Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water



Abstract
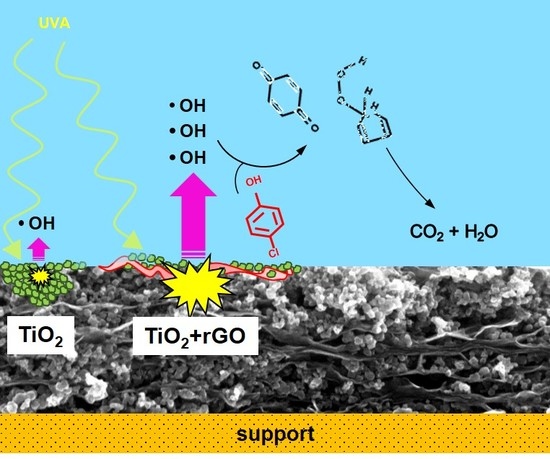
1. Introduction
2. Results
2.1. Preparation of Layers by EPD
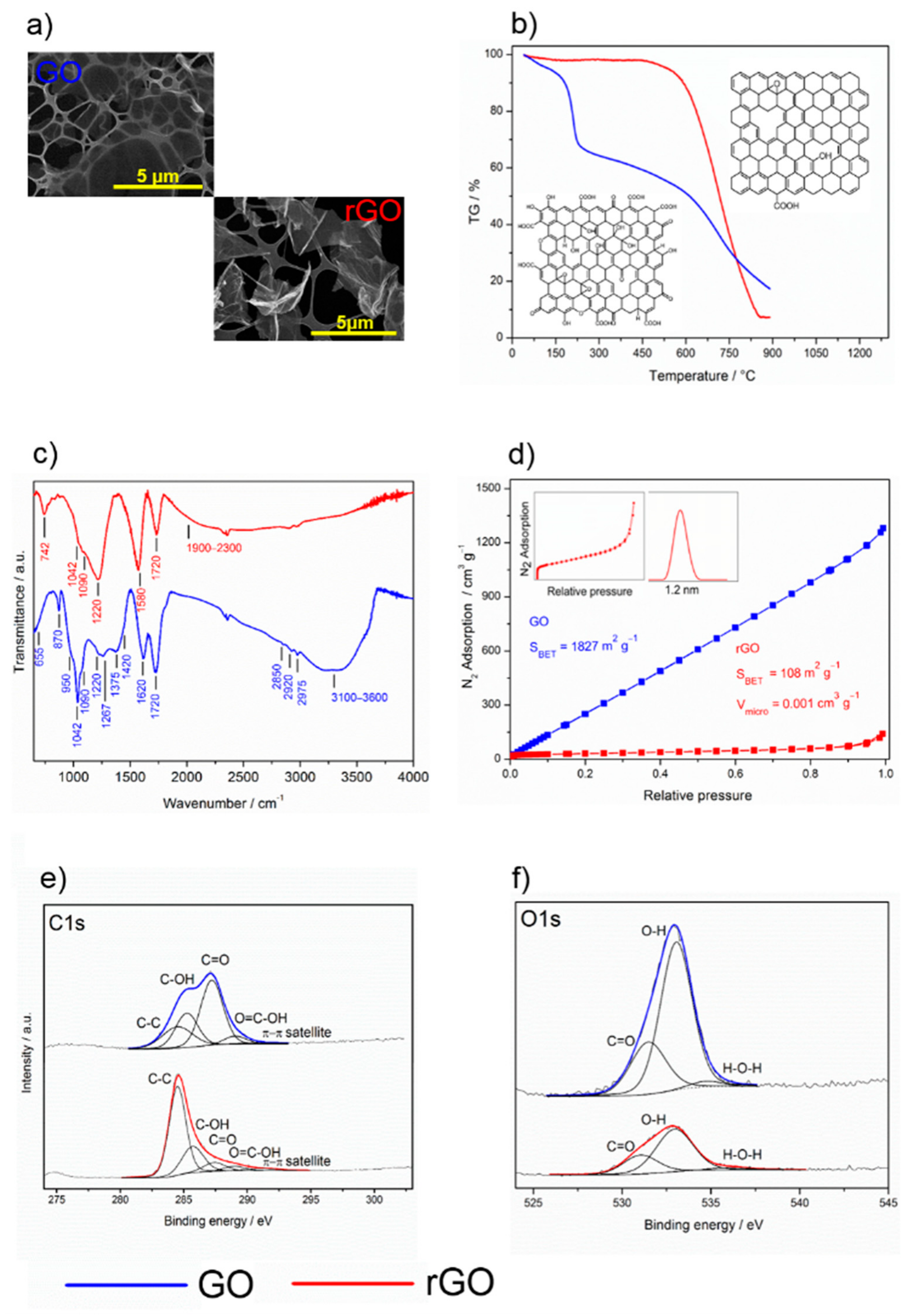
2.2. Properties of Graphene Materials
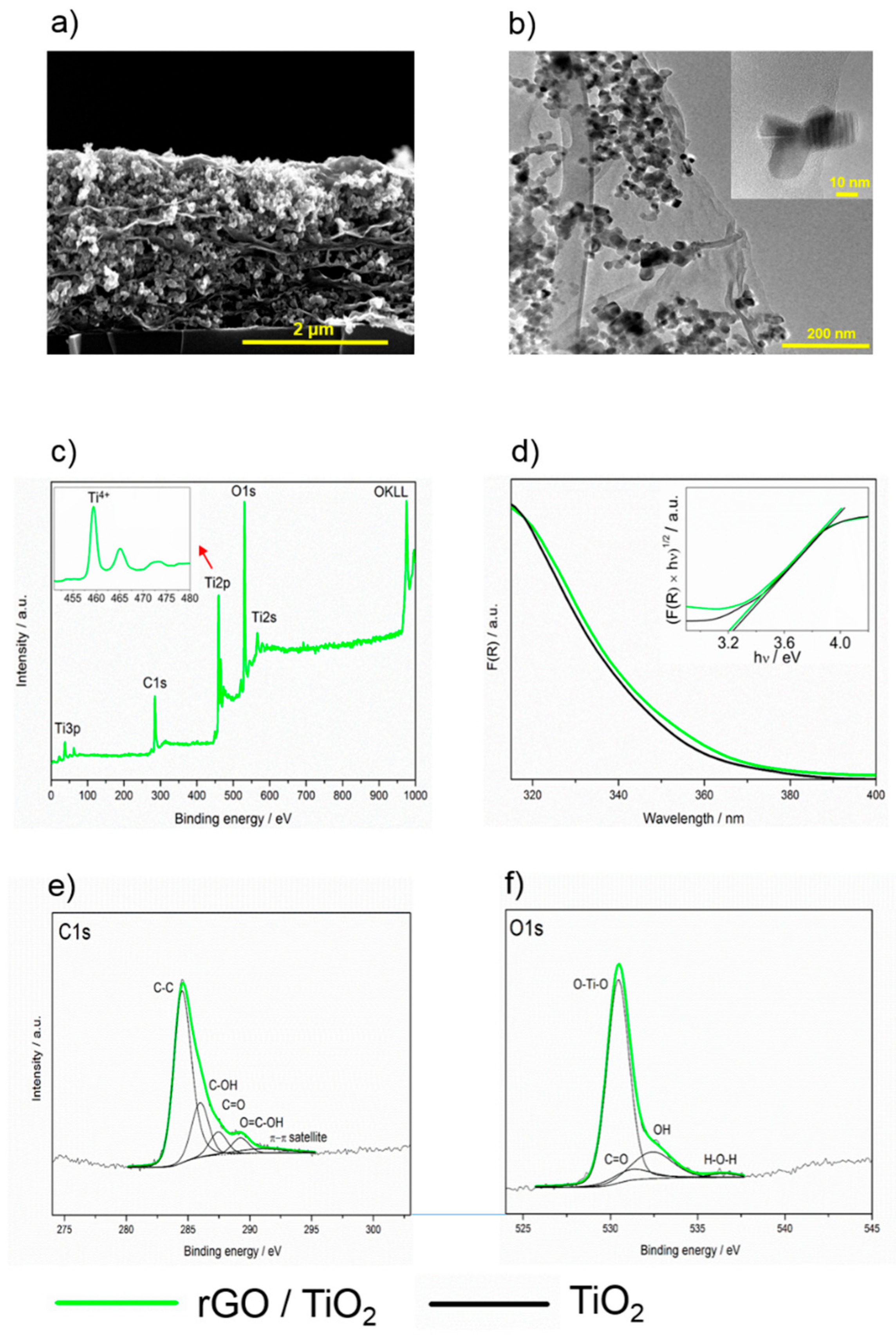
2.3. Properties of rGO/TiO2 Composite Layers
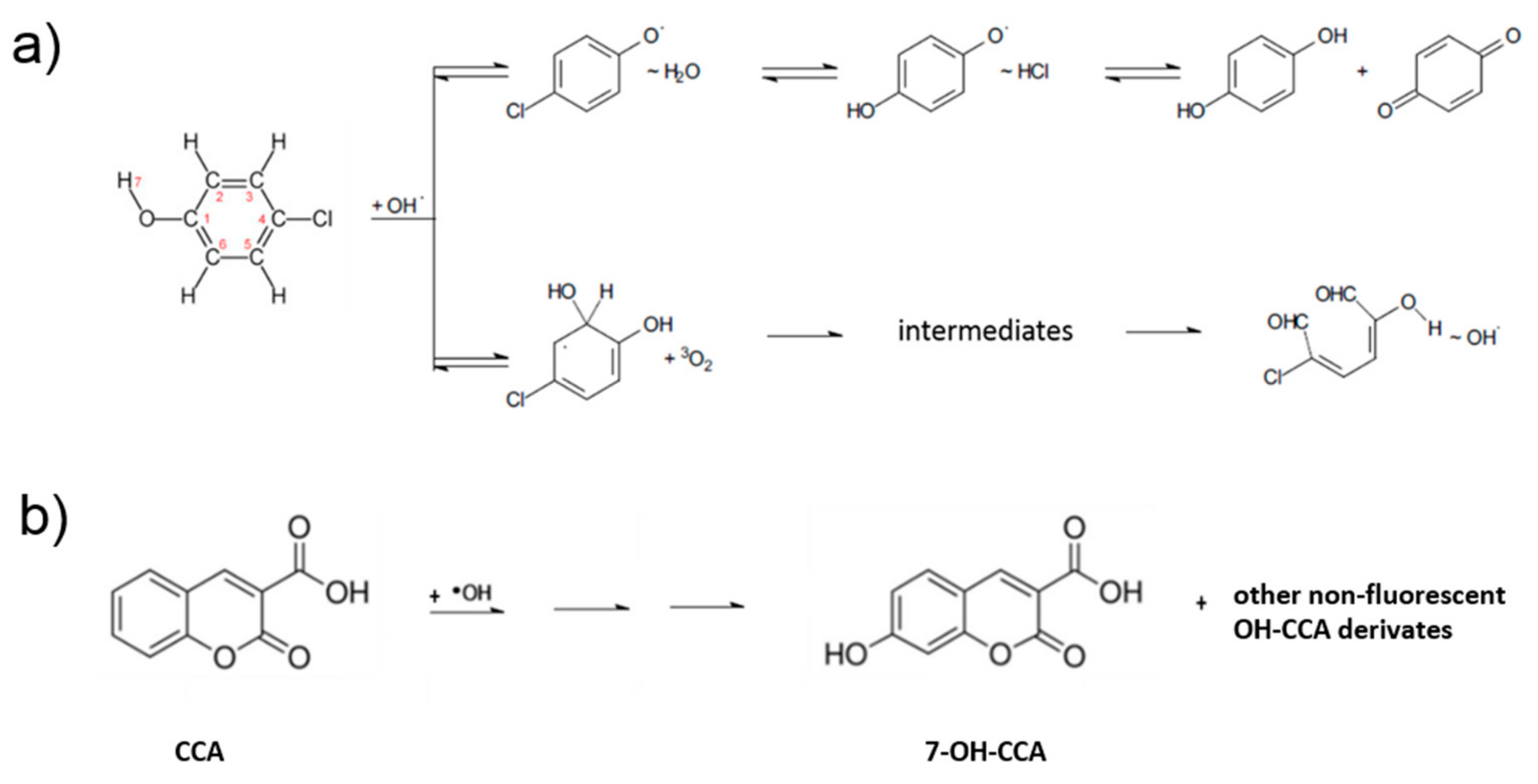
2.4. Photocatalytic Performance of rGO/TiO2 Layers
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niu, P.; Zhang, L.; Liu, G.; Cheng, H.M. Graphene-like carbon nitride nanosheets for improved photocatalytic activities. Adv. Funct. Mater. 2012, 22, 4763–4770. [Google Scholar] [CrossRef]
- Tu, W.; Zhou, Y.; Zou, Z. Versatile graphene-promoting photocatalytic performance of semiconductors: Basic principles, synthesis, solar energy conversion, and environmental applications. Adv. Funct. Mater. 2013, 23, 4996–5008. [Google Scholar] [CrossRef]
- Jeon, S.J.; Kang, T.W.; Ju, J.M.; Kim, M.J.; Park, J.H.; Raza, F.; Han, J.; Lee, H.R.; Kim, J.H. Modulating the Photocatalytic Activity of Graphene Quantum Dots via Atomic Tailoring for Highly Enhanced Photocatalysis under Visible Light. Adv. Funct. Mater. 2016, 26, 8211–8219. [Google Scholar] [CrossRef]
- Chabot, V.; Higgins, D.; Yu, A.; Xiao, X.; Chen, Z.; Zhang, J. A review of graphene and graphene oxide sponge: Material synthesis and applications to energy and the environment. Energy Environ. Sci. 2014, 7, 1564–1596. [Google Scholar] [CrossRef]
- Li, F.; Jiang, X.; Zhao, J.; Zhang, S. Graphene oxide: A promising nanomaterial for energy and environmental applications. Nano Energy 2015, 16, 488–515. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Avouris, P. Graphene: Electronic and photonic properties and devices. Nano Lett. 2010, 10, 4285–4294. [Google Scholar] [CrossRef]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Gao, W. The chemistry of graphene oxide. In Graphene Oxide Reduction Recipes, Spectroscope and Application; Springer: Cham, Switzerland, 2015; pp. 61–95. [Google Scholar]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Liu, L.; Qing, M.; Wang, Y.; Chen, S. Defects in Graphene: Generation, Healing, and Their Effects on the Properties of Graphene: A Review. J. Mater. Sci. Technol. 2015, 31, 599–606. [Google Scholar] [CrossRef]
- Huang, X.; Qi, X.; Boey, F.; Zhang, H. Graphene-based composites. Chem. Soc. Rev. 2012, 41, 666–686. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Yu, J.; Jaroniec, M. Graphene-based semiconductor photocatalysts. Chem. Soc. Rev. 2012, 41, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Eda, G.; Chhowalla, M. Graphene-based composite thin films for electronics. Nano Lett. 2009, 9, 814–818. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, J.C. Graphene-based photocatalytic composites. RSC Adv. 2011, 1, 1426–1434. [Google Scholar] [CrossRef]
- Li, N.; Liu, G.; Zhen, C.; Li, F.; Zhang, L.; Cheng, H.M. Battery performance and photocatalytic activity of mesoporous anatase TiO2 nanospheres/graphene composites by template-free self-assembly. Adv. Funct. Mater. 2011, 21, 1717–1722. [Google Scholar] [CrossRef]
- Oishi, I.; Kim, S.; Yoshii, K.; Esteban, C.R.; Izpisua Belmonte, J.C. Cre-LoxP-regulated expression of monoclonal antibodies driven by an ovalbumin promoter in primary oviduct cells. BMC Biotechnol. 2011, 11, 19. [Google Scholar] [CrossRef]
- Dąbrowski, A. Adsorption-from theory to practice. Adv. Colloid Interface Sci. 2001, 93, 135–224. [Google Scholar] [CrossRef]
- Zouzelka, R.; Remzova, M.; Brabec, L.; Rathousky, J. Photocatalytic performance of porous TiO2 layers prepared by quantitative electrophoretic deposition from organic solvents. Appl. Catal. B Environ. 2018, 227. [Google Scholar] [CrossRef]
- Liu, J.; Bai, H.; Wang, Y.; Liu, Z.; Zhang, X.; Sun, D.D. Self-assembling TiO2 nanorods on large graphene oxide sheets at a two-phase interface and their anti-recombination in photocatalytic applications. Adv. Funct. Mater. 2010, 20, 4175–4181. [Google Scholar] [CrossRef]
- Guérin, V.M.; Zouzelka, R.; Bibova-Lipsova, H.; Jirkovsky, J.; Rathousky, J.; Pauporté, T. Experimental and DFT study of the degradation of 4-chlorophenol on hierarchical micro-/nanostructured oxide films. Appl. Catal. B Environ. 2015, 168–169, 132–140. [Google Scholar] [CrossRef]
- Zouzelka, R.; Kusumawati, Y.; Remzova, M.; Rathousky, J.; Pauporté, T. Photocatalytic activity of porous multiwalled carbon nanotube-TiO2 composite layers for pollutant degradation. J. Hazard. Mater. 2016, 317, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Fattakhova-Rohlfing, D.; Kayaalp, B.E.; Rathouský, J.; Bein, T. Tailoring the morphology of mesoporous titania thin films through biotemplating with nanocrystalline cellulose. J. Am. Chem. Soc. 2014, 136, 5930–5937. [Google Scholar] [CrossRef] [PubMed]
- Zita, J.; Krýsa, J.; Černigoj, U.; Lavrenčič-Štangar, U.; Jirkovský, J.; Rathouský, J. Photocatalytic properties of different TiO2 thin films of various porosity and titania loading. Catal. Today 2011, 161, 29–34. [Google Scholar] [CrossRef]
- Hynek, J.; Kalousek, V.; Žouželka, R.; Bezdička, P.; Dzik, P.; Rathouský, J.; Demel, J.; Lang, K. High photocatalytic activity of transparent films composed of zno nanosheets. Langmuir 2014, 30, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Fravventura, M.C.; Fattakhova-Rohlfing, D.; Rathouský, J.; Movsesyan, L.; Ganter, P.; Savenije, T.J.; Bein, T. Nanocellulose-Templated Porous Titania Scaffolds Incorporating Presynthesized Titania Nanocrystals. Chem. Mater. 2015, 27, 6205–6212. [Google Scholar] [CrossRef]
- Kusumawati, Y.; Pauporté, T.; Viana, B.; Zouzelka, R.; Remzova, M.; Rathousky, J. Mesoporous TiO2/graphene composite films for the photocatalytic degradation of eco-persistent pollutants. In Proceedings of the Oxide-based Materials and Devices VIII, San Francisco, CA, USA, 29 January–1 February 2017; Volume 10105. [Google Scholar]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 183, 153–160. [Google Scholar] [CrossRef]
- Shahhriary, L.; Athawale, A.A. Graphene Oxide Synthesized by using Modified Hummers Approach. Int. J. Renew. Energy Environ. Eng. 2014, 2, 58–63. [Google Scholar]
- Rattana, T.; Chaiyakun, S.; Witit-Anun, N.; Nuntawong, N.; Chindaudom, P.; Oaew, S.; Kedkeaw, C.; Limsuwan, P. Preparation and characterization of graphene oxide nanosheets. Procedia Eng. 2012, 32, 759–764. [Google Scholar] [CrossRef]
- Li, L.; Yan, J.; Wang, T.; Zhao, Z.-J.; Zhang, J.; Gong, J.; Guan, N. Sub-10 nm rutile titanium dioxide nanoparticles for efficient visible-light-driven photocatalytic hydrogen production. Nat. Commun. 2015, 6, 5881. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the thermal deoxygenation of graphene oxide using high-resolution in situ X-ray-based spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef]
- Mattevi, C.; Eda, G.; Agnoli, S.; Miller, S.; Mkhoyan, K.A.; Celik, O.; Mastrogiovanni, D.; Granozzi, G.; Carfunkel, E.; Chhowalla, M. Evolution of electrical, chemical, and structural properties of transparent and conducting chemically derived graphene thin films. Adv. Funct. Mater. 2009, 19, 2577–2583. [Google Scholar] [CrossRef]
- Akhavan, O. The effect of heat treatment on formation of graphene thin films from graphene oxide nanosheets. Carbon 2010, 48, 509–519. [Google Scholar] [CrossRef]
- Jeong, H.K.; Yun, P.L.; Lahaye, R.J.W.E.; Park, M.H.; Kay, H.A.; Ick, J.K.; Yang, C.W.; Chong, Y.P.; Ruoff, R.S.; Young, H.L. Evidence of graphitic AB stacking order of graphite oxides. J. Am. Chem. Soc. 2008, 130, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Velamakanni, A.; Bozoklu, G.; Park, S.; Stoller, M.; Piner, R.D.; Stankovich, S.; Jung, I.; Field, D.A.; Ventrice, C.A.; et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon 2009, 47, 145–152. [Google Scholar] [CrossRef]
- Vanquickenborne, L.G.; Vranckx, J.; Görller-walrand, C. On the Kinetic Trans Effect in Square Planar Transition Metal Complexes. J. Am. Chem. Soc. 1974, 96, 4121–4125. [Google Scholar] [CrossRef]
- Kubelka, P. New Contributions to the Optics of Intensely Light-Scattering Materials. Part I. J. Opt. Soc. Am. 1948, 38, 448–457. [Google Scholar] [CrossRef]
- Ohtani, B. Photocatalysis A to Z-What we know and what we do not know in a scientific sense. J. Photochem. Photobiol. C Photochem. Rev. 2010, 11, 157–178. [Google Scholar] [CrossRef]
- Tauc, J. Optical Properties and electronic structure of amorphous. Mater. Res. Bull. 1968, 3, 37–46. [Google Scholar] [CrossRef]
- Motohashi, N.; Saito, Y. Competetive measurement of rate constants for hydroxyl radical reactions using radiolytic hydroxylation of benzoate. Chem. Pharm. Bull. (Tokyo) 1993, 41, 1842–1845. [Google Scholar] [CrossRef]
- Stafford, U.; Gray, K.A.; Kamat, P.V. Radiolytic and TiO2-assisted photocatalytic degradation of 4-chlorophenol. A comparative study. J. Phys. Chem. 1994, 98, 6343–6351. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C–C/% | C–OH/% | C=O/% | O=C–OH/% |
|---|---|---|---|---|
| GO | 22.7 | 24.6 | 46.8 | 5.8 |
| rGO | 63.5 | 23.9 | 8.2 | 4.3 |
| rGO/TiO2 | 66.7 | 19.4 | 8.2 | 5.6 |
| Sample | C=O/% | C–OH/% | H–O–H/% |
|---|---|---|---|
| GO | 27.6 | 69.8 | 2.6 |
| rGO | 28.5 | 67.7 | 3.8 |
| rGO/TiO2 | 26.6 | 66.5 | 6.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zouzelka, R.; Remzova, M.; Plsek, J.; Brabec, L.; Rathousky, J. Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water. Catalysts 2019, 9, 708. https://doi.org/10.3390/catal9090708
Zouzelka R, Remzova M, Plsek J, Brabec L, Rathousky J. Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water. Catalysts. 2019; 9(9):708. https://doi.org/10.3390/catal9090708
Chicago/Turabian StyleZouzelka, Radek, Monika Remzova, Jan Plsek, Libor Brabec, and Jiri Rathousky. 2019. "Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water" Catalysts 9, no. 9: 708. https://doi.org/10.3390/catal9090708
APA StyleZouzelka, R., Remzova, M., Plsek, J., Brabec, L., & Rathousky, J. (2019). Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water. Catalysts, 9(9), 708. https://doi.org/10.3390/catal9090708