Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation

 , ,
, , 


Abstract
1. Introduction
2. Results and Discussion
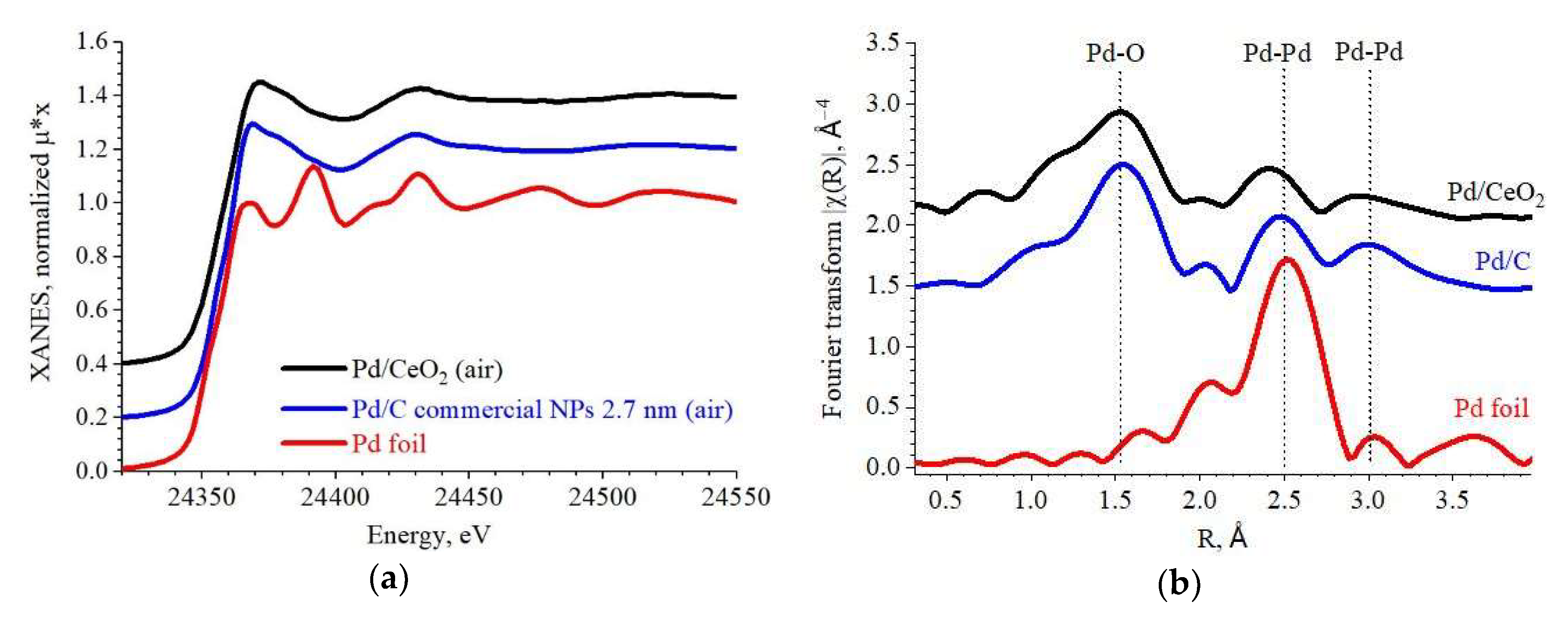
2.1. Elemental Composition
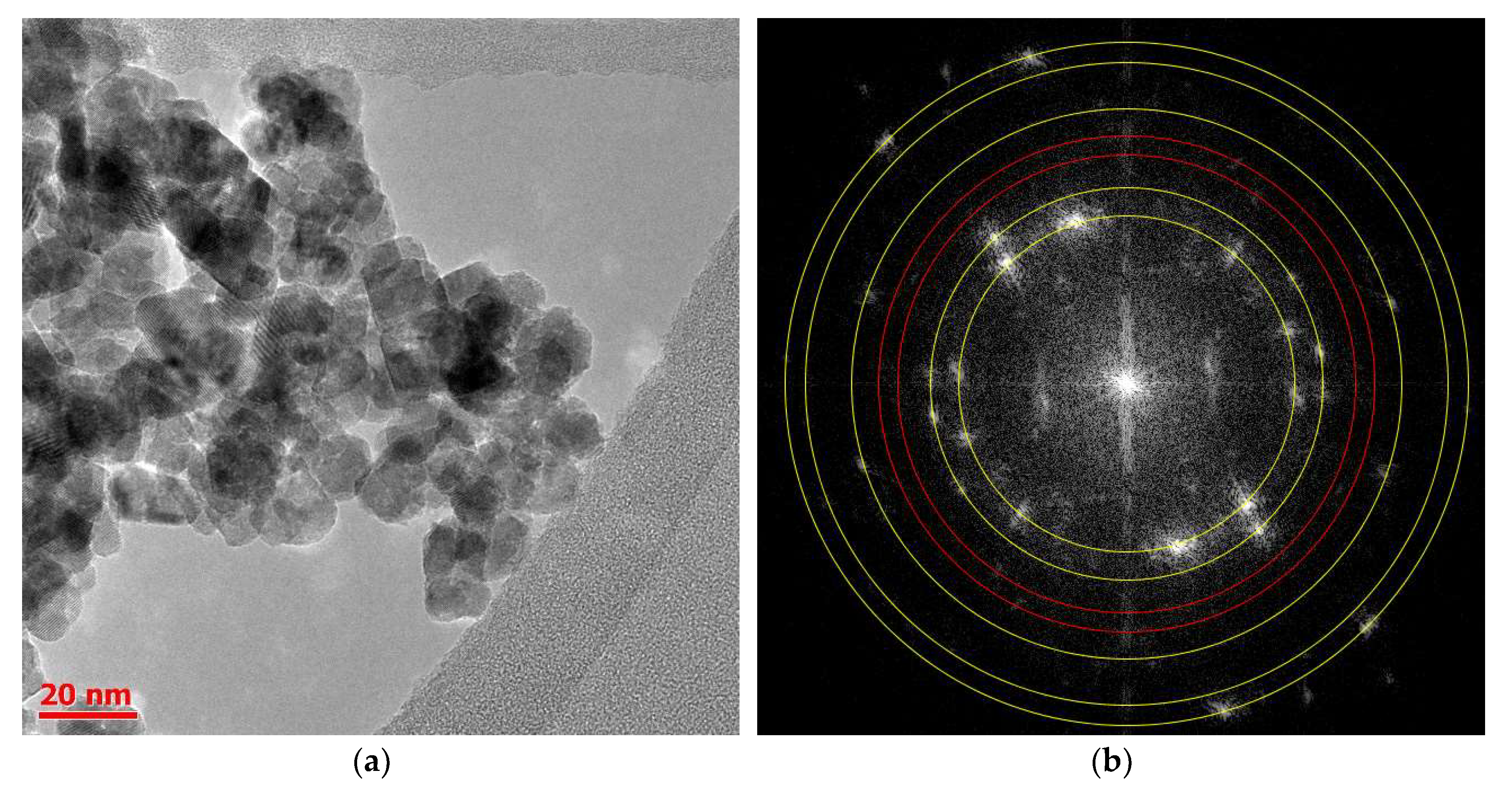
2.2. Structure and Size
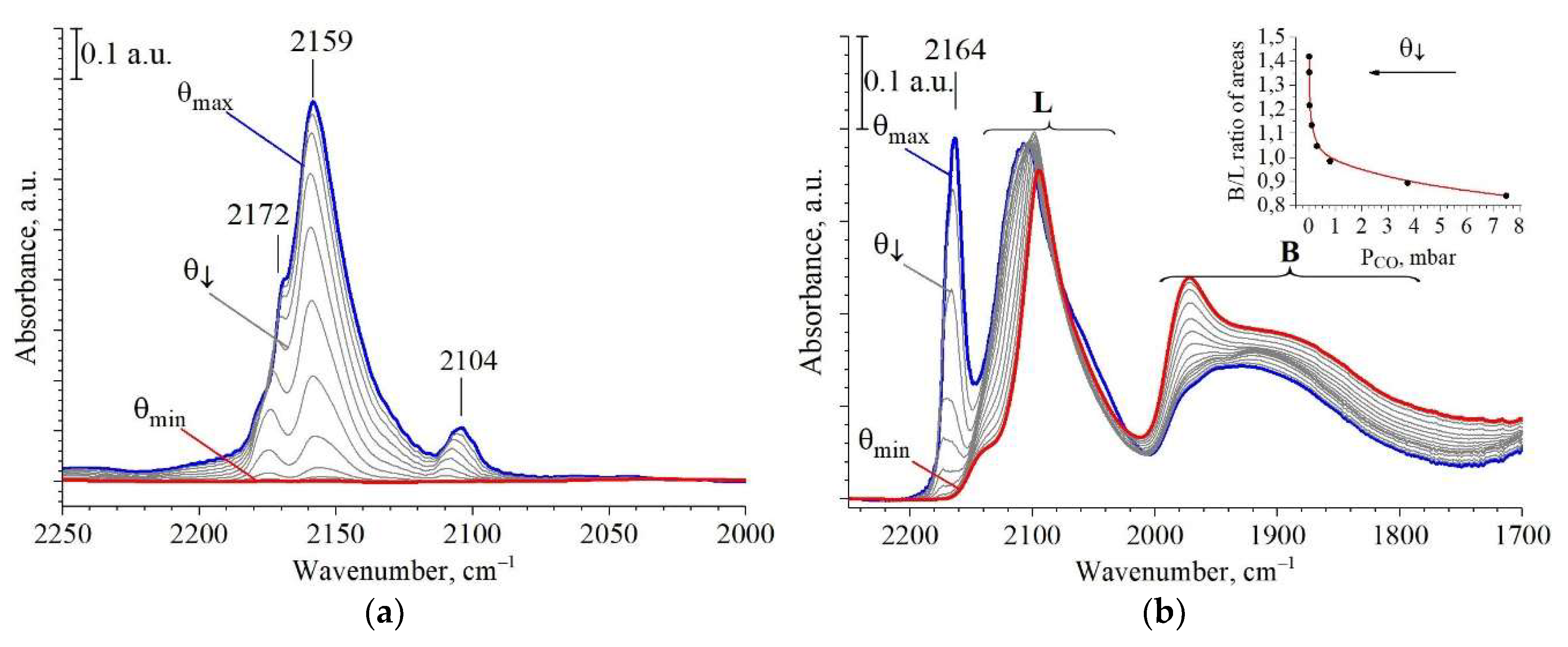
2.3. Active Sites Characterization
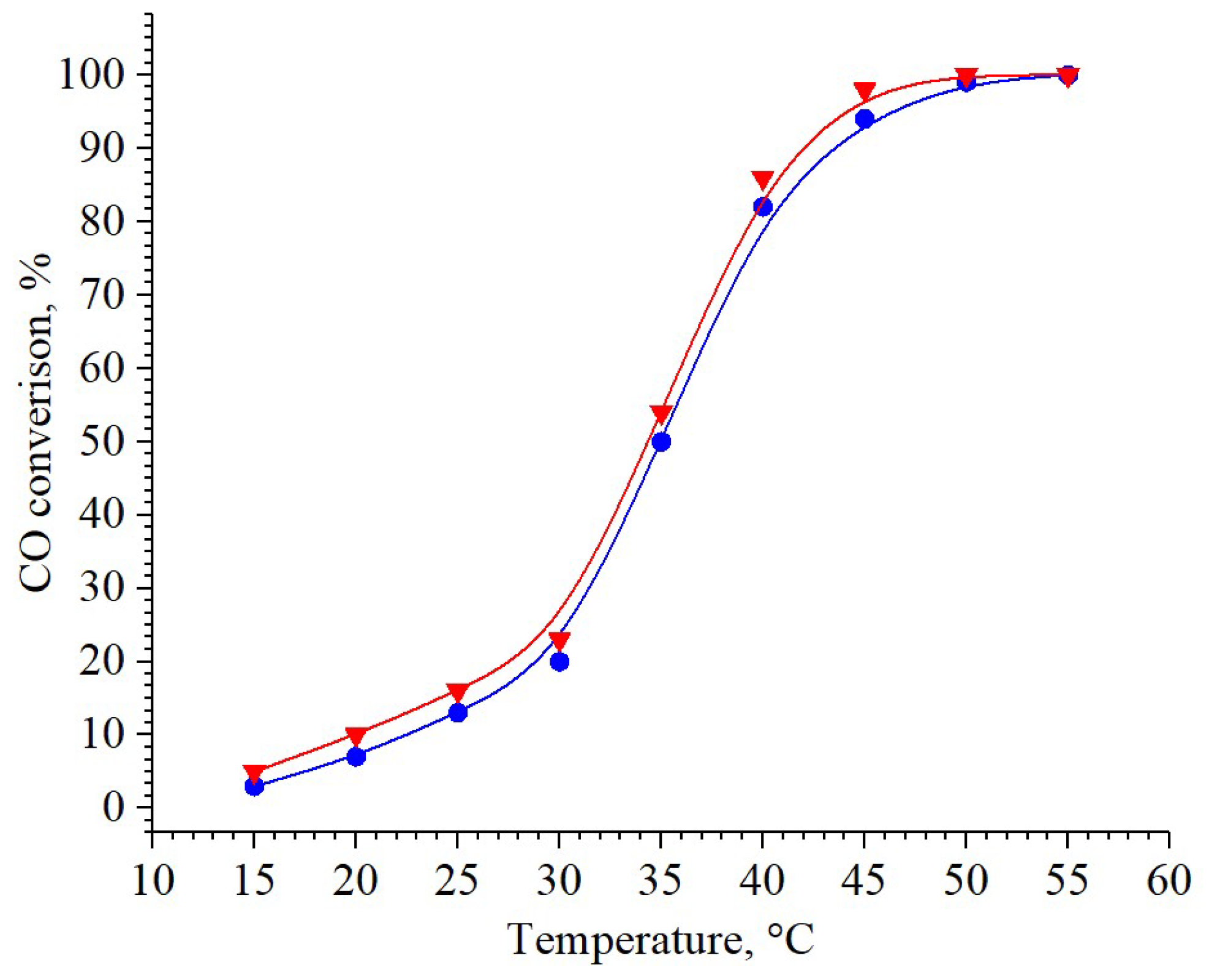
2.4. Catalytic Activity
3. Materials and Methods
3.1. Materials
3.1.1. Synthesis of a Modified Support
3.1.2. Synthesis of Supported Pd NPs
3.2. Methods
Catalytic Activity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A
References
- Tang, Z.; Edwards, J.; Bartley, J.; Taylor, S.; Carley, A.; Herzing, A.; Kiely, C.; Hutchings, G. Nanocrystalline cerium oxide produced by supercritical antisolvent precipitation as a support for high-activity gold catalysts. J. Catal. 2007, 249, 208–219. [Google Scholar] [CrossRef]
- Yang, Y.; Saoud, K.M.; Abdelsayed, V.; Glaspell, G.; Deevi, S.; El-Shall, M.S. Vapor phase synthesis of supported Pd, Au, and unsupported bimetallic nanoparticle catalysts for CO oxidation. Catal. Commun. 2006, 7, 281–284. [Google Scholar] [CrossRef]
- Glaspell, G.; Fuoco, L.; El-Shall, M.S. Microwave synthesis of supported Au and Pd nanoparticle catalysts for CO oxidation. J. Phys. Chem. B 2005, 109, 17350–17355. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Si, Z.; Cao, Y.; Ran, R.; Wu, X.; Weng, D. Localized surface plasmon resonance assisted photothermal catalysis of CO and toluene oxidation over Pd–CeO2 catalyst under visible light irradiation. J. Phys. Chem. C 2016, 120, 29116–29125. [Google Scholar] [CrossRef]
- Jeong, H.; Bae, J.; Han, J.W.; Lee, H. Promoting effects of hydrothermal treatment on the activity and durability of Pd/CeO2 catalysts for CO oxidation. ACS Catal. 2017, 7, 7097–7105. [Google Scholar] [CrossRef]
- Slavinskaya, E.M.; Gulyaev, R.V.; Zadesenets, A.V.; Stonkus, O.A.; Zaikovskii, V.I.; Shubin, Y.V.; Korenev, S.V.; Boronin, A.I. Low-temperature CO oxidation by Pd/CeO2 catalysts synthesized using the coprecipitation method. Appl. Catal. B Environ. 2015, 166, 91–103. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Environ. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Zhu, H.; Qin, Z.; Shan, W.; Shen, W.; Wang, J. Low-temperature oxidation of CO over Pd/CeO2–TiO2 catalysts with different pretreatments. J. Catal. 2005, 233, 41–50. [Google Scholar] [CrossRef]
- Kinoshita, K.J. Particle size effects for oxygen reduction on highly dispersed platinum in acid electrolytes. J. Electrochem. Soc. 1990, 137, 845–848. [Google Scholar] [CrossRef]
- Rojluechai, S.; Chavadej, S.; Schwank, J.W.; Meeyoo, V. Catalytic activity of ethylene oxidation over Au, Ag and Au–Ag catalysts: Support effect. Catal. Commun. 2007, 8, 57–64. [Google Scholar] [CrossRef]
- Buil, M.L.; Esteruelas, M.A.; Niembro, S.; Oliván, M.; Orzechowski, L.; Pelayo, C.; Vallribera, A. Dehalogenation and hydrogenation of aromatic compounds catalyzed by nanoparticles generated from rhodium bis(imino)pyridine complexes. Organometallics 2010, 29, 4375–4383. [Google Scholar] [CrossRef]
- Shim, J.; Yoo, D.-Y.; Lee, J.-S. Characteristics for electrocatalytic properties and hydrogen–oxygen adsorption of platinum ternary alloy catalysts in polymer electrolyte fuel cell. Electrochim. Acta 2000, 45, 1943–1951. [Google Scholar] [CrossRef]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 2017, 358, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Li, C.; Yang, Q. Accelerated catalytic activity of Pd NPs supported on amine-rich silica hollow nanospheres for quinoline hydrogenation. Catal. Sci. Technol. 2017, 7, 2221–2227. [Google Scholar] [CrossRef]
- Li, Z.; Liu, J.; Xia, C.; Li, F. Nitrogen-functionalized ordered mesoporous carbons as multifunctional supports of ultrasmall Pd nanoparticles for hydrogenation of phenol. ACS Catal. 2013, 3, 2440–2448. [Google Scholar] [CrossRef]
- Wong, A.; Liu, Q.; Griffin, S.; Nicholls, A.; Regalbuto, J.R. Synthesis of ultrasmall, homogeneously alloyed, bimetallic nanoparticles on silica supports. Science 2017, 358, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zou, L.; Li, Y. Facile synthesis of hollow structured mesoporous silica nanoreactors with confined ultra-small Pd NPs for efficient hydrogenation reactions. J. Porous Mater. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Chaudhary, S.; Sharma, P.; Singh, D.; Umar, A.; Kumar, R. Chemical and pathogenic cleanup of wastewater using surface-functionalized CeO2 nanoparticles. ACS Sustain. Chem. Eng. 2017, 5, 6803–6816. [Google Scholar] [CrossRef]
- Xu, J.; Li, L.; Li, G. A facile approach to well-dispersible CeO2 nanoparticles. J. Dispers. Sci. Technol. 2008, 29, 1072–1076. [Google Scholar] [CrossRef]
- Jorge, A.B.; Sakatani, Y.; Boissière, C.; Laberty-Roberts, C.; Sauthier, G.; Fraxedas, J.; Sanchez, C.; Fuertes, A. Nanocrystalline N-doped ceria porous thin films as efficient visible-active photocatalysts. J. Mater. Chem. 2012, 22, 3220. [Google Scholar] [CrossRef]
- Safonova, O.V.; Guda, A.A.; Paun, C.; Smolentsev, N.; Abdala, P.M.; Smolentsev, G.; Nachtegaal, M.; Szlachetko, J.; Soldatov, M.A.; Soldatov, A.V.; et al. Electronic and geometric structure of Ce3+ forming under reducing conditions in shaped ceria nanoparticles promoted by platinum. J. Phys. Chem. C 2014, 118, 1974–1982. [Google Scholar] [CrossRef]
- Groppo, E.; Bertarione, S.; Rotunno, F.; Agostini, G.; Scarano, D.; Pellegrini, R.; Leofanti, G.; Zecchina, A.; Lamberti, C. Role of the support in determining the vibrational properties of carbonyls formed on Pd supported on SiO2−Al2O3, Al2O3, and MgO. J. Phys. Chem. C 2007, 111, 7021–7028. [Google Scholar] [CrossRef]
- Lamberti, C.; Zecchina, A.; Groppo, E.; Bordiga, S. Probing the surfaces of heterogeneous catalysts by in situ ir spectroscopy. Chem. Soc. Rev. 2010, 39, 4951–5001. [Google Scholar] [CrossRef] [PubMed]
- Abbott, H.L.; Aumer, A.; Lei, Y.; Asokan, C.; Meyer, R.J.; Sterrer, M.; Shaikhutdinov, S.; Freund, H.J. CO adsorption on monometallic and bimetallic Au−Pd nanoparticles supported on oxide thin films. J. Phys. Chem. C 2010, 114, 17099–17104. [Google Scholar] [CrossRef]
- Baylet, A.; Marécot, P.; Duprez, D.; Castellazzi, P.; Groppi, G.; Forzatti, P. In situ raman and in situ XRD analysis of Pdo reduction and Pd° oxidation supported on γ-Al2O3 catalyst under different atmospheres. Phys. Chem. Chem. Phys. 2011, 13, 4607–4613. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lee, J.Y. Particle size effects in Pd-catalyzed electrooxidation of formic acid. J. Phys. Chem. C 2008, 112, 3789–3793. [Google Scholar] [CrossRef]
- Xia, Y.; Ye, J.; Cheng, D.-G.; Chen, F.; Zhan, X. Identification of a flattened pd–ce oxide cluster as a highly efficient catalyst for low-temperature Co oxidation. Catal. Sci. Technol. 2018, 8, 5137–5147. [Google Scholar] [CrossRef]
- Cargnello, M.; Montini, T.; Polizzi, S.; Wieder, N.L.; Gorte, R.J.; Graziani, M.; Fornasiero, P. Novel embedded Pd@CeO2 catalysts: A way to active and stable catalysts. Dalton Trans. 2010, 39, 2122–2127. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Agostini, G.; Lamberti, C.; Pellegrini, R.; Leofanti, G.; Giannici, F.; Longo, A.; Groppo, E. Effect of pre-reduction on the properties and the catalytic activity of Pd/carbon catalysts: A comparison with Pd/Al2O3. ACS Catal. 2014, 4, 187–194. [Google Scholar] [CrossRef]
- Qi, W.; Huang, B.; Wang, M. Structure of unsupported small palladium nanoparticles. Nanoscale Res. Lett. 2009, 4, 269. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Binet, C.; Daturi, M.; Lavalley, J.-C. Ir study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Zaki, M.I.; Knözlngem, H. Characterization of oxide surfaces by adsorption of carbon monoxide—a low temperature infrared spectroscopy study. Spectrochim. Acta Part A Mol. Spectrosc. 1987, 43, 1455–1459. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. Metal–support interaction in Pd/CeO2 catalysts. Part 2.—ceria textural effects. J. Chem. Soc. Faraday Trans. 1996, 92, 1603–1608. [Google Scholar] [CrossRef]
- Bozon-Verduraz, F.; Bensalem, A. Ir studies of cerium dioxide: Influence of impurities and defects. J. Chem. Soc. Faraday Trans. 1994, 90, 653–657. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by fourier-transform infrared spectroscopy. Part 1.—formation of carbonate species on dehydroxylated CeO2, at room temperature. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 929–943. [Google Scholar] [CrossRef]
- Mudiyanselage, K.; Kim, H.Y.; Senanayake, S.D.; Baber, A.E.; Liu, P.; Stacchiola, D. Probing adsorption sites for CO on ceria. Phys. Chem. Chem. Phys. 2013, 15, 15856–15862. [Google Scholar] [CrossRef]
- Chen, S.; Cao, T.; Gao, Y.; Li, D.; Xiong, F.; Huang, W. Probing surface structures of CeO2, TiO2, and Cu2O nanocrystals with CO and CO2 chemisorption. J. Phys. Chem. C 2016, 120, 21472–21485. [Google Scholar] [CrossRef]
- Bensalem, A.; Bozon-Verduraz, F.; Perrichon, V. Palladium–ceria catalysts: Reversibility of hydrogen chemisorption and redox phenomena. J. Chem. Soc. Faraday Trans. 1995, 91, 2185–2189. [Google Scholar] [CrossRef]
- Padilla, J.M.; Del Angel, G.; Navarrete, J. Improved Pd/γ-Al2O3–Ce catalysts for benzene combustion. Catal. Today 2008, 133, 541–547. [Google Scholar] [CrossRef]
- Di Gregorio, F.; Bisson, L.; Armaroli, T.; Verdon, C.; Lemaitre, L.; Thomazeau, C. Characterization of well faceted palladium nanoparticles supported on alumina by transmission electron microscopy and ft-ir spectroscopy of CO adsorption. Appl. Catal. A Gen. 2009, 352, 50–60. [Google Scholar] [CrossRef]
- Yudanov, I.V.; Sahnoun, R.; Neyman, K.M.; Rösch, N.; Hoffmann, J.; Schauermann, S.; Johánek, V.; Unterhalt, H.; Rupprechter, G.; Libuda, J.; et al. CO adsorption on Pd nanoparticles: Density functional and vibrational spectroscopy studies. J. Phys. Chem. B 2003, 107, 255–264. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Guan, N.; Li, L. Supported Pd catalysts for solvent-free benzyl alcohol selective oxidation: Effects of calcination pretreatments and reconstruction of Pd sites. Appl. Catal. B Environ. 2012, 115, 7–15. [Google Scholar] [CrossRef]
- Baidya, T.; Bera, P.; Mukri, B.; Kumar Parida, S.; Kröcher, O.; Elsener, M.; Hegde, M.S. Drifts studies on CO and NO adsorption and NO plus CO reaction over Pd2+-substituted CeOSS and Ce0.75Sn0.25O2 catalysts. J. Catal. 2013, 303, 117–129. [Google Scholar] [CrossRef]
- Spezzati, G.; Su, Y.; Hofmann, J.P.; Benavidez, A.D.; DeLaRiva, A.T.; McCabe, J.; Datye, A.K.; Hensen, E.J.M. Atomically dispersed Pd–O species on CeO2(111) as highly active sites for low-temperature CO oxidation. ACS Catal. 2017, 7, 6887–6891. [Google Scholar] [CrossRef] [PubMed]
- Sheu, L.; Karpinski, Z.; Sachtler, W.M.H. Effects of palladium particle size and palladium silicide formation on fourier transform infrared spectra of CO adsorbed on Pd/SiO2 catalysts. J. Phys. Chem. (USA) 1989, 93, 4890–4894. [Google Scholar] [CrossRef]
- Fan, Q.; He, S.; Hao, L.; Liu, X.; Zhu, Y.; Xu, S.; Zhang, F. Photodeposited Pd nanoparticles with disordered structure for phenylacetylene semihydrogenation. Sci. Rep. 2017, 7, 42172. [Google Scholar] [CrossRef]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au–CeO2 catalysts for low-temperature water–gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Iglesias-Juez, A.; Hungría, A.B.; Martínez-Arias, A.; Anderson, J.A.; Fernández-García, M. Pd-based (Ce,Zr)ox-supported catalysts: Promoting effect of base metals (Cr, Cu, Ni) in CO and NO elimination. Catal. Today 2009, 143, 195–202. [Google Scholar] [CrossRef]
- Qi, J.; Chen, J.; Li, G.; Li, S.; Gao, Y.; Tang, Z. Facile synthesis of core–shell Au@CeO2 nanocomposites with remarkably enhanced catalytic activity for CO Oxidation. Energy Environ. Sci. 2012, 5, 8937. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Benmouhoub, C.; Kadri, A.; Benbrahim, N.; Hadji, S. Synthesis and characterization of cerium Oxide (CeO2) nanoparticles. Mater. Sci. Forum 2009, 609, 189–194. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic computing system jana2006: General features. Zeitschrift für Kristallographie-Crystalline Materials 2014, 229, 345. [Google Scholar] [CrossRef]
- Chernyshov, A.A.; Veligzhanin, A.; Zubavichus, Y. Structural materials science end-station at the kurchatov synchrotron radiation source: Recent instrumentation upgrades and experimental results. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2009, 603, 95–98. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Data analysis for X-ray absorption spectroscopy using ifeffit. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Max. Achieved CO Conversion, % | Temperature of max. CO Conversion, °C | Reference |
|---|---|---|---|
| 5 wt% Pd/CeO2 | 100 | 108 | [2] |
| 5 wt% Pd/ZrO2 | 98 | 150 | |
| 5 wt% Pd/SiO2 | 92 | 240 | |
| 5 wt% Pd/CeO2 | 100 | 151 | [3] |
| 10 wt% Pd/CeO2 | 100 | 173 | |
| 5 wt% Pd/ZnO | 98 | 200 | |
| 10 wt% Pd/ZnO | 99 | 194 | |
| 5 wt% Pd/CuO | 98 | 186 | |
| 10 wt% Pd/CuO | 97 | 192 | |
| 0.7% Pd-CeO2/Al2O3 (PCA-R) | 100 | 180 | [4] |
| 2 wt% Pd/CeO2 | 100 | 125 | [5] |
| 2 wt% Pd/CeO2 (T) | 143 | ||
| 2 wt% Pd/CeO2 (HT) | 75 | ||
| 2 wt% Pd/CeO2 (H2O) | 105 | ||
| 0.74 wt% Pd/CeO2 (450 °C) | 100 | 190 | [6] |
| 0.74 wt% Pd/CeO2 (600 °C) | 190 | ||
| 0.74 wt% Pd/CeO2 (800 °C) | 190 | ||
| 3.8 wt% Pd/CeO2 (450 °C) | 145 | ||
| 3.8 wt% Pd/CeO2 (600 °C) | 90 | ||
| 3.8 wt% Pd/CeO2 (800 °C) | 175 | ||
| 7.7 wt% Pd/CeO2 (450 °C) | 115 | ||
| 7.7 wt% Pd/CeO2 (600 °C) | 70 | ||
| 7.7 wt% Pd/CeO2 (800 °C) | 170 | ||
| 1 wt% Pd/CeO2-rod | 100 | 150 | [7] |
| 1 wt% Pd/CeO2-cube | 200 | ||
| 5 wt% Pd/SiO2 | 60 | 300 | |
| 1 wt% Pd/CeO2 | 100 | 204 | [8] |
| 1 wt% Pd/TiO2 | 120 | ||
| 1 wt% Pd/CeO2–TiO2 | 97 | ||
| 1 wt% Pd/CeO2 (LTR) | 97 | ||
| 1 wt% Pd/TiO2 (LTR) | 70 | ||
| 1 wt% Pd/CeO2–TiO2 (LTR) | 54 | ||
| 3 wt% Pd/CeO2 | 100 | 50 | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tereshchenko, A.; Polyakov, V.; Guda, A.; Lastovina, T.; Pimonova, Y.; Bulgakov, A.; Tarasov, A.; Kustov, L.; Butova, V.; Trigub, A.; et al. Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation. Catalysts 2019, 9, 385. https://doi.org/10.3390/catal9040385
Tereshchenko A, Polyakov V, Guda A, Lastovina T, Pimonova Y, Bulgakov A, Tarasov A, Kustov L, Butova V, Trigub A, et al. Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation. Catalysts. 2019; 9(4):385. https://doi.org/10.3390/catal9040385
Chicago/Turabian StyleTereshchenko, Andrei, Vladimir Polyakov, Alexander Guda, Tatiana Lastovina, Yulia Pimonova, Alexey Bulgakov, Andrey Tarasov, Leonid Kustov, Vera Butova, Alexander Trigub, and et al. 2019. "Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation" Catalysts 9, no. 4: 385. https://doi.org/10.3390/catal9040385
APA StyleTereshchenko, A., Polyakov, V., Guda, A., Lastovina, T., Pimonova, Y., Bulgakov, A., Tarasov, A., Kustov, L., Butova, V., Trigub, A., & Soldatov, A. (2019). Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation. Catalysts, 9(4), 385. https://doi.org/10.3390/catal9040385