PPh3-Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT



Abstract
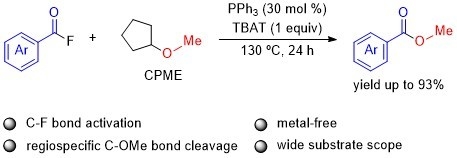
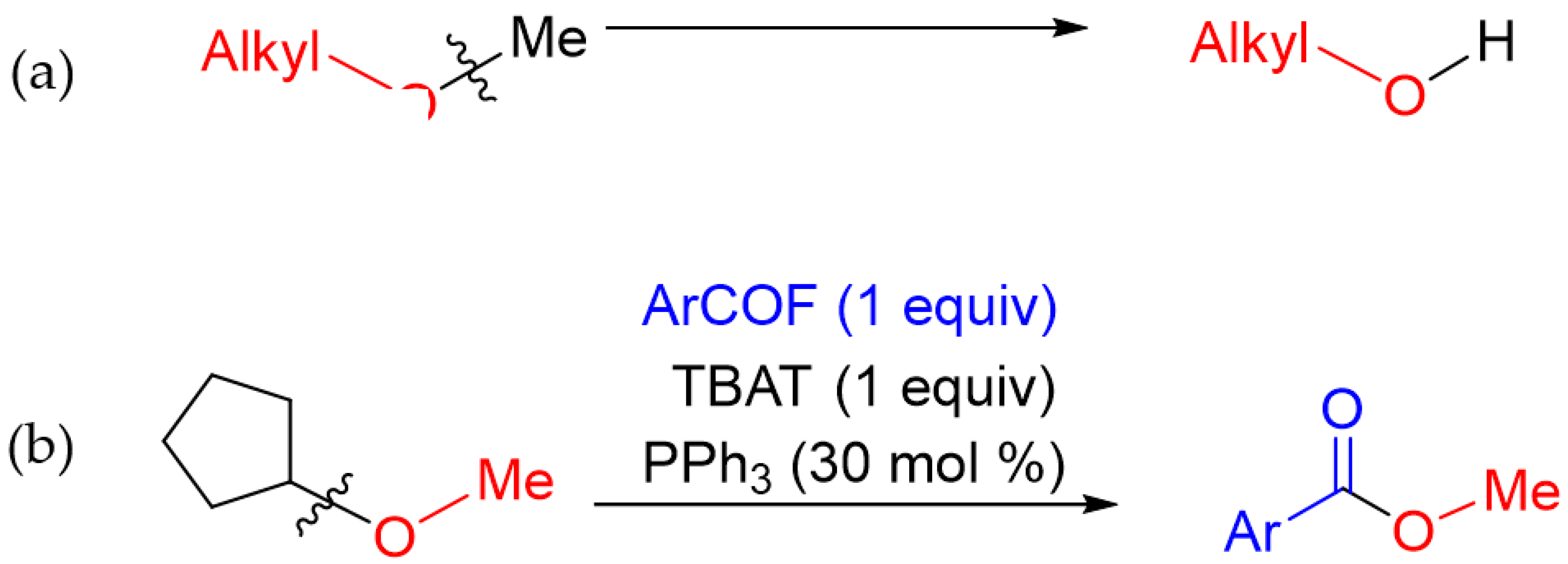
1. Introduction
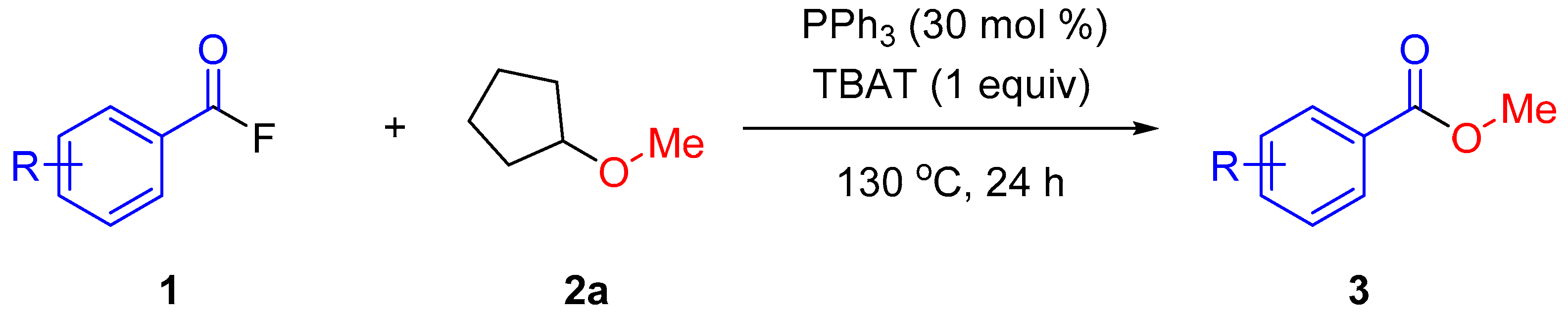
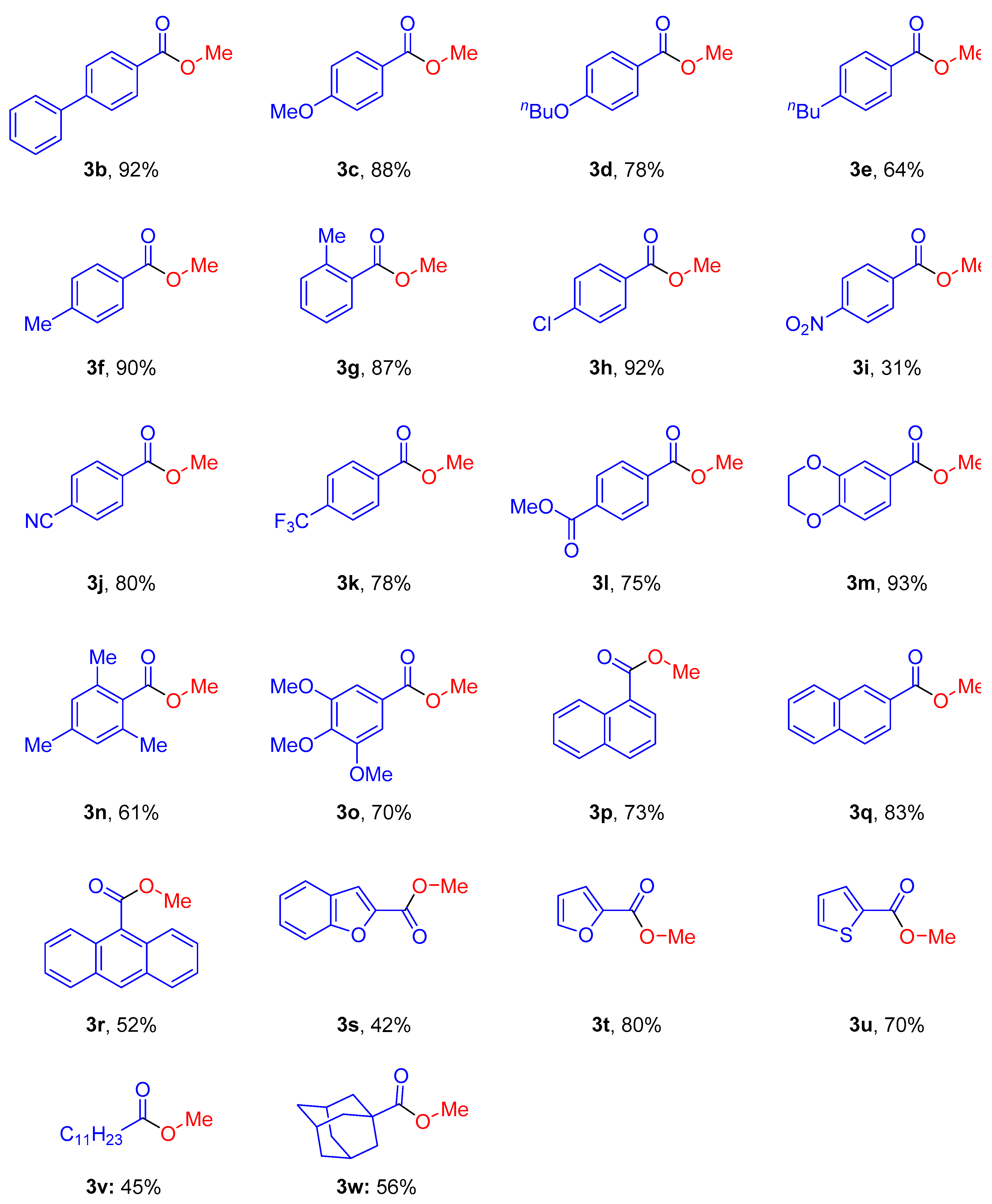
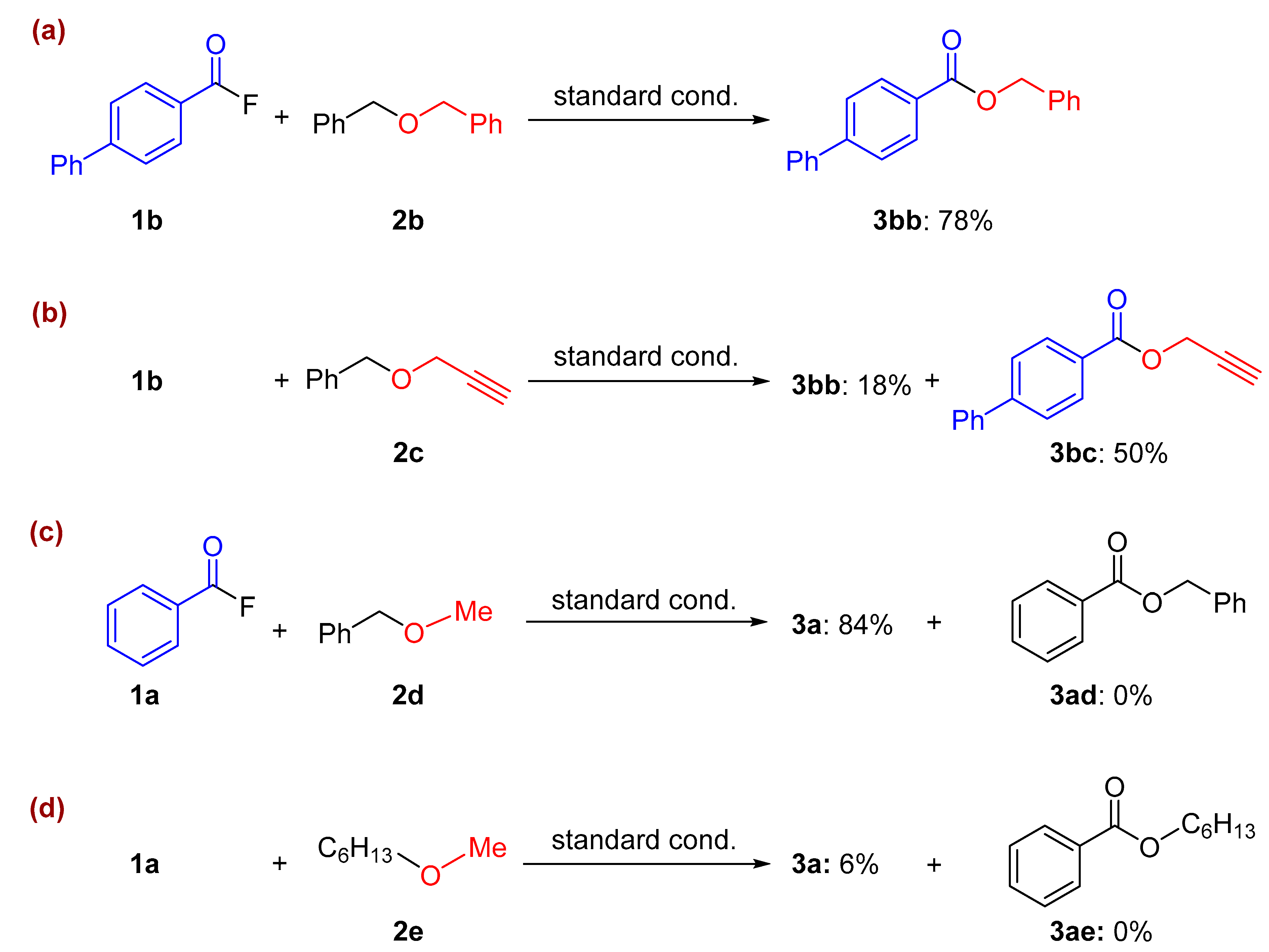
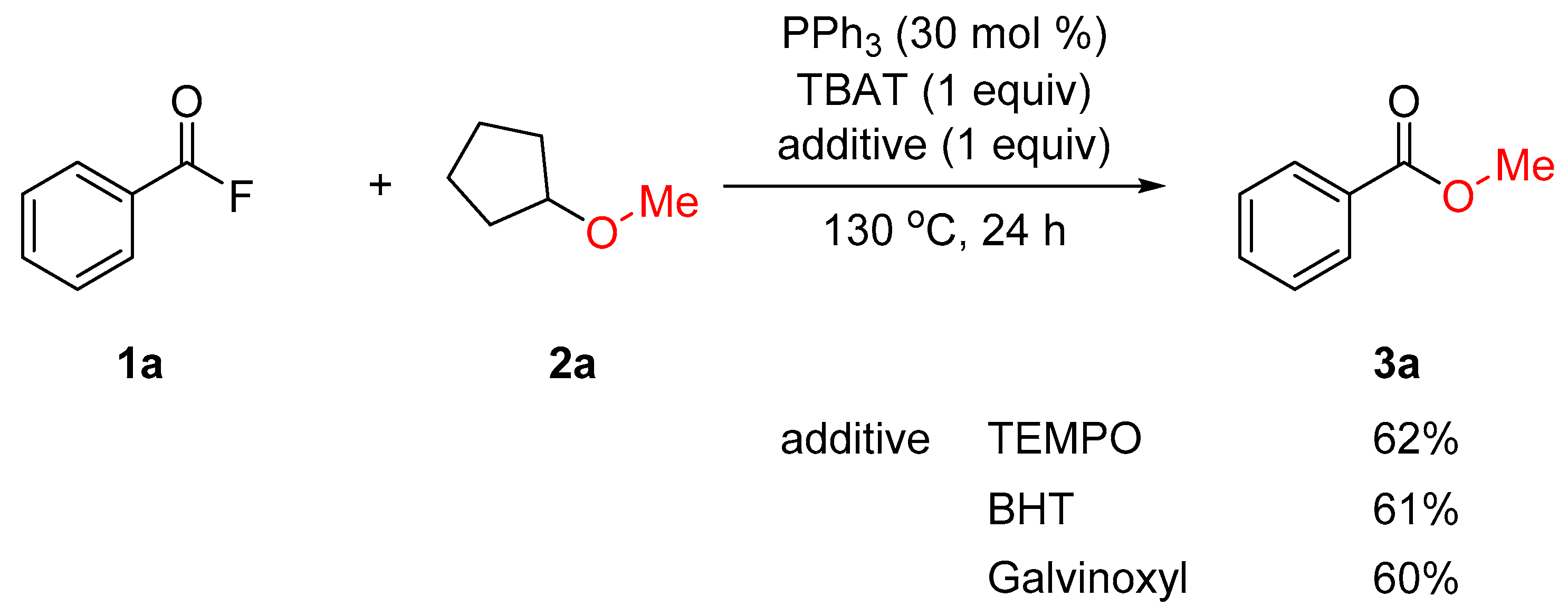
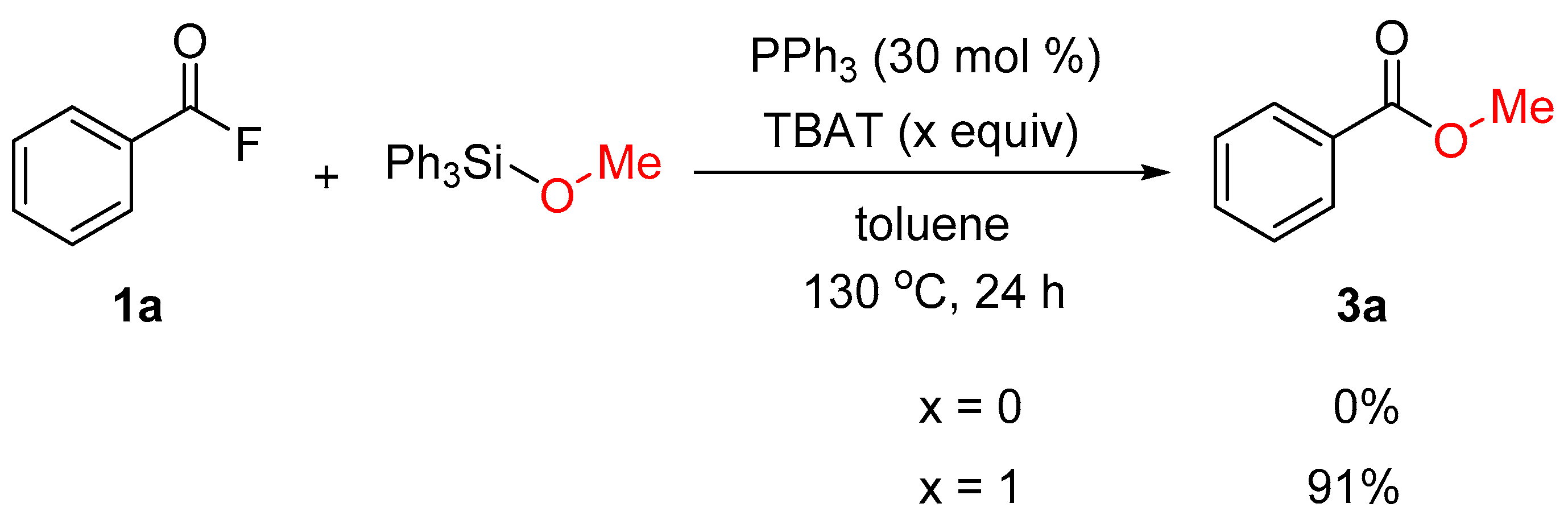
2. Results and Discussion
3. Experimental Sections
3.1. General
3.2. Experimental Method
3.2.1. Representative Procedure for the Synthesis of Acyl Fluorides from Acyl Chlorides
3.2.2. Representative Procedure for the Synthesis of Acyl Fluorides from Carboxylic Acids
3.2.3. Synthesis of Methoxytriphenylsilane
3.2.4. General Methods for the Synthesis of Benzyl Ethers 2b–2d
3.2.5. Representative Procedure for Methoxylation of Acyl Fluorides 1 with CPME (2a)
3.3. Characterization Data of Starting Materials and Products
4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burwell, R.L., Jr. The Cleavage of Ethers. Chem. Rev. 1954, 54, 615–685. [Google Scholar] [CrossRef]
- Maercker, A. Ether Cleavage with Organo-Alkali-Metal Compounds and Alkali Metals. Angew. Chem. Int. Ed. Engl. 1987, 26, 972–989. [Google Scholar] [CrossRef]
- Bhatt, M.V.; Kulkarni, S.U. Cleavage of Ethers. Synthesis 1983, 1983, 249–282. [Google Scholar] [CrossRef]
- Tiecco, M. Selective Dealkylations of Aryl Alkyl Ethers, Thioethers, and Selenoethers. Synthesis 1988, 1988, 749–759. [Google Scholar] [CrossRef]
- Ranu, B.C.; Bhar, S. Dealkylation of Ethers. A Review. Org. Prep. Proced. Int. 1996, 28, 371–409. [Google Scholar] [CrossRef]
- Minamikawa, J.; Brossi, A. Selective o-demethylation of an aromatic methylether in the presence of an aromatic methylenedioxy group with trimethylisilyl iodide in quinoline. Tetrahedron Lett. 1978, 19, 3085–3086. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Moore, J.L.; Cregge, R.J. A convenient new method for converting aromatic methyl ethers to phenols with sodium cyanide-dimethyl sulfoxide. Tetrahedron Lett. 1978, 19, 5183–5186. [Google Scholar] [CrossRef]
- Kamal, A.; Gayatri, N.L. An efficient method for 4-β-anilino-4′-demethylepipodophyllotoxins: Synthesis of NPF and W-68. Tetrahedron Lett. 1996, 37, 3359–3362. [Google Scholar] [CrossRef]
- Brooks, P.R.; Wirtz, M.C.; Vetelino, M.G.; Rescek, D.M.; Woodworth, G.F.; Morgan, B.P.; Coe, J.W. Boron Trichloride/Tetra-n-Butylammonium Iodide: A Mild, Selective Combination Reagent for the Cleavage of Primary Alkyl Aryl Ethers. J. Org. Chem. 1999, 64, 9719–9721. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Nayak, M.K.; Sharma, L. Diphenyl Disulfide and Sodium in NMP as an Efficient Protocol for in Situ Generation of Thiophenolate Anion: Selective Deprotection of Aryl Alkyl Ethers and Alkyl/Aryl Esters under Nonhydrolytic Conditions. J. Org. Chem. 2002, 67, 1776–1780. [Google Scholar] [CrossRef]
- Narayanan, C.R.; Iyer, K.N. Regeneration of Steroid Alcohols from Their Methyl Ethers. J. Org. Chem. 1965, 30, 1734–1736. [Google Scholar] [CrossRef]
- Géro, S.D. The preparation of 1-0-tosyl-(-)-inositol from quebrachitol. Tetrahedron Lett. 1966, 7, 591–595. [Google Scholar] [CrossRef]
- Ayer, W.A.; Bowman, W.R.; Joseph, T.C.; Smith, P. Synthesis of dl-lycopodine. J. Am. Chem. Soc. 1968, 90, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Node, M.; Hori, H.; Fujita, E. Demethylation of aliphatic methyl ethers with a thiol and boron trifluoride. J. Chem. Soc. Perkin Trans. 1 1976, 1, 2237–2240. [Google Scholar] [CrossRef]
- Jung, M.E.; Lyster, M.A. Quantitative dealkylation of alkyl ethers via treatment with trimethylsilyl iodide. A new method for ether hydrolysis. J. Org. Chem. 1977, 42, 3761–3764. [Google Scholar] [CrossRef]
- Landani, D.; Montanari, F.; Rolla, F. Cleavage of Dialkyl and Aryl Alkyl Ethers with Hydrobromic Acid in the Presence of Phase-Transfer Catalysts. Synthesis 1978, 1978, 771–773. [Google Scholar] [CrossRef]
- Niwa, H.; Hida, T.; Yamada, K. A new method for cleavage of aliphatic methyl ethers. Tetrahedron Lett. 1981, 22, 4239–4240. [Google Scholar] [CrossRef]
- Guindon, Y.; Yoakim, C.; Morton, H.E. Cleavage of carbonoxygen bonds. Dimethylboron bromide. A new reagent for ether cleavage. Tetrahedron Lett. 1983, 24, 2969–2972. [Google Scholar] [CrossRef]
- Node, M.; Ohta, K.; Kajimoto, T.; Nishide, K.; Fujita, E.; Fuji, K. Selective Demethylation of Aliphatic Methyl Ether in the Presence of Aromatic Methyl Ether with the Aluminum Chloride-Sodium Iodide-Acetonitrile System. Chem. Pharm. Bull. 1983, 31, 4178–4180. [Google Scholar] [CrossRef][Green Version]
- Narayana, C.; Padmanabhan, S.; Kabalka, G.W. Cleavage of ethers and geminal diacetates using the boron triiodide-N,N-diethylaniline complex. Tetrahedron Lett. 1990, 31, 6977–6978. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamagiwa, N.; Torisawa, Y. Cyclopentyl Methyl Ether as a New and Alternative Process Solvent. Org. Process. Res. Dev. 2007, 11, 251–258. [Google Scholar] [CrossRef]
- Antonucci, V.; Coleman, J.; Ferry, J.B.; Johnson, N.; Mathe, M.; Scott, J.P.; Xu, J. Toxicological Assessment of 2-Methyltetrahydrofuran and Cyclopentyl Methyl Ether in Support of Their Use in Pharmaceutical Chemical Process Development. Org. Process. Res. Dev. 2011, 15, 939–941. [Google Scholar] [CrossRef]
- Ochiai, H.; Uetake, Y.; Niwa, T.; Hosoya, T. Rhodium-Catalyzed Decarbonylative Borylation of Aromatic Thioesters for Facile Diversification of Aromatic Carboxylic Acids. Angew. Chem. Int. Ed. 2017, 56, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Majdanski, T.C.; Vitz, J.; Meier, A.; Brunzel, M.; Schubert, S.; Nischang, I.; Schubert, U.S. “Green” ethers as solvent alternatives for anionic ring-opening polymerizations of ethylene oxide (EO): In-situ kinetic and advanced characterization studies. Polymer 2018, 159, 86–94. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, C.-Y.; Yang, C.; Chen, J.-P. Ag-Catalyzed Chemoselective Decarboxylative Mono- and gem-Difluorination of Malonic Acid Derivatives. J. Am. Chem. Soc. 2019, 141, 5617–5622. [Google Scholar] [CrossRef]
- Atienza, B.J.P.; Truong, N.; Williams, F.J. Reliably Regioselective Dialkyl Ether Cleavage with Mixed Boron Trihalides. Org. Lett. 2018, 20, 6332–6335. [Google Scholar] [CrossRef] [PubMed]
- Bhar, S.; Ranu, B.C. Zinc-Promoted Selective Cleavage of Ethers in Presence of Acyl Chloride. J. Org. Chem. 1995, 60, 745–747. [Google Scholar] [CrossRef]
- Craig, R.; Litvajova, M.; Cronin, S.A.; Connon, S.J. Enantioselective acyl-transfer catalysis by fluoride ions. Chem. Commun. 2018, 54, 10108–10111. [Google Scholar]
- Blanchard, N.; Bizet, V. Acid Fluorides in Transition-Metal Catalysis: A Good Balance between Stability and Reactivity. Angew. Chem. Int. Ed. 2019, 58, 6814–6817. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, Y.; Sakai, N. Acyl Fluorides in Late Transition-Metal Catalysis. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef]
- Zhang, Y.D.; Rovis, T. A Unique Catalyst Effects the Rapid Room-Temperature Cross-Coupling of Organozinc Reagents with Carboxylic Acid Fluorides, Chlorides, Anhydrides, and Thioesters. J. Am. Chem. Soc. 2004, 126, 15964–15965. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, Y.; Sakino, D.; Sakurai, Y.; Sakai, N. Acid Fluorides as Acyl Electrophiles in Suzuki-Miyaura Coupling. Eur. J. Org. Chem. 2017, 4324–4327. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Sakurai, Y.; Hattori, H.; Sakai, N. Palladium-Catalyzed Reductive Conversion of Acyl Fluorides via Ligand-Controlled Decarbonylation. Org. Lett. 2018, 20, 4204–4208. [Google Scholar] [CrossRef]
- Keaveney, S.T.; Schoenebeck, F. Palladium-Catalyzed Decarbonylative Trifluoromethylation of Acid Fluorides. Angew. Chem. Int. Ed. 2018, 57, 4073–4077. [Google Scholar] [CrossRef] [PubMed]
- Malapit, C.A.; Bour, J.R.; Brigham, C.E.; Sanford, M.S. Base-free nickel-catalysed decarbonylative Suzuki-Miyaura coupling of acid fluorides. Nature 2018, 563, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Yoshida, T.; Tobisu, M. Iridium-catalyzed Decarbonylative Coupling of Acyl Fluorides with Arenes and Heteroarenes via C–H Activation. Chem. Lett. 2019, 48, 94–97. [Google Scholar] [CrossRef]
- Okuda, Y.; Xu, J.; Ishida, T.; Wang, C.; Nishihara, Y. Nickel-Catalyzed Decarbonylative Alkylation of Aroyl Fluorides Assisted by Lewis-Acidic Organoboranes. ACS Omega 2018, 3, 13129–13140. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Nishihara, Y. Nickel-catalysed decarbonylative borylation of aroyl fluorides. Chem. Commun. 2018, 54, 13969–13972. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Asanuma, Y.; Nishihara, Y. Synthesis of 2-Substituted Propenes by Bidentate Phosphine Assisted Methylenation of Acyl Fluorides and Acyl Chlorides with AlMe3. Org. Lett. 2019, 21, 3640–3643. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Liu, L.; Asanuma, Y.; Nishihara, Y. Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions. Molecules 2019, 24, 1671. [Google Scholar] [CrossRef]
- Pilcher, A.S.; Ammon, H.L.; DeShong, P. Utilization of Tetrabutylammonium Triphenylsilyldifluoride as a Fluoride Source for Nucleophilic Fluorination. J. Am. Chem. Soc. 1995, 117, 5166–5167. [Google Scholar] [CrossRef]
- Gazizov, T.K.; Belyalov, R.U.; Pudovik, A.N. Reaction of phosphorus acid esters with carboxylic acid chlorides and fluorides. Chem. Inf. 1982, 52, 776–780. [Google Scholar]
- Bou, V.; Vilarrasa, J. New synthetic ‘tricks’. Trimethylsilyl triflate mediated cleavage of hindered silyl ethers. Tetrahedron Lett. 1990, 31, 567–568. [Google Scholar] [CrossRef]
- Tamao, K.; Yoshida, J.; Takahashi, M.; Yamamoto, H.; Kakui, T.; Matsumoto, H.; Kurita, A.; Kumada, M. Organofluorosilicates in organic synthesis. 1. A novel general and practical method for anti-Markownikoff hydrohalogenation of olefins via organopentafluorosilicates derived from hydrosilylation products. J. Am. Chem. Soc. 1978, 100, 290–292. [Google Scholar] [CrossRef]
- Nakao, Y.; Hiyama, T. Silicon-based cross-coupling reaction: An environmentally benign version. Chem. Soc. Rev. 2011, 40, 4893–4901. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Shim, C.S.; Chung, S.Y.; Kim, H.Y.; Lee, H.W. Cross-interaction constants as a measure of the transition-state structure. Part 1. The degree of bond formation in nucleophilic substitution reactions. J. Chem. Soc. Perkin Trans. 2 1988, 11, 1919–1923. [Google Scholar] [CrossRef]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H.S. Bis(2-methoxyethyl)aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999, 64, 7048–7054. [Google Scholar] [CrossRef]
- Savela, R.; Zawartka, W.; Leino, R. Iron-Catalyzed Chlorination of Silanes. Organometallics 2012, 31, 3199–3206. [Google Scholar] [CrossRef]
- Yasukawa, N.; Kanie, T.; Kuwata, M.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Palladium on Carbon-Catalyzed Benzylic Methoxylation for Synthesis of Mixed Acetals and Orthoesters. Chem. Eur. J. 2017, 23, 10974–10977. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, J.; Yang, H.; Xia, C.; Jiang, G. Rhodium-Catalyzed Double Alkyl-Oxygen Bond Cleavage: An Alkyl Transfer Reaction from Bis/Tris(o-alkyloxyphenyl)phosphine to Aryl Acids. Organometallics 2016, 35, 3406–3412. [Google Scholar] [CrossRef]
- Riggleman, S.; DeShong, P. Application of Silicon-Based Cross-Coupling Technology to Triflates. J. Org. Chem. 2003, 68, 8106–8109. [Google Scholar] [CrossRef] [PubMed]
- Torraca, K.E.; Huang, X.; Parrish, C.A.; Buchwald, S.L. An Efficient Intermolecular Palladium-Catalyzed Synthesis of Aryl Ethers. J. Am. Chem. Soc. 2001, 123, 10770–10771. [Google Scholar] [CrossRef] [PubMed]
- Ebert, G.W.; Rieke, R.D. Preparation of aryl, alkynyl, and vinyl organocopper compounds by the oxidative addition of zerovalent copper to carbon-halogen bonds. J. Org. Chem. 1988, 53, 4482–4488. [Google Scholar] [CrossRef]
- Mane, R.S.; Sasakib, T.; Bhanage, B.M. Silica supported palladium-phosphine as a reusable catalyst for alkoxycarbonylation and aminocarbonylation of aryl and heteroaryl iodides. RSC Adv. 2015, 5, 94776–94785. [Google Scholar] [CrossRef]
- Chng, L.L.; Yang, J.; Ying, J.Y. Efficient Synthesis of Amides and Esters from Alcohols under Aerobic Ambient Conditions Catalyzed by a Au/Mesoporous Al2O3 Nanocatalyst. ChemSusChem 2015, 8, 1916–1925. [Google Scholar] [CrossRef]
- Leduc, A.B.; Jamison, T.F. Continuous Flow Oxidation of Alcohols and Aldehydes Utilizing Bleach and Catalytic Tetrabutylammonium Bromide. Org. Process. Res. Dev. 2012, 16, 1082–1089. [Google Scholar] [CrossRef]
- Sun, J.; Ren, S.-Z.; Lu, X.-Y.; Li, J.-J.; Shen, F.-Q.; Xu, C.; Zhu, H.-L. Discovery of a series of 1,3,4-oxadiazole-2(3H)-thione derivatives containing piperazine skeleton as potential FAK inhibitors. Bioorg. Med. Chem. 2017, 25, 2593–2600. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, R.; Negrerie, D.Z.; Du, Y.; Zhao, K. Direct Conversion of N-Alkoxyamides to Carboxylic Esters through Tandem NBS-Mediated Oxidative Homocoupling and Thermal Denitrogenation. J. Org. Chem. 2013, 78, 8705–8711. [Google Scholar] [CrossRef]
- Hirose, T.; Takai, H.; Watabe, M.; Minamikawa, H.; Tachikawa, T.; Kodama, K.; Yasutake, M. Effect of alkoxy terminal chain length on mesomorphism of 1,6-disubstituted pyrene-based hexacatenar liquid crystals: Columnar phase control. Tetrahedron 2014, 70, 5100–5108. [Google Scholar] [CrossRef]
- Zhu, Y.; Yan, H.; Lu, L.; Liu, D.; Rong, G.; Mao, J. Copper-Catalyzed Methyl Esterification Reactions via C–C Bond Cleavage. J. Org. Chem. 2013, 78, 9898–9905. [Google Scholar] [CrossRef]
- Marco, P.; Elisa, A.; Nicoletta, B.; Silvia, P.; Michal, Z.; Claudia, B.; Giannamaria, A.; Agostino, B.; Federica, V.; Gabriele, C. Accepting the Invitation to Open Innovation in Malaria Drug Discovery: Synthesis, Biological Evaluation, and Investigation on the Structure–Activity Relationships of Benzo[b]thiophene-2-carboxamides as Antimalarial Agents. J. Med. Chem. 2017, 60, 1959–1970. [Google Scholar]
- Liu, C.; Wang, J.; Meng, L.; Deng, Y.; Li, Y.; Lei, A. Palladium-Catalyzed Aerobic Oxidative Direct Esterification of Alcohols. Angew. Chem. Int. Ed. 2011, 50, 5144–5148. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Tabata, Y.; Yoshida, Y.; Toh, K.; Yamashita, K.; Ogura, Y.; Ishihara, K. Metal-free transesterification catalyzed by tetramethylammonium methyl carbonate. Green Chem. 2018, 20, 1193–1198. [Google Scholar] [CrossRef]
- Rodriguez, A.; Nomen, M.; Spur, B.W.; Godfroid, J.J. A selective method for the preparation of aliphatic methyl esters in the presence of aromatic carboxylic acids. Tetrahedron Lett. 1998, 39, 8563–8566. [Google Scholar] [CrossRef]
- Rout, S.K.; Guin, S.; Ghara, K.K.; Banerjee, A.; Patel, B.K. Copper Catalyzed Oxidative Esterification of Aldehydes with Alkylbenzenes via Cross Dehydrogenative Coupling. Org. Lett. 2012, 14, 3982–3985. [Google Scholar] [CrossRef] [PubMed]
- Ramanjaneyulu, B.T.; Reddy, V.; Arde, P.; Mahesh, S.; Anand, R.V. Combining Oxidative N-Heterocyclic Carbene Catalysis with Click Chemistry: A Facile One-Pot Approach to 1,2,3-Triazole Derivatives. Chem. Asian J. 2013, 8, 1489–1496. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | [P] | Additive | Yield of 3a (%) 1 |
|---|---|---|---|
| 1 | PPh3 | TBAT | 74 (74) |
| 2 | P(OPh)3 | TBAT | 40 |
| 3 | PCy3 | TBAT | 50 |
| 4 | PtBu3 | TBAT | 45 |
| 5 | P(4-F-C6H4)3 | TBAT | 55 |
| 6 | PPh3 | NBu4F | 46 |
| 7 | PPh3 | NBu4Cl | 30 |
| 8 | PPh3 | NBu4Br | 14 |
| 9 | PPh3 | NBu4I | 25 |
| 10 | PPh3 | NBu4OTf | 0 |
| 11 | PPh3 | KF | 12 |
| 12 | PPh3 | CsF | 14 |
| 13 | PPh3 | 18-crown-6/KF | 34 |
| 14 | PPh3 | Ph3SiF | 0 |
| 15 | PPh3 | - | 0 |
| 16 | - | TBAT | 53 |
| 17 2 | PPh3 | TBAT | 50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wang, X.; Nishihara, Y. PPh3-Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT. Catalysts 2019, 9, 574. https://doi.org/10.3390/catal9070574
Wang Z, Wang X, Nishihara Y. PPh3-Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT. Catalysts. 2019; 9(7):574. https://doi.org/10.3390/catal9070574
Chicago/Turabian StyleWang, Zhenhua, Xiu Wang, and Yasushi Nishihara. 2019. "PPh3-Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT" Catalysts 9, no. 7: 574. https://doi.org/10.3390/catal9070574
APA StyleWang, Z., Wang, X., & Nishihara, Y. (2019). PPh3-Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT. Catalysts, 9(7), 574. https://doi.org/10.3390/catal9070574