |
| 8 | - | 68 | 44 | 2 |
| 9 | Xantphos | 84 | 77 | 6 |
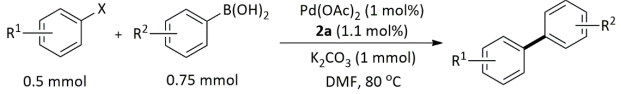
| Entry | R1 | X | R2 | Yield (%) b |
|---|---|---|---|---|
| 1 | 4-Me | Br | H | 94 |
| 2 | 4-CF3 | Br | H | 82 |
| 3 | 4-Ac | Br | H | 92 |
| 4 | 4-Ome | Br | H | 91 |
| 5 | 2-bromonaphthalene | H | 91 | |
| 6 | 4-Me | Br | 4-OMe | 82 |
| 7 | 4-Me | Br | 4-Ac | 78 |
| 8 | 4-OMe | Br | 4-Ac | 73 |
| 9 | 4-Ac | Br | 4-Me | 81 |
| 10 | 2-Me | Br | H | 58 |
| 11 | 4-Me | Br | 2-Me | 54 |
| 12 | 4-Me | Br | 2,6-diMe | 46 |
| 13 | 4-CF3 | I | H | 78 |
| 14 | 4-OMe | I | H | 80 |
| 15 | 4-Ac | Cl | H | 75 |
| 16 | 4-Ac | Cl | H | 3 c |
| 17 | 4-Me | Cl | H | 25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, Z.; Schwalm, C.; Rambo, R.; Dias, R.; Stieler, R.; Monteiro, A. Synthesis of Mono- and Bis-Pyrazoles Bearing Flexible p-Tolyl Ether and Rigid Xanthene Backbones, and Their Potential as Ligands in the Pd-Catalysed Suzuki–Miyaura Cross-Coupling Reaction. Catalysts 2019, 9, 718. https://doi.org/10.3390/catal9090718
Hussain Z, Schwalm C, Rambo R, Dias R, Stieler R, Monteiro A. Synthesis of Mono- and Bis-Pyrazoles Bearing Flexible p-Tolyl Ether and Rigid Xanthene Backbones, and Their Potential as Ligands in the Pd-Catalysed Suzuki–Miyaura Cross-Coupling Reaction. Catalysts. 2019; 9(9):718. https://doi.org/10.3390/catal9090718
Chicago/Turabian StyleHussain, Zahid, Cristiane Schwalm, Raoní Rambo, Renieidy Dias, Rafael Stieler, and Adriano Monteiro. 2019. "Synthesis of Mono- and Bis-Pyrazoles Bearing Flexible p-Tolyl Ether and Rigid Xanthene Backbones, and Their Potential as Ligands in the Pd-Catalysed Suzuki–Miyaura Cross-Coupling Reaction" Catalysts 9, no. 9: 718. https://doi.org/10.3390/catal9090718
APA StyleHussain, Z., Schwalm, C., Rambo, R., Dias, R., Stieler, R., & Monteiro, A. (2019). Synthesis of Mono- and Bis-Pyrazoles Bearing Flexible p-Tolyl Ether and Rigid Xanthene Backbones, and Their Potential as Ligands in the Pd-Catalysed Suzuki–Miyaura Cross-Coupling Reaction. Catalysts, 9(9), 718. https://doi.org/10.3390/catal9090718