Intrinsic Kinetics Study of Biogas Methanation Coupling with Water Gas Shift over Re-Promoted Ni Bifunctional Catalysts

Abstract
:
1. Introduction
2. Results and Discussion
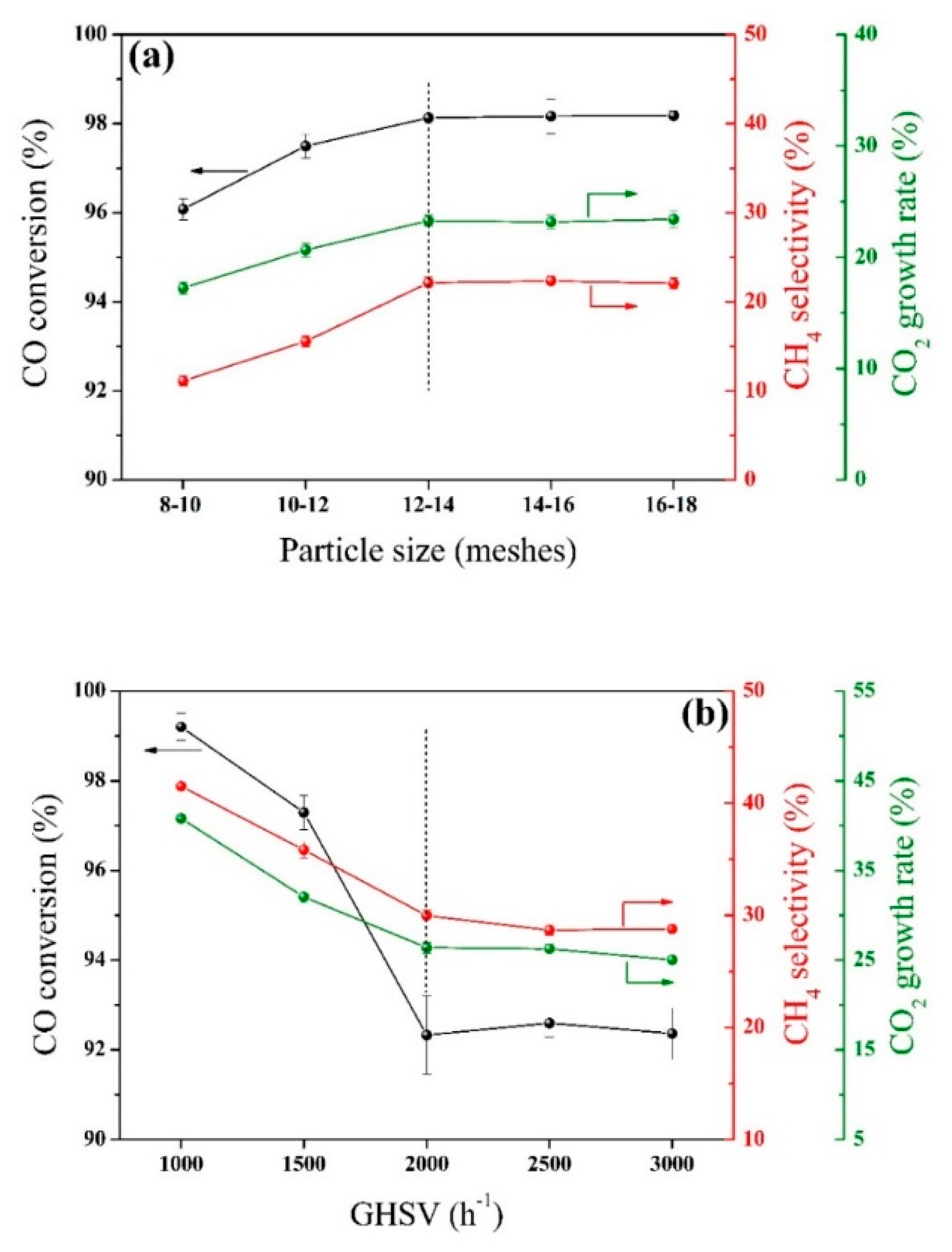
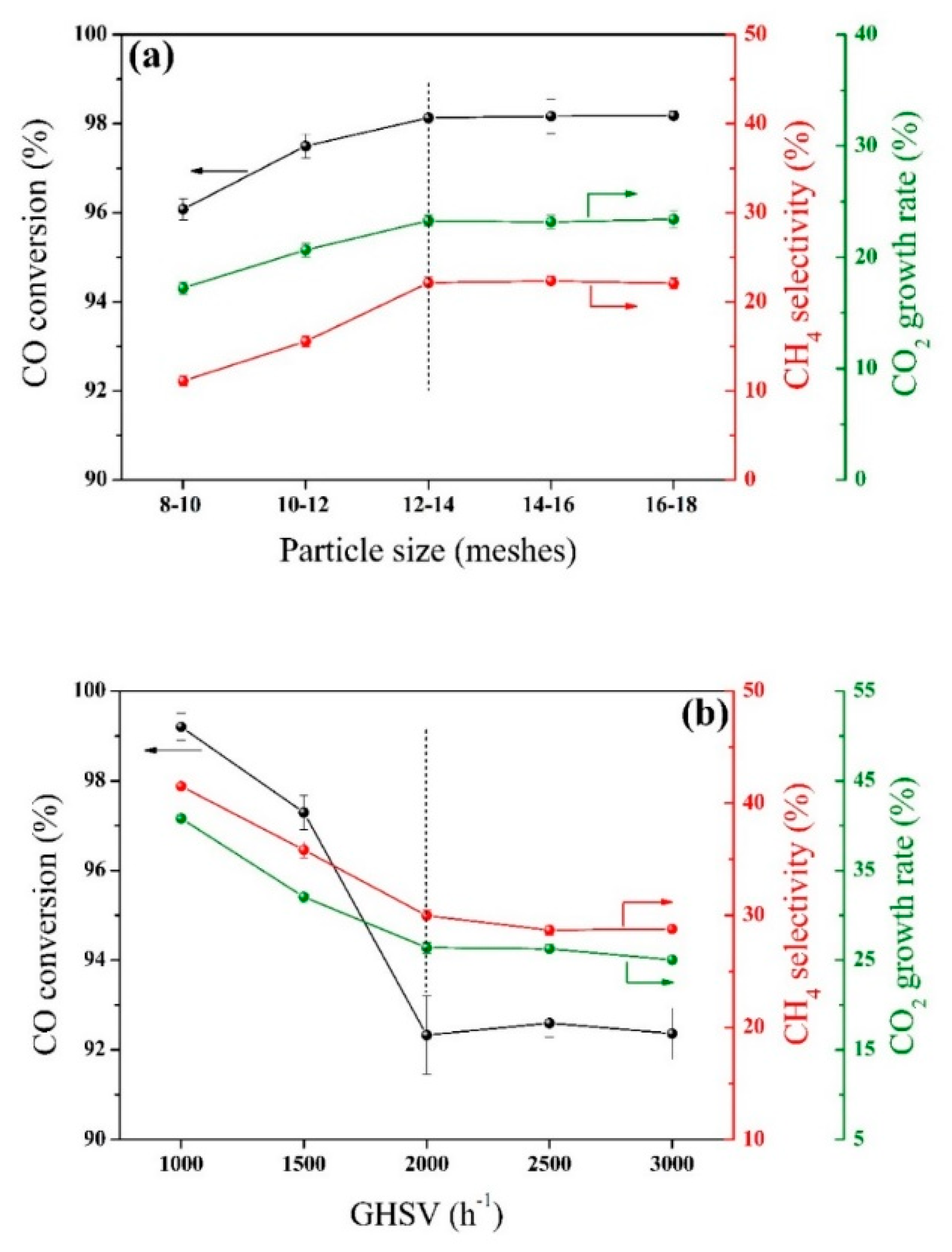
2.1. Internal/External Diffusion Elimination
2.2. Intrinsic Kinetic Models
2.2.1. Empirical Power-Law Model
2.2.2. Synergetic Model
2.2.3. Independent Model
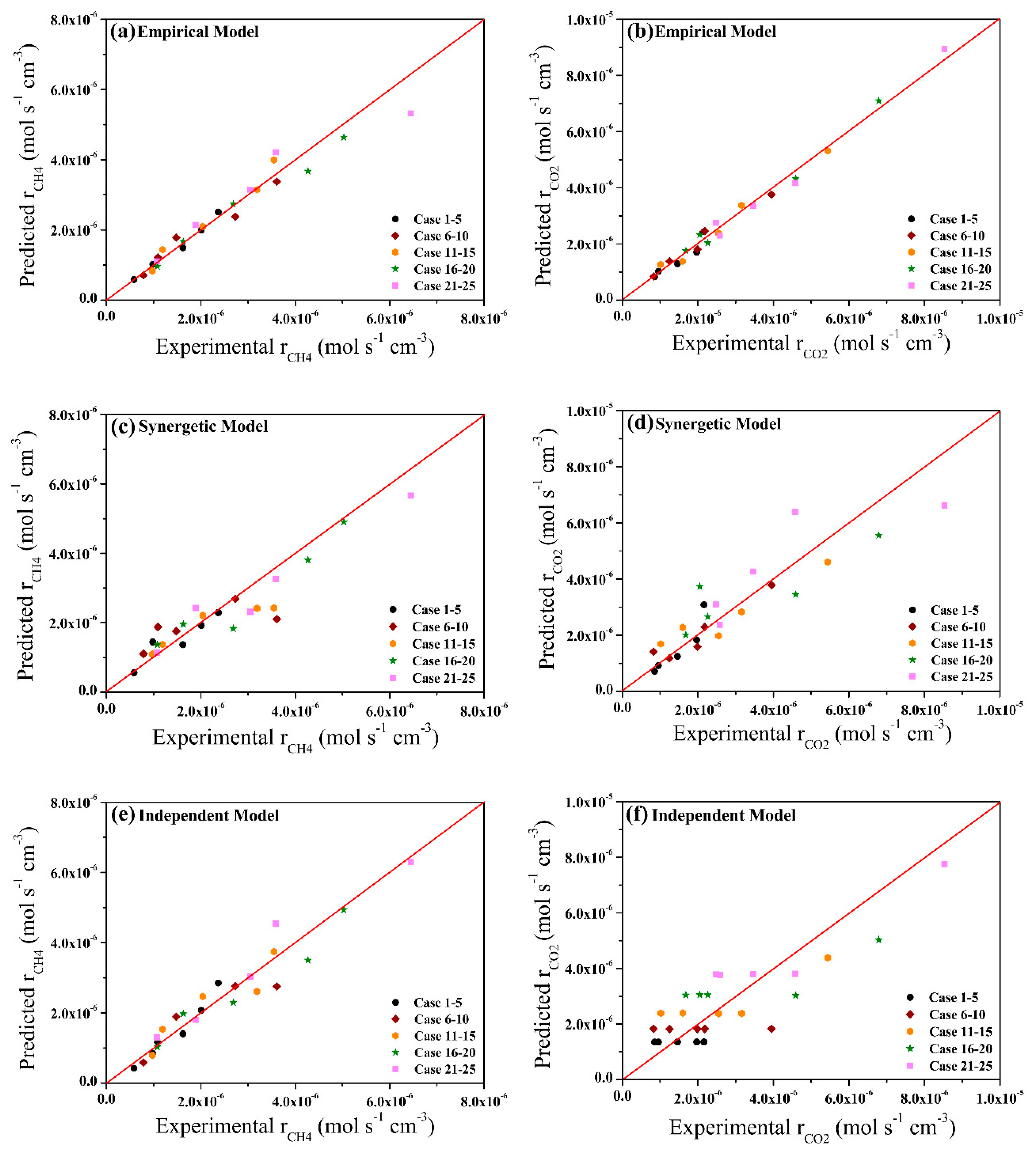
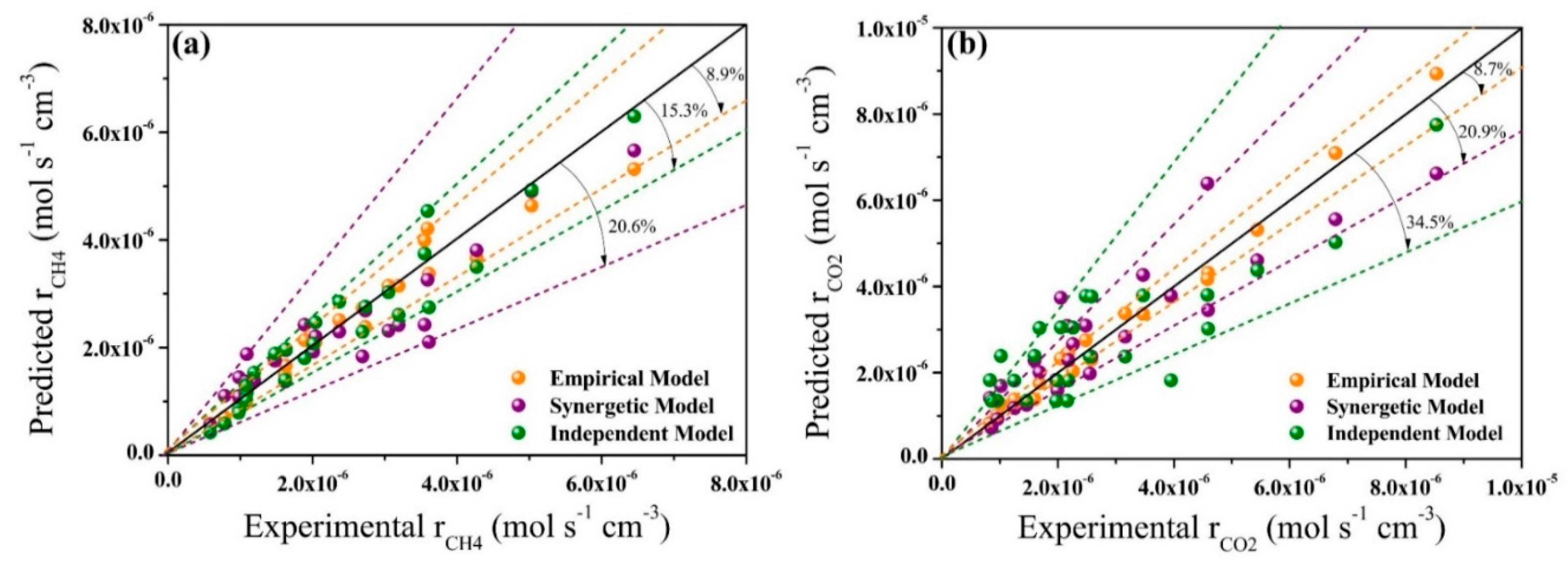
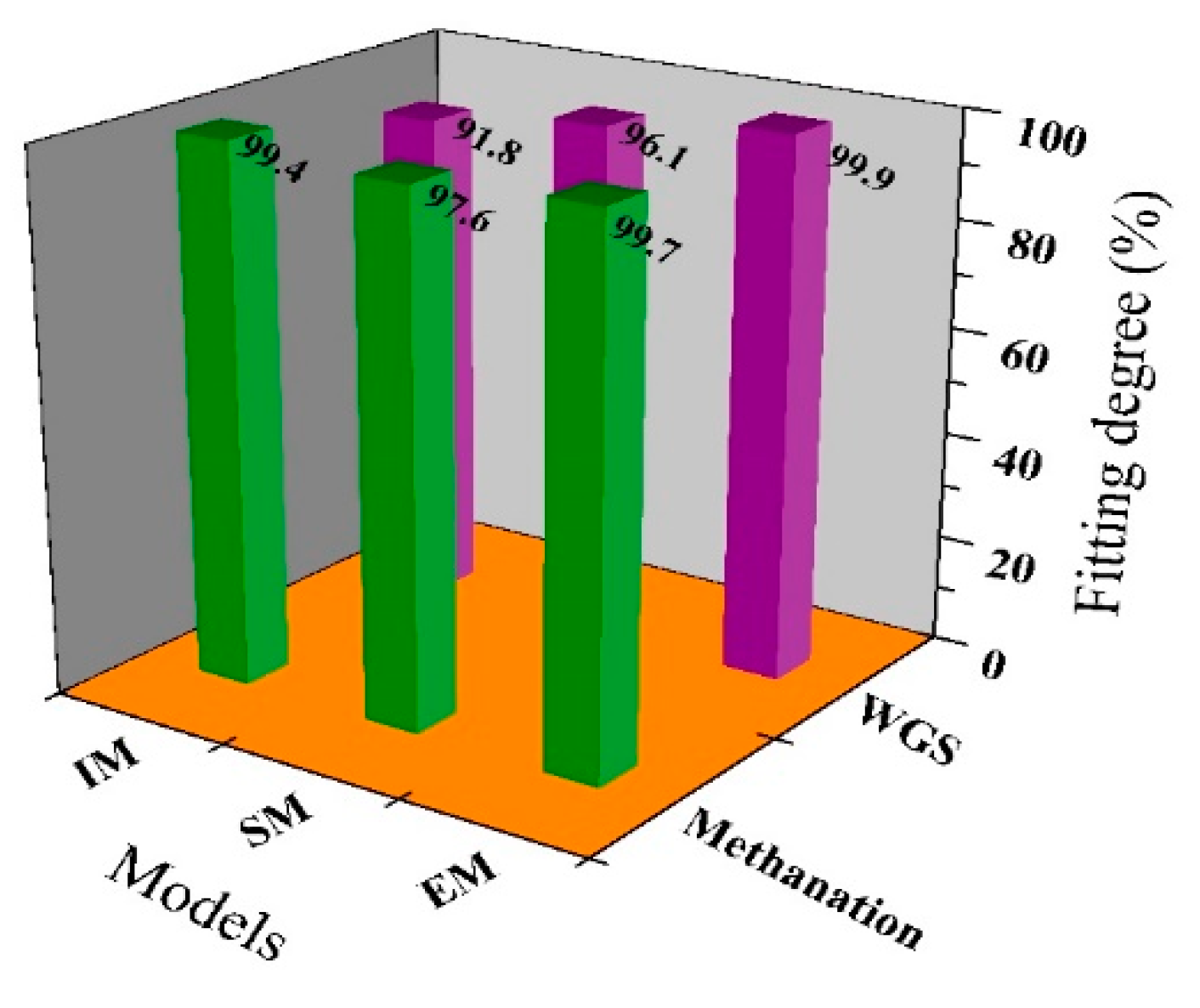
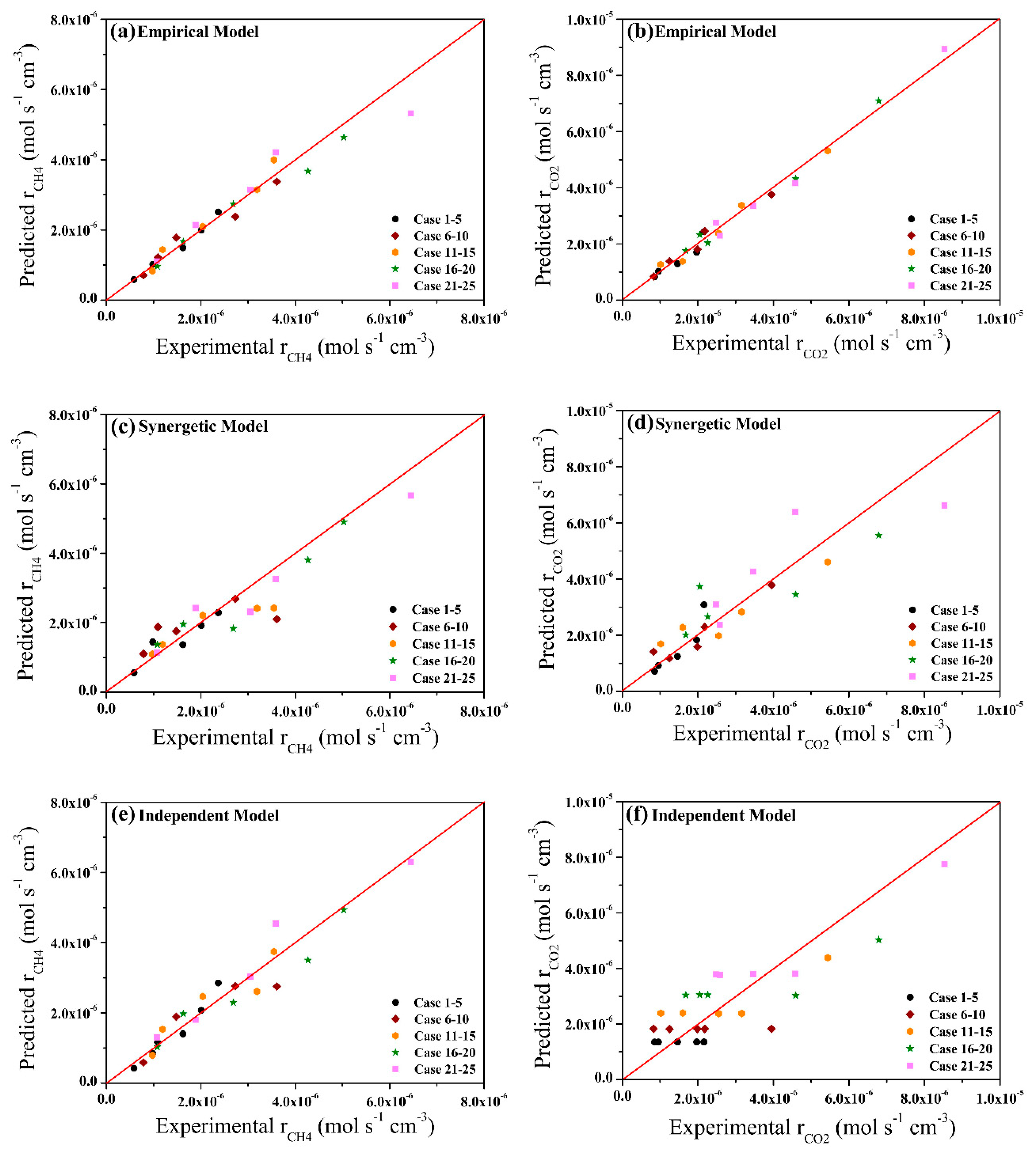
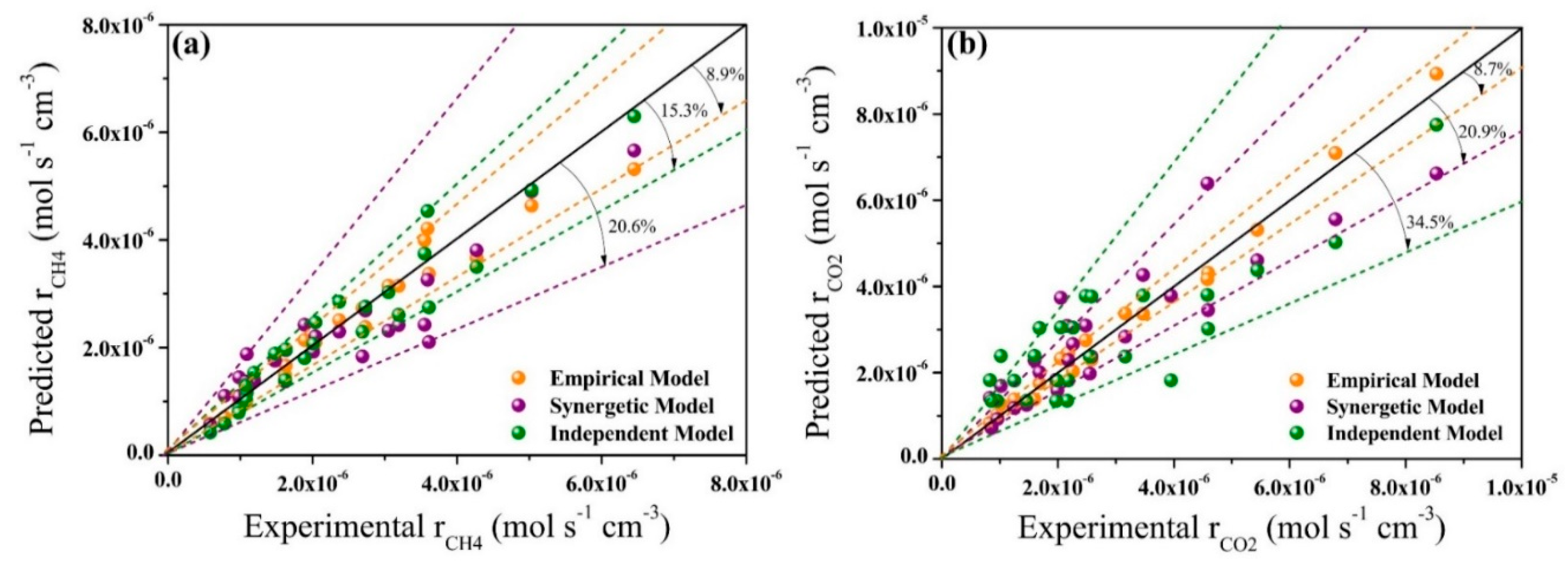
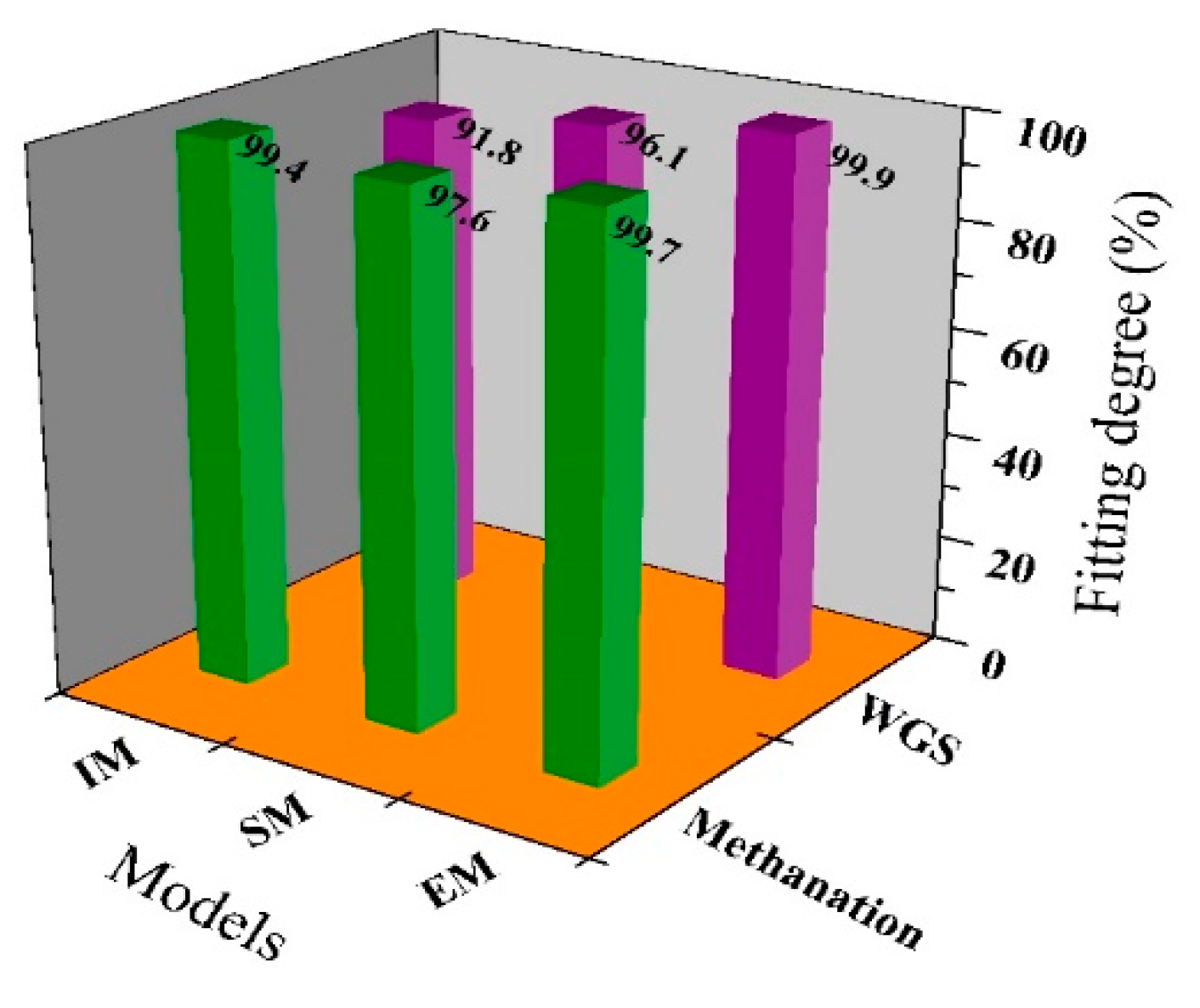
2.3. Model Validation and Comparison
3. Materials and Methods
3.1. Catalyst Preparation
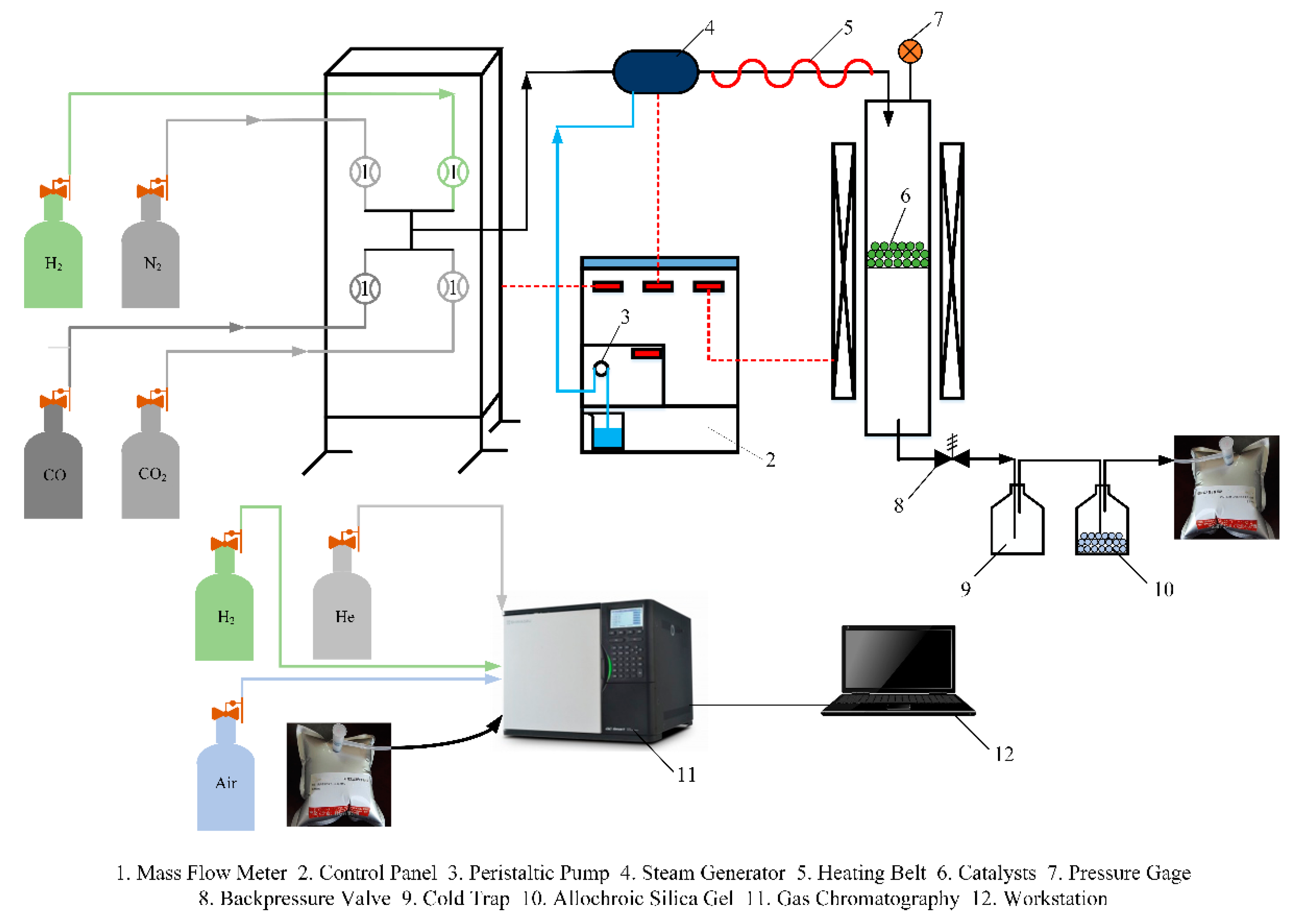
3.2. Reactor System
3.3. Intrinsic Kinetics Measurements
3.3.1. Inlet Biogas Composition and Operating Conditions
3.3.2. Internal/External Diffusion Elimination and Verification
3.3.3. Intrinsic Kinetics Test Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| F | molar flow of species, mol s−1 | Keq | reaction equilibrium constant |
| r | reaction rate per volume of catalyst, mol s−1 cm−3 | Ki | adsorption equilibrium constant of component i |
| R | catalyst particle radius, cm | Ki0 | adsorption pre-exponential factor |
| Cs | reactant concentration at the particle surface, mol cm−3 | ∆H | enthalpy change of adsorption, kJ mol−1 |
| Cb | reactant concentration in the bulk phase, mol cm−3 | Greek symbols | |
| Deff | effective diffusivity, cm2 s−1 | ε | catalyst porosity |
| DAB | binary gas phase diffusion coefficient, cm2 s−1 | τ | tortuosity factor |
| T | reaction temperature, K | ν | kinetic viscosity, cm2 s−1 |
| M | molecular weight | Subscript | |
| P | total reaction pressure, bar | in | inlet |
| n | reaction order | out | outlet |
| kc | mass transfer coefficient, cm s−1 | ave | average |
| L | characteristic length, cm | Superscript | |
| u | fluid velocity, cm s−1 | exp | experimental |
| k0 | pre-exponential factor | pre | predicted |
| E | activation energy, kJ mol−1 | ||
| Pi | partial pressure of component i, kPa |
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhuang, Q.; Cai, X.; He, Y.; Huang, Y.; Jiang, D.; Lin, E.; Liu, Y.; Tang, Y.; Wang, M.Q. Biomass and biofuels in China: toward bioenergy resource potentials and their impacts on the environment. Renew. Sustain. Energy Rev. 2018, 82, 2387–2400. [Google Scholar] [CrossRef]
- Zhang, J.; Fatah, N.; Capela, S.; Kara, Y.; Guerrini, O.; Khodakov, A.Y. Kinetic investigation of carbon monoxide hydrogenation under realistic conditions of methanation of biomass derived syngas. Fuel 2013, 111, 845–854. [Google Scholar] [CrossRef]
- Yu, L.; Song, M.; Wei, Y.; Xiao, J. Combining carbon fibers with Ni/γ–Al2O3 used for syngas production: part A: preparation and evaluation of complex carrier catalysts. Catalysts 2018, 8, 658. [Google Scholar] [CrossRef]
- Wei, Y.; Song, M.; Yu, L.; Gao, R.; Meng, F.; Xiao, J.; Zhang, Y. Promotion effect of SiO2 on the catalytic performance of Ni/CF for biomass derived gas reforming. Ind. Eng. Chem. Res. 2018, 57, 10851–10858. [Google Scholar] [CrossRef]
- Khan, I.U.; Othman, M.H.D.; Hashim, H.; Matsuura, T.; Ismail, A.F.; Rezaei-DashtArzhandi, M.; Azelee, I.W. Biogas as a renewable energy fuel-a review of biogas upgrading, utilisation and storage. Energy Convers. Manage. 2017, 150, 277–294. [Google Scholar] [CrossRef]
- Angelidaki, I.; Treu, L.; Tsapekos, P.; Luo, G.; Campanaro, S.; Wenzel, H.; Kougias, P.G. Biogas upgrading and utilization: current status and perspectives. Biotechnol. Adv. 2018, 36, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.B.; Sun, D.; Kidon, D.; Siddiqui, A.; Kuner, A.; Fowler, M.; Simakov, D.S.A. Upgrading biogas produced at dairy farms into renewable natural gas by methanation. Int. J. Energy Res. 2018, 42, 1714–1728. [Google Scholar] [CrossRef]
- Dong, X.; Song, M.; Jin, B.; Zhou, Z.; Yang, X. The synergy effect of Ni-M (M=Mo, Fe, Co, Mn or Cr) bicomponent catalysts on partial methanation coupling with water gas shift under low H2/CO conditions. Catalysts 2017, 7, 51. [Google Scholar] [CrossRef]
- Yuan, C.; Yao, N.; Wang, X.; Wang, J.; Lv, D.; Li, X. The SiO2 supported bimetallic Ni-Ru particles: a good sulfur-tolerant catalyst for methanation reaction. Chem. Eng. J. 2015, 260, 1–10. [Google Scholar] [CrossRef]
- Mei, Z.; Li, Y.; Fan, M.; Argyle, M.D.; Tang, J. The effects of bimetallic Co-Ru nanoparticles on Co/RuO2/Al2O3 catalysts for the water gas shift and methanation. Int. J. Hydrogen Energy 2014, 39, 14808–14816. [Google Scholar] [CrossRef]
- Huo, X.; Wang, Z.; Huang, J.; Zhang, R.; Fang, Y. Bulk Mo and Co-Mo carbides as catalysts for methanation. Catal. Commun. 2016, 79, 39–44. [Google Scholar] [CrossRef]
- Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L. Re-promoted Ni-Mn bifunctional catalysts prepared by microwave heating for partial methanation coupling with water gas shift under low H2/CO conditions. Appl. Catal. A 2018, 552, 105–116. [Google Scholar] [CrossRef]
- Dong, X.; Jin, B.; Sun, Y.; Yu, L. Urban gas production from low H2/CO biogas using Re-promoted Ni catalysts supported on modified manganese sand. Fuel 2018, 220, 60–71. [Google Scholar] [CrossRef]
- Ding, M.; Tu, J.; Zhang, Q.; Wang, M.; Tsubaki, N.; Wang, T.; Ma, L. Enhancement of methanation of bio-syngas over CeO2-modified Ni/Al2O3 catalysts. Biomass Bioenergy 2016, 85, 12–17. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, K.; Wang, W.; Qu, J.; Tian, Y.; Wang, B.; Ma, X. Kinetics of sulfur-resistant methanation over supported molybdenum-based catalyst. J. Taiwan Inst. Chem. E. 2016, 68, 239–245. [Google Scholar] [CrossRef]
- Hla, S.S.; Park, D.; Duffy, G.J.; Edwards, J.H.; Roberts, D.G.; Ilyushechkin, A.; Morpeth, L.D.; Nguyen, T. Kinetics of high-temperature water-gas shift reaction over two iron-based commercial catalysts using simulated coal-derived syngases. Chem. Eng. J. 2009, 146, 148–154. [Google Scholar] [CrossRef]
- Lefebvre, J.; Bajohr, S.; Kolb, T. A comparison of two-phase and three phase CO2 methanation reaction kinetics. Fuel 2019, 239, 896–904. [Google Scholar] [CrossRef]
- Lim, J.Y.; McGregor, J.; Sederman, A.J.; Dennis, J.S. Kinetic studies of the methanation of CO over a Ni/γ-Al2O3 catalyst using a batch reactor. Chem. Eng. Sci. 2016, 146, 316–336. [Google Scholar] [CrossRef]
- Queiroz, G.A.; Menezes Barbosa, C.M.B.; Abreu, C.A.M. Mechanism-based kinetics of the water-gas shift reaction at low temperature with a ruthenium catalysts. React. Kinet. Mech. Catal. 2018, 123, 573–583. [Google Scholar] [CrossRef]
- Roensch, S.; Koechermann, J.; Schneider, J.; Matthischke, S. Global reaction kinetics of CO and CO2 methanation for dynamic process modeling. Chem. Eng. Technol. 2016, 39, 208–218. [Google Scholar] [CrossRef]
- Hla, S.S.; Sun, Y.; Duffy, G.J.; Morpeth, L.D.; Ilyushechkin, A.; Cousins, A.; Roberts, D.G.; Edwards, J.H. Kinetics of the water-gas shift reaction over a La0.7Ce0.2FeO3 perovskite-like catalyst using simulated coal-derived syngas at high temperature. Int. J. Hydrogen Energy 2011, 36, 518–527. [Google Scholar] [CrossRef]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Smith, B.R.J.; Loganathan, M.; Shantha, M.S. A review of the water gas shift reaction kinetics. Int. J. Chem. Reactor Eng. 2010, 8. [Google Scholar] [CrossRef]
- Guichard, B.; Gaulier, F.; Barbier, J.; Corre, T.; Bonneau, J.; Levitz, P.; Espinat, D. Asphaltenes diffusion/adsorption through catalyst alumina supports-influence on catalytic activity. Catal. Today 2018, 305, 49–57. [Google Scholar] [CrossRef]
- Vernuccio, S.; Dempfle, D.; Goy, R.; Medlock, J.; Rudolf Von Rohr, P. External mass transfer in a laser sintered structured reactor for continuous hydrogenation of alkynes. Chem. Eng. Process. 2018, 126, 74–80. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B. Levulinic acid hydrodeoxygenation, decarboxylation and oligmerization over NiMo/Al2O3 catalyst to bio-based value-added chemicals: modelling of mass transfer, thermodynamics and micro-kinetics. Chem. Eng. J. 2017, 330, 383–397. [Google Scholar] [CrossRef]
- Choi, Y.; Stenger, H.G. Water gas shift reaction kinetics and reactor modeling for fuel cell grade hydrogen. J. Power Sources 2003, 123, 432–439. [Google Scholar] [CrossRef]
- Koryabkina, N.A.; Phatak, A.A.; Ruettinger, W.F.; Farrauto, R.J.; Ribeiro, F.H. Determination of kinetic parameters for the water-gas shift reaction on copper catalysts under realistic conditions for fuel cell applications. J. Catal. 2003, 217, 233–239. [Google Scholar] [CrossRef]
- Lei, Y.; Cant, N.W.; Trimm, D.L. Kinetics of the water-gas shift reaction over a rhodium-promoted iron–chromium oxide catalyst. Chem. Eng. J. 2005, 114, 81–85. [Google Scholar] [CrossRef]
- Hla, S.S.; Duffy, G.J.; Morpeth, L.D.; Cousins, A.; Roberts, D.G.; Edwards, J.H. Investigation of the effect of total pressure on performance of the catalytic water-gas shift reaction using simulated coal-derived syngases. Catal. Commun. 2009, 11, 272–275. [Google Scholar] [CrossRef]
- Chein, R.; Yu, C.; Wang, C. Numerical simulation on the effect of operating conditions and syngas compositions for synthetic natural gas production via methanation reaction. Fuel 2016, 185, 394–409. [Google Scholar] [CrossRef]
- Molino, A.; Braccio, G. Synthetic natural gas SNG production from biomass gasification-thermodynamics and processing aspects. Fuel 2015, 139, 425–429. [Google Scholar] [CrossRef]
- Lim, J.Y.; McGregor, J.; Sederman, A.J.; Dennis, J.S. The role of the Boudouard and water–gas shift reactions in the methanation of CO or CO2 over Ni/γ-Al2O3 catalyst. Chem. Eng. Sci. 2016, 152, 754–766. [Google Scholar] [CrossRef]
- He, Q.; Yu, G.; Yan, S.; Dumée, L.F.; Zhang, Y.; Strezov, V.; Zhao, S. Renewable CO2 absorbent for carbon capture and biogas upgrading by membrane contactor. Sep. Purif. Technol. 2018, 194, 207–215. [Google Scholar] [CrossRef]
- Baccioli, A.; Antonelli, M.; Frigo, S.; Desideri, U.; Pasini, G. Small scale bio-LNG plant: comparison of different biogas upgrading techniques. Appl. Energy 2018, 217, 328–335. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, X.; Xie, Y.; Lu, X. Thermodynamic analysis of CO2 separation from biogas with conventional ionic liquids. Appl. Energy 2018, 217, 75–87. [Google Scholar] [CrossRef]
- Xu, J.G.; Froment, G.F. Methane steam reforming, methanation and water-gas shift: I. intrinsic kinetics. AIChE J. 1989, 35, 88–96. [Google Scholar]
- Materazzi, M.; Grimaldi, F.; Foscolo, P.U.; Cozens, P.; Taylor, R.; Chapman, C. Analysis of syngas methanation for bio-SNG production from wastes: kinetic model development and pilot scale validation. Fuel Process. Technol. 2017, 167, 292–305. [Google Scholar] [CrossRef]
- Parlikkad, N.R.; Chambrey, S.; Fongarland, P.; Fatah, N.; Khodakov, A.; Capela, S.; Guerrini, O. Modeling of fixed bed methanation reactor for syngas production: operating window and performance characteristics. Fuel 2013, 107, 254–260. [Google Scholar] [CrossRef]
- Klose, J.; Baerns, M. Kinetics of the methanation of carbon monoxide on an alumina-supported nickel catalyst. J. Catal. 1984, 85, 105–116. [Google Scholar] [CrossRef]
- Mendes, D.; Chibante, V.; Mendes, A.; Madeira, L.M. Determination of the low-temperature water-gas shift reaction kinetics using a Cu-based catalyst. Ind. Eng. Chem. Res. 2010, 49, 11269–11279. [Google Scholar] [CrossRef]
- Engelbrecht, N.; Chiuta, S.; Everson, R.C.; Neomagus, H.W.J.P.; Bessarabov, D.G. Experimentation and CFD modelling of a microchannel reactor for carbon dioxide methanation. Chem. Eng. J. 2017, 313, 847–857. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Valle, B.; Aramburu, B.; Montero, C.; Bilbao, J. Kinetic model considering catalyst deactivation for the steam reforming of bio-oil over Ni/La2O3-αAl2O3. Chem. Eng. J. 2018, 332, 192–204. [Google Scholar] [CrossRef]
- Falbo, L.; Martinelli, M.; Visconti, C.G.; Lietti, L.; Bassano, C.; Deiana, P. Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Appl. Catal. B 2018, 225, 354–363. [Google Scholar] [CrossRef]
- Link, S.; Tran, K.; Bach, Q.; Yrjas, P.; Lindberg, D.; Arvelakis, S.; Rosin, A. Catalytic effect of oil shale ash on CO2 gasification of leached wheat straw and reed chars. Energy 2018, 152, 906–913. [Google Scholar] [CrossRef]
- Jin, F.; Sun, H.; Wu, C.; Ling, H.; Jiang, Y.; Williams, P.T.; Huang, J. Effect of calcium addition on Mg-AlOx supported Ni catalysts for hydrogen production from pyrolysis-gasification of biomass. Catal. Today 2018, 309, 2–10. [Google Scholar] [CrossRef]
- Liu, L.; Huang, Y.; Cao, J.; Liu, C.; Dong, L.; Xu, L.; Zha, J. Experimental study of biomass gasification with oxygen-enriched air in fluidized bed gasifier. Sci. Total Environ. 2018, 626, 423–433. [Google Scholar] [CrossRef]
- Duan, P.; Li, S.; Jiao, J.; Wang, F.; Xu, Y. Supercritical water gasification of microalgae over a two-component catalyst mixture. Sci. Total Environ. 2018, 630, 243–253. [Google Scholar] [CrossRef]
- Bui, P.P.; Oyama, S.T.; Takagaki, A.; Carrow, B.P.; Nozaki, K. Reactions of 2-methyltetrahydropyran on silica-supported nickel phosphide in comparison with 2-methyltetrahydrofuran. ACS Catal. 2016, 6, 4549–4558. [Google Scholar] [CrossRef]
- Mohagheghi, M.; Bakeri, G.; Saeedizad, M. Study of the effects of external and internal diffusion on the propane dehydrogenation reaction over Pt-Sn/Al2O3 catalyst. Chem. Eng. Technol. 2007, 30, 1721–1725. [Google Scholar] [CrossRef]
- Lee, W.; Wang, Z.; Zheng, W.; Vlachos, D.G.; Bhan, A. Vapor phase hydrodeoxygenation of furfural to 2–methylfuran on molybdenum carbide catalysts. Catal. Sci. Technol. 2014, 4, 2340. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Number | PCO (kPa) | PH2 (kPa) | PH2O (kPa) | PCO2 (kPa) | PN2 (kPa) | rCH4 (mol s−1 cm−3) | rCO2 (mol s−1 cm−3) |
|---|---|---|---|---|---|---|---|
| 1 | 33.33 | 20.00 | 33.33 | 8.00 | 5.33 | 5.84 × 10−7 | 2.16 × 10−6 |
| 2 | 48.03 | 33.60 | 48.03 | 12.24 | 8.16 | 9.84 × 10−7 | 1.97 × 10−6 |
| 3 | 61.50 | 49.30 | 61.50 | 16.62 | 11.08 | 1.62 × 10−6 | 1.46 × 10−6 |
| 4 | 74.08 | 66.68 | 74.08 | 21.10 | 14.08 | 2.01 × 10−6 | 9.55 × 10−7 |
| 5 | 85.71 | 85.71 | 85.71 | 25.71 | 17.13 | 2.37 × 10−6 | 8.55 × 10−7 |
| 6 | 32.02 | 22.40 | 32.02 | 8.16 | 5.44 | 7.85 × 10−7 | 3.95 × 10−6 |
| 7 | 46.13 | 36.98 | 46.13 | 12.47 | 8.31 | 1.09 × 10−6 | 2.18 × 10−6 |
| 8 | 59.26 | 53.34 | 59.26 | 16.88 | 11.26 | 1.48 × 10−6 | 1.99 × 10−6 |
| 9 | 71.43 | 71.43 | 71.43 | 21.43 | 14.28 | 2.73 × 10−6 | 1.25 × 10−6 |
| 10 | 99.99 | 60.00 | 99.99 | 24.00 | 15.99 | 3.61 × 10−6 | 8.28 × 10−7 |
| 11 | 30.75 | 24.65 | 30.75 | 8.31 | 5.54 | 9.76 × 10−7 | 5.44 × 10−6 |
| 12 | 44.45 | 40.01 | 44.45 | 12.66 | 8.45 | 1.19 × 10−6 | 3.16 × 10−6 |
| 13 | 57.14 | 57.14 | 57.14 | 17.14 | 11.42 | 2.04 × 10−6 | 2.55 × 10−6 |
| 14 | 83.33 | 50.00 | 83.33 | 20.00 | 13.33 | 3.19 × 10−6 | 1.60 × 10−6 |
| 15 | 96.06 | 67.20 | 96.06 | 24.48 | 16.32 | 3.55 × 10−6 | 1.02 × 10−6 |
| 16 | 29.63 | 26.67 | 29.63 | 8.44 | 5.63 | 1.08 × 10−6 | 6.79 × 10−6 |
| 17 | 42.86 | 42.86 | 42.86 | 12.86 | 8.57 | 1.63 × 10−6 | 4.59 × 10−6 |
| 18 | 66.66 | 40.00 | 66.66 | 16.00 | 10.66 | 2.69 × 10−6 | 2.05 × 10−6 |
| 19 | 80.05 | 56.00 | 80.05 | 20.40 | 13.60 | 4.27 × 10−6 | 2.26 × 10−6 |
| 20 | 92.25 | 73.95 | 92.25 | 24.93 | 16.62 | 5.03 × 10−6 | 1.68 × 10−6 |
| 21 | 28.57 | 28.57 | 28.57 | 8.57 | 5.71 | 1.07 × 10−6 | 8.53 × 10−6 |
| 22 | 50.00 | 30.00 | 50.00 | 12.00 | 8.00 | 1.89 × 10−6 | 4.58 × 10−6 |
| 23 | 64.04 | 44.80 | 64.04 | 16.32 | 10.88 | 3.05 × 10−6 | 3.47 × 10−6 |
| 24 | 76.88 | 61.63 | 76.88 | 20.78 | 13.85 | 3.59 × 10−6 | 2.48 × 10−6 |
| 25 | 88.89 | 80.01 | 88.89 | 25.32 | 16.89 | 6.45 × 10−6 | 2.58 × 10−6 |
| Reaction | Reaction Orders a | Reaction Orders b | Reaction Orders c | Reaction Orders d | Pre-exponential Factor k0 | Activation Energy E (kJ mol−1) |
|---|---|---|---|---|---|---|
| Methanation | 0.5803 | 0.2468 | 0 | 0.5803 | 6.290 × 10−7 | 23.25 |
| Water gas shift | 1.9645 | 1.9645 | 4.2979 | −9.4584 | 2.550 × 10−6 | 33.48 |
| Number | Elementary Reaction | Instruction |
|---|---|---|
| 1 | H2 + 2* ⇋ 2H* | |
| 2 | CO + * ⇋ CO* | |
| 3 | CO* + * ⇋ C* + O* | |
| 4 | C* + H* ⇋ CH* + * | |
| 5 | CH* + H* ⇋ CH2* + * | RDS |
| 6 | CH2* + H* ⇋ CH3* + * | |
| 7 | CH3* + H* ⇋ CH4 + 2* | |
| 8 | H2O + * ⇋ H2 + O* | |
| 9 | CO* + O* ⇋ CO2* + * | RDS |
| 10 | CO2* ⇋ CO2 + * |
| Parameter | Value | Unit |
|---|---|---|
| k10 | 1.216 × 1016 | mol s−1 cm−3 kPa0.5 |
| E1 | 221.10 | kJ mol−1 |
| k20 | 18.32 | mol s−1 cm−3 kPa−1 |
| E2 | 14.90 | kJ mol−1 |
| KCO0 | 11.67 | kPa−1 |
| ∆HCO | −10.85 | kJ mol−1 |
| KH20 | 8.32 × 10−3 | kPa−1 |
| ∆HH2 | 26.68 | kJ mol−1 |
| KH2O0 | 7.95 × 10−2 | - |
| ∆HH2O | 17.33 | kJ mol−1 |
| Number | Dissociative Methanation Mechanism | Langmuir–Hinshelwood Water Gas Shift Mechanism |
|---|---|---|
| 1 | CO + 2* ⇋ C* + O* | CO + * ⇋ CO* |
| 2 | H2 + 2* ⇋ 2H* | H2O + * ⇋ H2O* |
| 3 | C* + 2H* → H2C* + 2* or C* + H*⇋*C…H* + H* → H2C* + 2* | CO* + H2O* ⇋ CO2* + H2* |
| 4 | H2C* + 2H* → CH4 + 3* | H2* ⇋ H2 + * |
| 5 | O* + 2H* → H2O + 3* | CO2* ⇋ CO2 + * |
| Parameter | Value | Unit |
|---|---|---|
| kCH20 | 12.92 | mol s−1 cm−3 |
| ∆ECH2 | 1.42 × 10−2 | kJ mol−1 |
| kWGS0 | 0.35 | mol s−1 cm−3 kPa−2 |
| ∆EWGS | 15.18 | kJ mol−1 |
| KC0 | 4.61 × 10−3 | kPa−0.5 |
| ∆HC | 15.98 | kJ mol−1 |
| KH0 | 4.41 × 10−3 | kPa−0.5 |
| ∆HH | 5.67 | kJ mol−1 |
| KCO0 | 35.22 | kPa−1 |
| ∆HCO | 2.60 | kJ mol−1 |
| KH20 | 2.20 × 10−2 | kPa−1 |
| ∆HH2 | 95.30 | kJ mol−1 |
| KCO20 | 3.17 × 10−3 | kPa−1 |
| ∆HCO2 | 83.86 | kJ mol−1 |
| KH2O0 | 5.25 | kPa−1 |
| ∆HH2O | −12.98 | kJ mol−1 |
| H2/CO Ratio | Dry Base Compositions | H2O/CO Molar Flow Ratio | |||
|---|---|---|---|---|---|
| H2 (%) | CO (%) | CO2 (%) | N2 (%) | ||
| 0.6 | 30.0 | 50.0 | 12.0 | 8.0 | 1.0 |
| 0.7 | 32.9 | 47.1 | 12.0 | 8.0 | 1.0 |
| 0.8 | 35.6 | 44.4 | 12.0 | 8.0 | 1.0 |
| 0.9 | 37.9 | 42.1 | 12.0 | 8.0 | 1.0 |
| 1.0 | 40.0 | 40.0 | 12.0 | 8.0 | 1.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, X.; Jin, B.; Kong, Z.; Dong, L. Intrinsic Kinetics Study of Biogas Methanation Coupling with Water Gas Shift over Re-Promoted Ni Bifunctional Catalysts. Catalysts 2019, 9, 422. https://doi.org/10.3390/catal9050422
Dong X, Jin B, Kong Z, Dong L. Intrinsic Kinetics Study of Biogas Methanation Coupling with Water Gas Shift over Re-Promoted Ni Bifunctional Catalysts. Catalysts. 2019; 9(5):422. https://doi.org/10.3390/catal9050422
Chicago/Turabian StyleDong, Xinxin, Baosheng Jin, Zhiwei Kong, and Lu Dong. 2019. "Intrinsic Kinetics Study of Biogas Methanation Coupling with Water Gas Shift over Re-Promoted Ni Bifunctional Catalysts" Catalysts 9, no. 5: 422. https://doi.org/10.3390/catal9050422
APA StyleDong, X., Jin, B., Kong, Z., & Dong, L. (2019). Intrinsic Kinetics Study of Biogas Methanation Coupling with Water Gas Shift over Re-Promoted Ni Bifunctional Catalysts. Catalysts, 9(5), 422. https://doi.org/10.3390/catal9050422