Continuous-Flow Process for Glycerol Conversion to Solketal Using a Brönsted Acid Functionalized Carbon-Based Catalyst


 ,
, 


Abstract
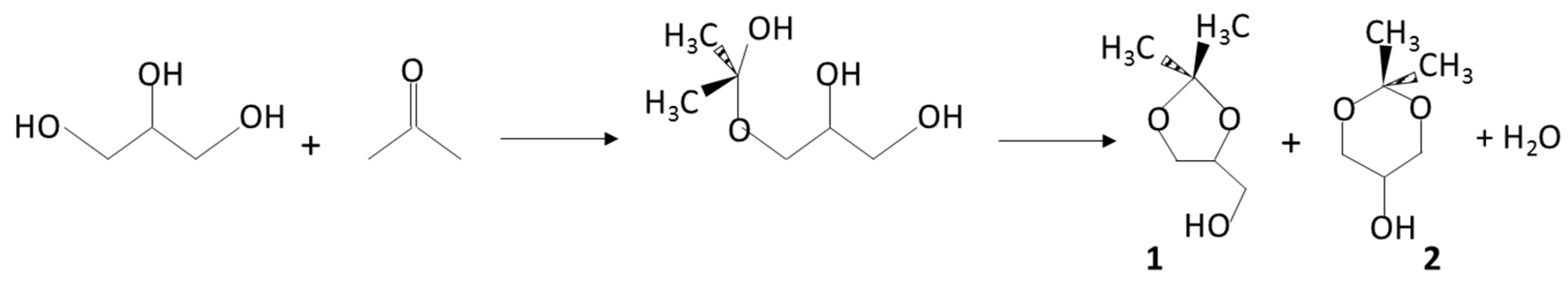
1. Introduction
2. Results and Discussion
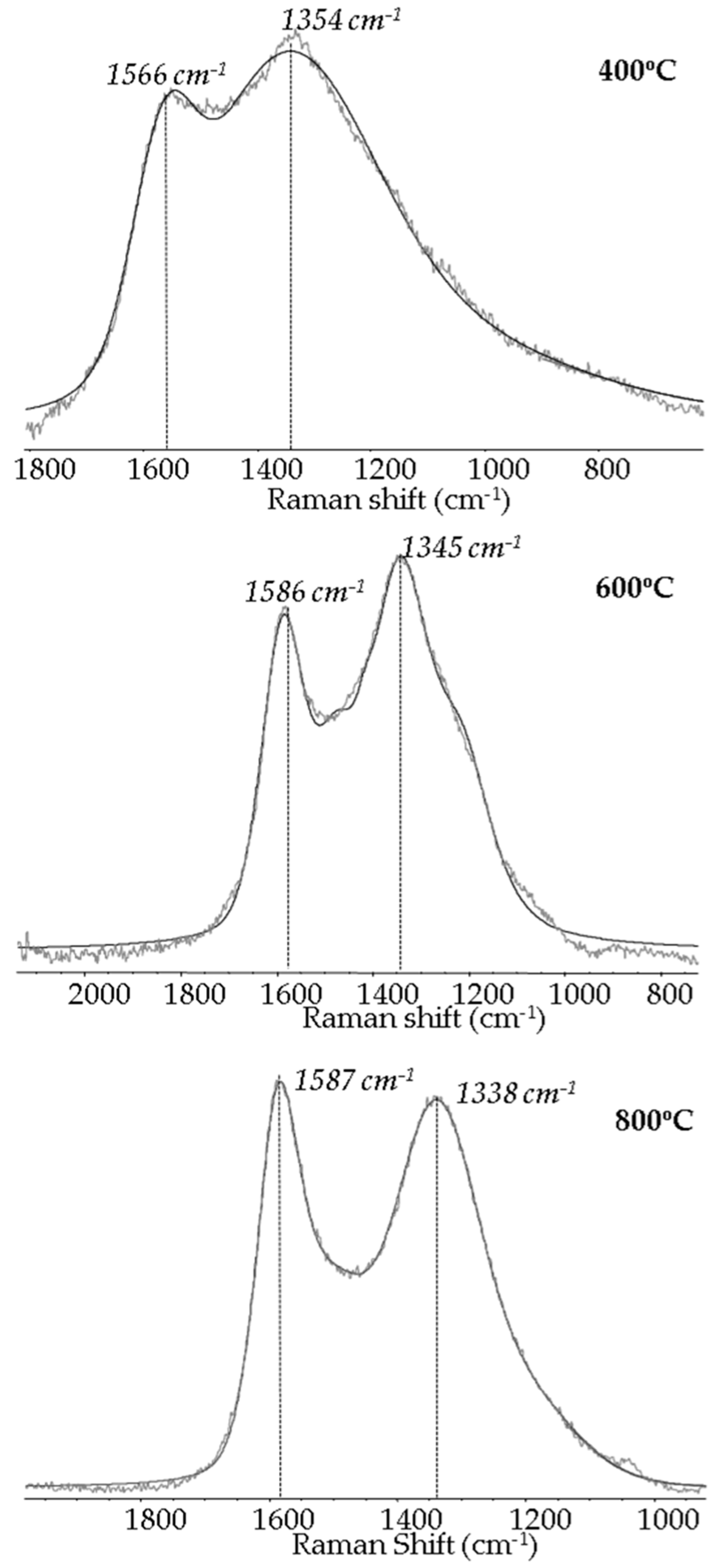
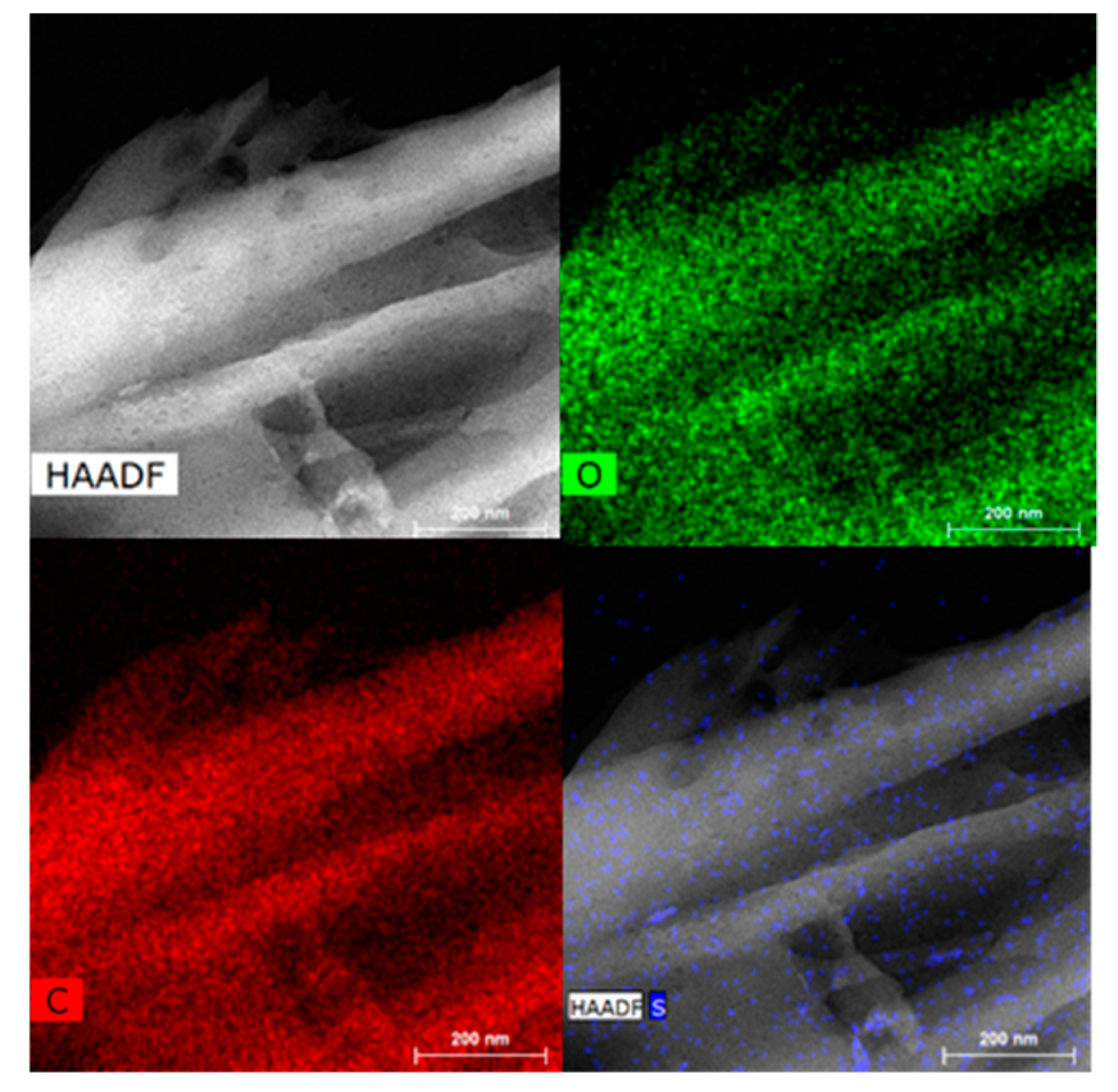
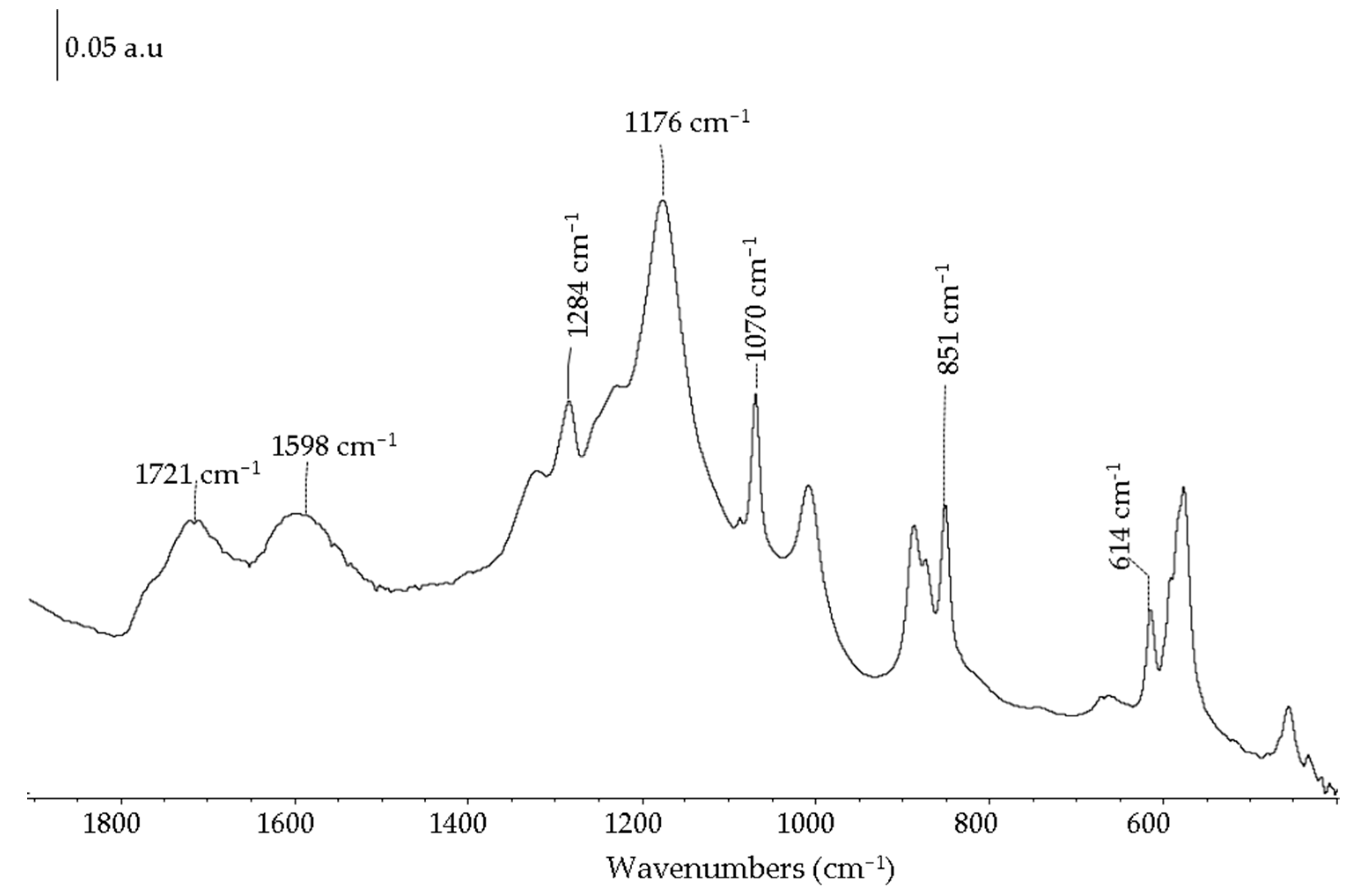
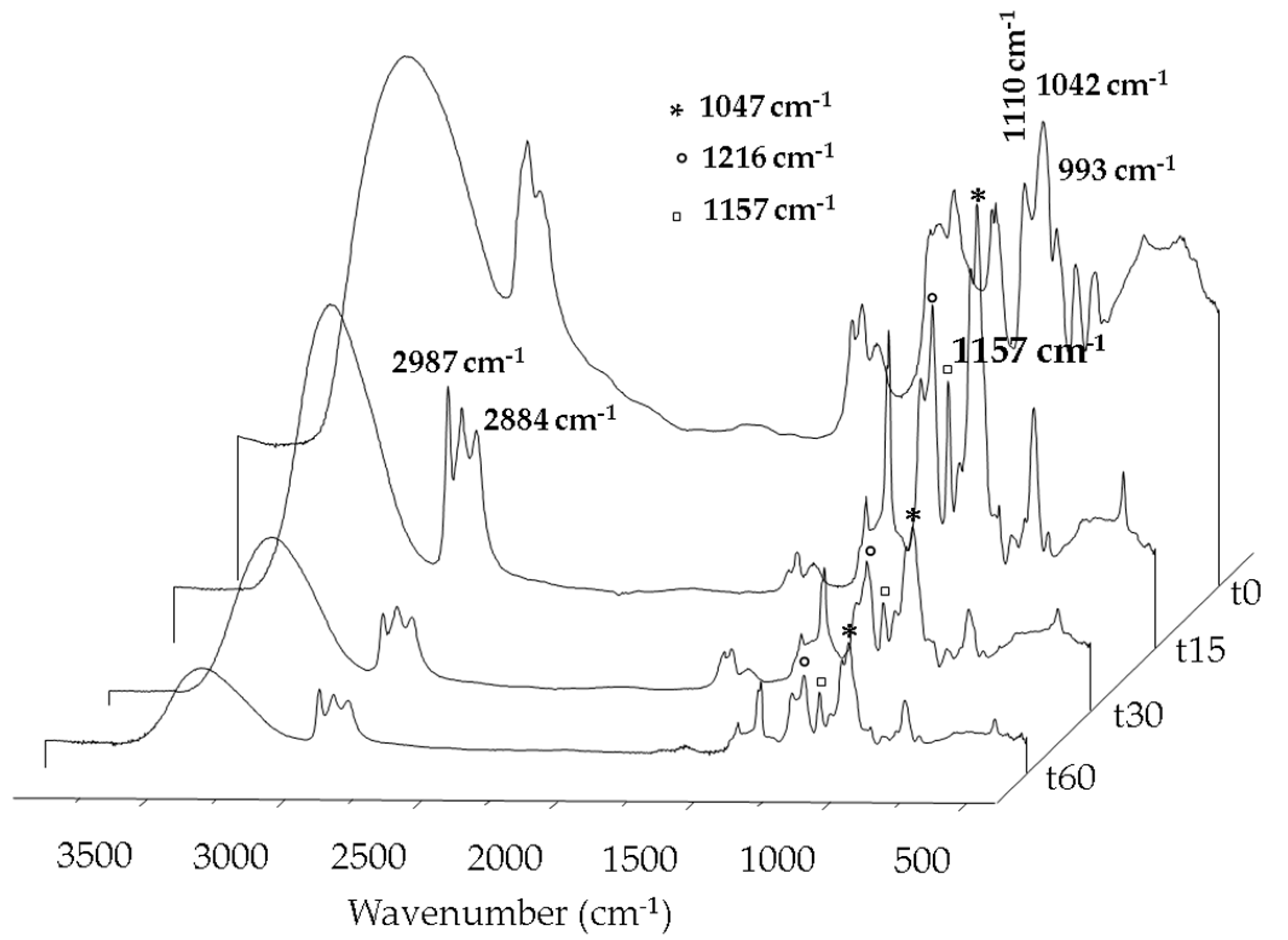
2.1. Carbon-Based Support and Catalyst Characterization
2.2. Catalytic Test
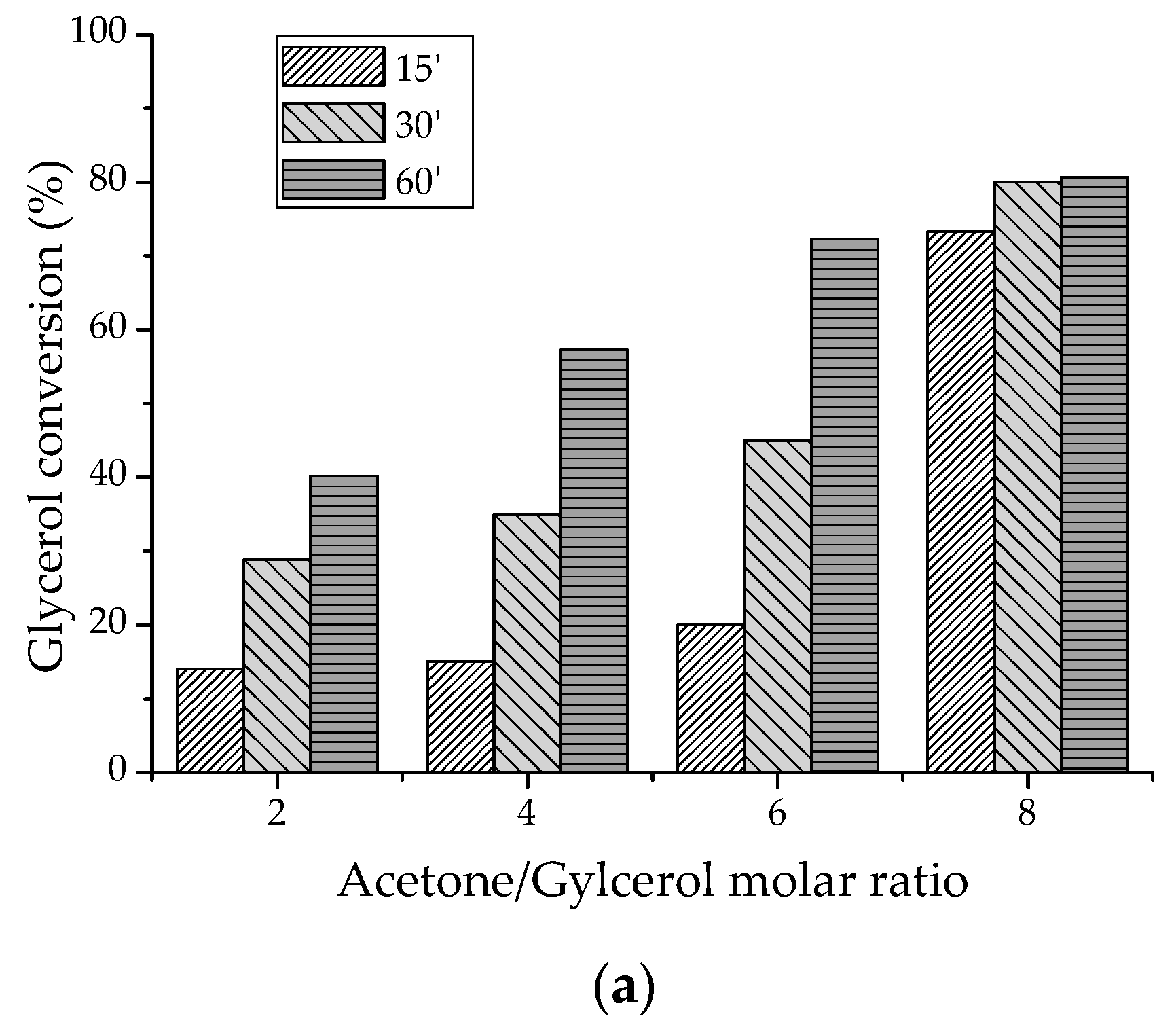
2.2.1. Batch-Reactor Experiments
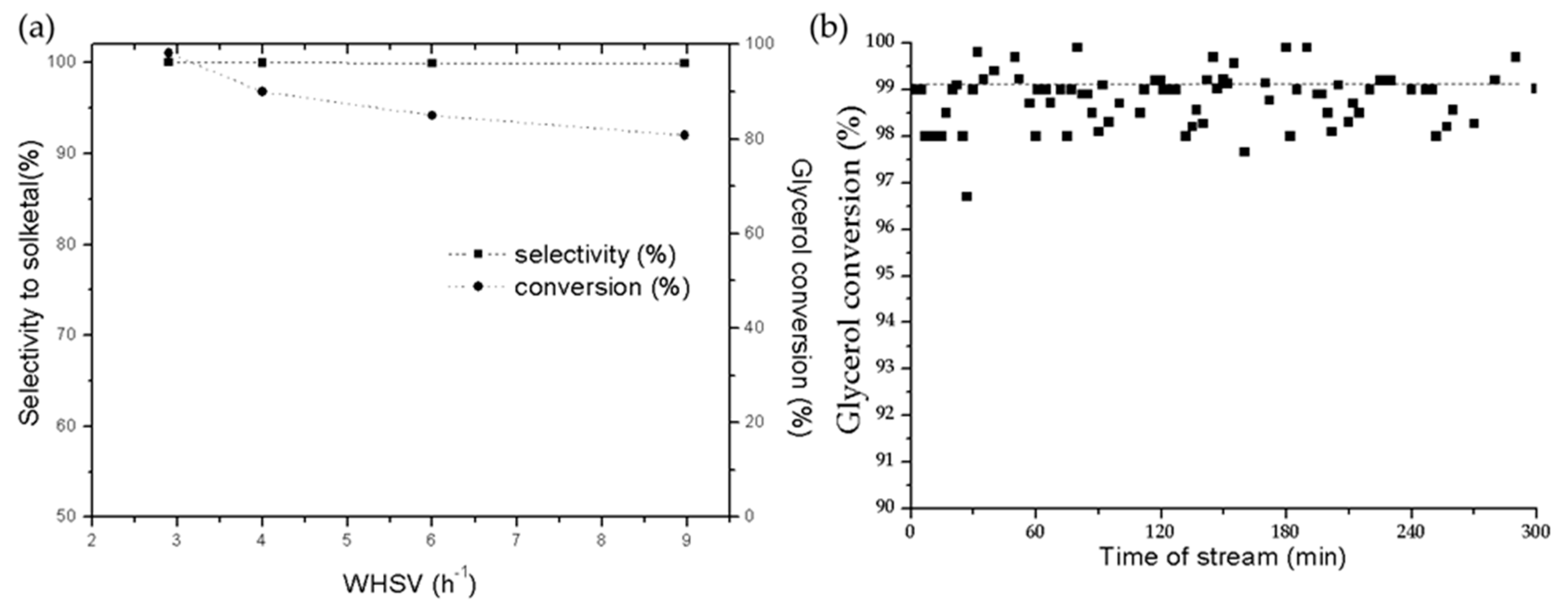
2.2.2. Continuous-Flow Reactor Experiments
3. Materials and Methods
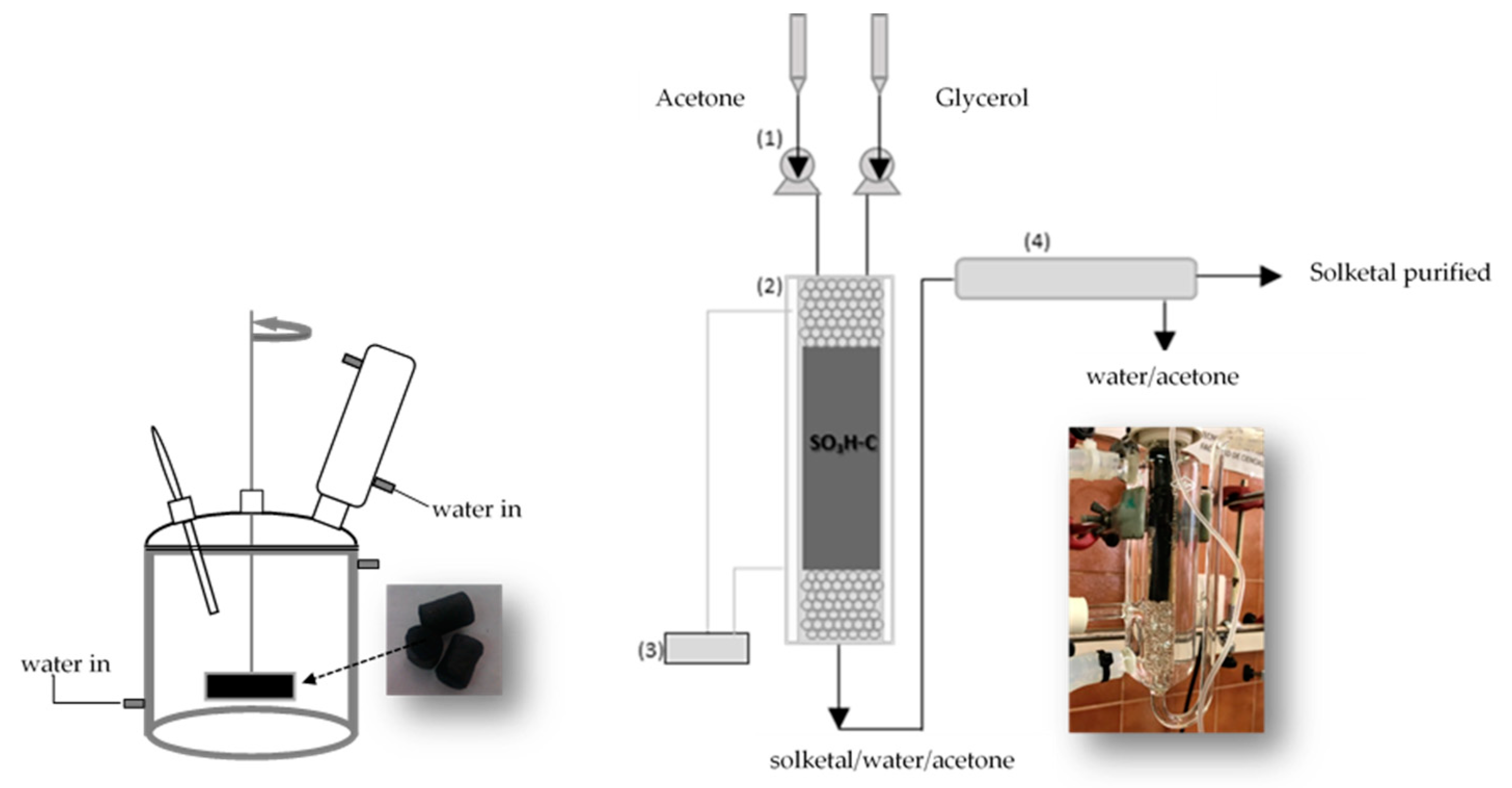
3.1. Catalyst Preparation and Characterization
3.2. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smirnov, A.A.; Selishcheva, S.A.; Yakovlev, V.A. Acetalization catalysts for synthesis of valuable oxygenated fuel additives from glycerol. Catalysts 2018, 8, 598. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z. Critical review on the current scenario and significance of crude glycerol resulting from biodiesel industry towards more sustainable renewable energy industry. Renew. Sustain. Energy Rev. 2012, 16, 2671–2686. [Google Scholar] [CrossRef]
- Gu, Y.; Jerome, F. Glycerol as a sustainable solvent for green chemistry. Green Chem. 2010, 12, 1127–1138. [Google Scholar] [CrossRef]
- Garcia, E.; Laca, M.; Perez, E.; Garrido, A.; Peinado, J. New class of acetal derived from glycerine as a biodiesel fuel component. Energy Fuels 2008, 22, 4274–4280. [Google Scholar] [CrossRef]
- Ntumba Tshibalonza, N.; Monbaliu, J.C.M. Revisiting the deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions. Green Chem. 2017, 19, 3006–3013. [Google Scholar] [CrossRef]
- Moreira, A.B.F.; Bruno, A.M.; Souza, M.M.V.M.; Manfro, R.L. Continuous production of lactic acid from glycerol in alkaline medium using supported copper catalysts. Fuel Process. Technol. 2016, 144, 170–180. [Google Scholar] [CrossRef]
- Bruno, A.M.; Chagas, C.A.; Souza, M.M.V.M.; Manfro, R.L. Lactic acid production from glycerol in alkaline medium using Pt-based catalysts in continuous flow reaction system. Renew. Energy 2018, 118, 160–171. [Google Scholar] [CrossRef]
- Rastegari, H.; Ghaziaskar, H.S.; Yalpani, M.; Shafiei, A. Development of a continuous system based on azeotropic reactive distillation to enhance triacetin selectivity in glycerol esterification with acetic acid. Energy Fuels 2017, 31, 8256–8262. [Google Scholar] [CrossRef]
- Calmanti, R.; Galvan, M.; Amadi, E.; Perosa, A.; Selva, M. High-temperature batch and continuous-flow transesterification of alkyl and enol esters with glycerol and its acetal derivatives. ACS Sustain. Chem. Eng. 2018, 6, 3964–3973. [Google Scholar] [CrossRef]
- Li, Z.; Miao, Z.; Wang, X.; Zhao, J.; Zhou, J.; Si, W.; Zhuo, S. One-pot synthesis of ZrMo-KIT-6 solid acid catalyst for solvent-free conversion of glycerol to solketal. Fuel 2018, 233, 377–387. [Google Scholar] [CrossRef]
- Samoilov, L.; Maximov, V.O.; Stolonogova, T.I.; Chernysheva, E.A.; KapustinbA, O.K.; Arpunina, V.M. Glycerol to renewable fuel oxygenates. Part I: Comparison between solketal and its methyl ether. Fuel 2019, 249, 486–495. [Google Scholar] [CrossRef]
- Mallesham, B.; Sudarsanam, P.; Raju, G.; Reddy, B.M. Design of highly efficient Mo and W promoted SnO2 solid acids for heterogeneous catalysis: Acetalization of bioglycerol. Green Chem. 2013, 15, 478–489. [Google Scholar] [CrossRef]
- Pinto, B.P.; De Lyra, J.T.; Nascimento, J.A.C.; Mota, C.J.A. Ethers of glycerol and ethanol as bioadditives for biodiesel. Fuel 2016, 168, 76–80. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Poirier, M.-A.; Xu, C.C. A new continuous-flow process for catalytic conversion of glycerol to oxygenated fuel additive: Catalyst screening. Appl. Energy 2014, 123, 75–81. [Google Scholar] [CrossRef]
- Roldan, L.; Mallada, R.; Fraile, J.M.; Mayoral, J.A.; Menendez, M. Glycerol upgrading by ketalization in a zeolite membrane reactor. Asia-Pac J Chem. Eng. 2009, 4, 279–284. [Google Scholar] [CrossRef]
- Chen, L.; Nohair, B.; Zhao, D.; Kaliaguine, S. Glycerol acetalization with formaldehyde using heteropolyacid salts supported on mesostructured silica. Appl. Catal. A Gen. 2018, 549, 207–215. [Google Scholar] [CrossRef]
- Fechete, I.; Wang, Y.; Védrine, J.C. The past, present and future of heterogeneous catalysis. Catal. Today 2012, 189, 2–27. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.; Yu, W.J.; Li, L.D.; Zhang, M.H.; Zhang, Z.B. A swelling-changeful catalyst for glycerol acetylation with controlled acid concentration. Fuel Process. Technol. 2016, 142, 228–234. [Google Scholar] [CrossRef]
- Zhou, L.; Nguyen, T.-H.; Adesina, A.A. The acetylation of glycerol over amberlyst-15: Kinetic and product distribution. Fuel Process. Technol. 2012, 104, 310–318. [Google Scholar] [CrossRef]
- Nda-Umar, U.I.; Ramli, I.; Taufiq-Yap, Y.H.; Muhamad, E.N. An overview of recent research in the conversion of glycerol into biofuels, fuel additives and other Bio-Based chemicals. Catalysts 2019, 5, 15. [Google Scholar] [CrossRef]
- Reddy, P.S.; Sudarsanam, P.; Mallesham, B.; Raju, G.; Reddy, B.M. Acetalisation of glycerol with acetone over zirconia and promoted zirconia catalysts under mild reaction conditions. J. Ind. Eng. Chem. 2011, 17, 377–381. [Google Scholar] [CrossRef]
- Esteban, J.; Vorholt, A.J.; Behr, A.; Ladero, M.; Garcia-Ochoa, F.J. Liquid–Liquid Equilibria for the System Acetone + Solketal + Glycerol at (303.2, 313.2, and 323.2) K. Chem. Eng. Data. 2014, 59, 2850–2855. [Google Scholar] [CrossRef]
- Nanda, M.R.; Zhang, Y.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Xu, C. Catalytic conversion of glycerol for sustainable production of solketal as a fuel additive: A review. Renew. Sustain. Energy Rev. 2016, 56, 1022–1031. [Google Scholar] [CrossRef]
- Shirani, M.; Ghaziaskar, H.S.; Xu, C. Optimization of glycerol ketalization to produce solketal as biodiesel additive in a continuous reactor with subcritical acetone using Purolite® PD206 as catalyst. Fuel Process. Technol. 2014, 124, 206–211. [Google Scholar] [CrossRef]
- Krishnamachari, P.; Hashaikeh, R.; Materials, M.T. Modified cellulose morphologies and its composites; SEM and TEM analysis. Micron 2011, 42, 751–761. [Google Scholar] [CrossRef]
- Wang, Y.; Serrano, S.; Avilés, J.J. Raman characterization of carbon nanofibers prepared using Electrospinning. Synth. Met. 2003, 138, 423–427. [Google Scholar] [CrossRef]
- Ulla, M.A.; Valera, A.; Ubieto, T.; Latorre, N.; Romeo, E.; Milt, V.G.; Monzón, A. Carbon nanofiber growth onto a cordierite monolith coated with Co-mordenite. Catal. Today 2008, 133–135, 7–12. [Google Scholar] [CrossRef]
- Boyano, A.; Herrera, C.; Larrubia, M.A.; Alemany, L.J.; Moliner, R.; Lázaro, M.J. Vanadium loaded carbon-based monoliths for the on-board no reduction: Influence of temperature and period of the oxidation treatment. Chem. Eng. J. 2010, 160, 623–633. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, X.; Lin, D.; Chun, Y.; Xu, Q. Preparation of shaped magnesium oxide/carbon catalysts using rice grains. Appl. Catal. A Gen. 2013, 460–461, 26–35. [Google Scholar] [CrossRef]
- Fraile, J.M.; García-Bordeje, E.; Pires, E.; Roldán, L. New insights into the strength and accessibility of acid sites of sulfonated hydrothermal carbon. Carbon 2014, 77, 1157–1167. [Google Scholar] [CrossRef]
- NIST X-ray Photoelectron Spectroscopy Database; NIST Standard Reference Database Number 20; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2000. [CrossRef]
- Petit, C.; Seredych, M.; Bandosz, T.J. Revisiting the chemistry of graphite oxides and its effect on ammonia adsorption. J. Mater. Chem. 2009, 19, 9176–9185. [Google Scholar] [CrossRef]
- Takagaki, A.; Toda, M.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Esterification of higher fatty acids by a novel strong solid acid. Catal. Today 2006, 116, 157–161. [Google Scholar] [CrossRef]
- Rodrigues, R.; Gonçalves, M.; Mandelli, D.; Pescarmona, P.P.; Carvalho, W.A. Solvent-free conversion of glycerol to Solketal catalysed by activated carbons functionalized with acid groups. Catal. Sci. Technol. 2014, 4, 2293–2301. [Google Scholar] [CrossRef]
- Kolvari, E.; Koukabi, N.; Hosseini, M.M. Perlite: A cheap natural support for immobilization of sulfonic acid as a heterogeneous solid acid catalyst for the heterocyclic multicomponent reaction. J. Mol. Catal. A Chem. 2015, 397, 68–75. [Google Scholar] [CrossRef]
- Fraile, J.M.; García-Bordejé, E.; Roldán, L. Deactivation of sulfonated hydrothermal carbons in the presence of alcohols: Evidences for sulfonic esters formation. J. Catal. 2012, 289, 73–79. [Google Scholar] [CrossRef]
- Santos, E.M.; de Carvalho Teixeira, A.P.; Silva, F.G.; Cibaka, T.E.; Araújo, M.H.; Oliveira, W.X.C.; Medeiros, F.; N.Brasil, A.; de Oliveira, L.S.; Lago, R.M. New heterogeneous catalyst for the esterification of fatty acid produced by surface aromatization/sulfonation of oilseed cake. Fuel 2015, 150, 408–414. [Google Scholar] [CrossRef]
- Esteban, J.; Ladero, M.; García-Ochoa, F. Kinetic modelling of the solventless syntesis of Solketal with a sulphonic ion exchange resin. Chem. Eng. J. 2015, 269, 194–202. [Google Scholar] [CrossRef]
- Serafim, H.; Fonseca, I.M.; Ramos, A.M.; Vital, J.; Castanheiro, J.E. Valorization of glycerol into fuel additives over zeolites as catalysts. Chem. Eng. J. 2011, 178, 291–296. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Poirier, M.-A.; Xu, C.C. Catalytic conversion of glycerol to oxygenated fuel additive in a continuous flow reactor: Process optimization. Fuel 2014, 128, 113–119. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raman Data a | Temperature of Pyrolysis (°C) | |||||||
| 400 °C | 600 °C | 800 °C | ||||||
| D peak position (cm−1) | 1354 | 1345 | 1338 | |||||
| G peak position (cm−1) | 1566 | 1586 | 1587 | |||||
| R = ID/IG | 4.501 | 1.859 | 1.453 | |||||
| Ln = 4.4 nm/R | 0.98 | 2.37 | 3.03 | |||||
| xG = 1/(1 + R) | 0.181 | 0.350 | 0.408 | |||||
| XPS Data | Relative Amount (%) | Atomic Ratio | Deconvolution of C1s and O1s | |||||
| C1s | O1s | N1s | S2p | O/C | C1s | O1s | ||
| C support b | 94.78 | 5.22 | n.d. | - | 0.06 | 284.8 (78) | 532.9 (100) | |
| 286.4 (15) | - | |||||||
| C support c | 80.00 | 17.10 | 2.82 | - | 0.21 | 284.7 (65) | 532.0 (47) | |
| 286.2 (29) 288.9 (6) | 534.1 (53) | |||||||
| Elemental Analysis | Composition (%) | Ratio (%) | ||||||
| C | N | H | S | O | O/C | |||
| C support b | 95.15 | n.d. | 0 | - | 4.68 | 0.05 | ||
| C support c | 78.27 | 2.32 | 7.58 | - | 11.67 | 0.15 | ||
| Raman Data | Peak Position (cm−1) | R = ID/IG | La = 4.4 nm/R | XG = 1/(1 + R) | |||||
| D | 1338 | 2.26 | 1.95 | 0.31 | |||||
| G | 1589 | ||||||||
| XPS Data | Relative Amount (%) | Atomic Ratio | Deconvolution of C1s O1s and S2p | ||||||
| C1s | O1s | N1s | S2p | O/C | C1s | O1s | S2p | ||
| 62.42 | 30.55 | 1.39 | 5.65 | 0.50 | 284.7 (65) | 532.0 (55) | 169.5 (100) | ||
| 286.2 (29) 288.9 (6) | 533.4 (40) 534.6 (6) | ||||||||
| Elemental Analysis | Composition (%) | Ratio (%) | Acid Density (mmol H+ g−1) | ||||||
| C | O | H | S | N | O/C | S/C | |||
| 51.9 | 38.51 | 3.82 | 4.51 | 1.17 | 0.61 | 0.12 | 1.54 a | 2.93 b | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Barroso, V.; Herrera, C.; Larrubia, M.Á.; González-Gil, R.; Cortés-Reyes, M.; Alemany, L.J. Continuous-Flow Process for Glycerol Conversion to Solketal Using a Brönsted Acid Functionalized Carbon-Based Catalyst. Catalysts 2019, 9, 609. https://doi.org/10.3390/catal9070609
Domínguez-Barroso V, Herrera C, Larrubia MÁ, González-Gil R, Cortés-Reyes M, Alemany LJ. Continuous-Flow Process for Glycerol Conversion to Solketal Using a Brönsted Acid Functionalized Carbon-Based Catalyst. Catalysts. 2019; 9(7):609. https://doi.org/10.3390/catal9070609
Chicago/Turabian StyleDomínguez-Barroso, Vanesa, Concepción Herrera, María Ángeles Larrubia, Rafael González-Gil, Marina Cortés-Reyes, and Luis J. Alemany. 2019. "Continuous-Flow Process for Glycerol Conversion to Solketal Using a Brönsted Acid Functionalized Carbon-Based Catalyst" Catalysts 9, no. 7: 609. https://doi.org/10.3390/catal9070609
APA StyleDomínguez-Barroso, V., Herrera, C., Larrubia, M. Á., González-Gil, R., Cortés-Reyes, M., & Alemany, L. J. (2019). Continuous-Flow Process for Glycerol Conversion to Solketal Using a Brönsted Acid Functionalized Carbon-Based Catalyst. Catalysts, 9(7), 609. https://doi.org/10.3390/catal9070609