Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates




Abstract
1. Introduction
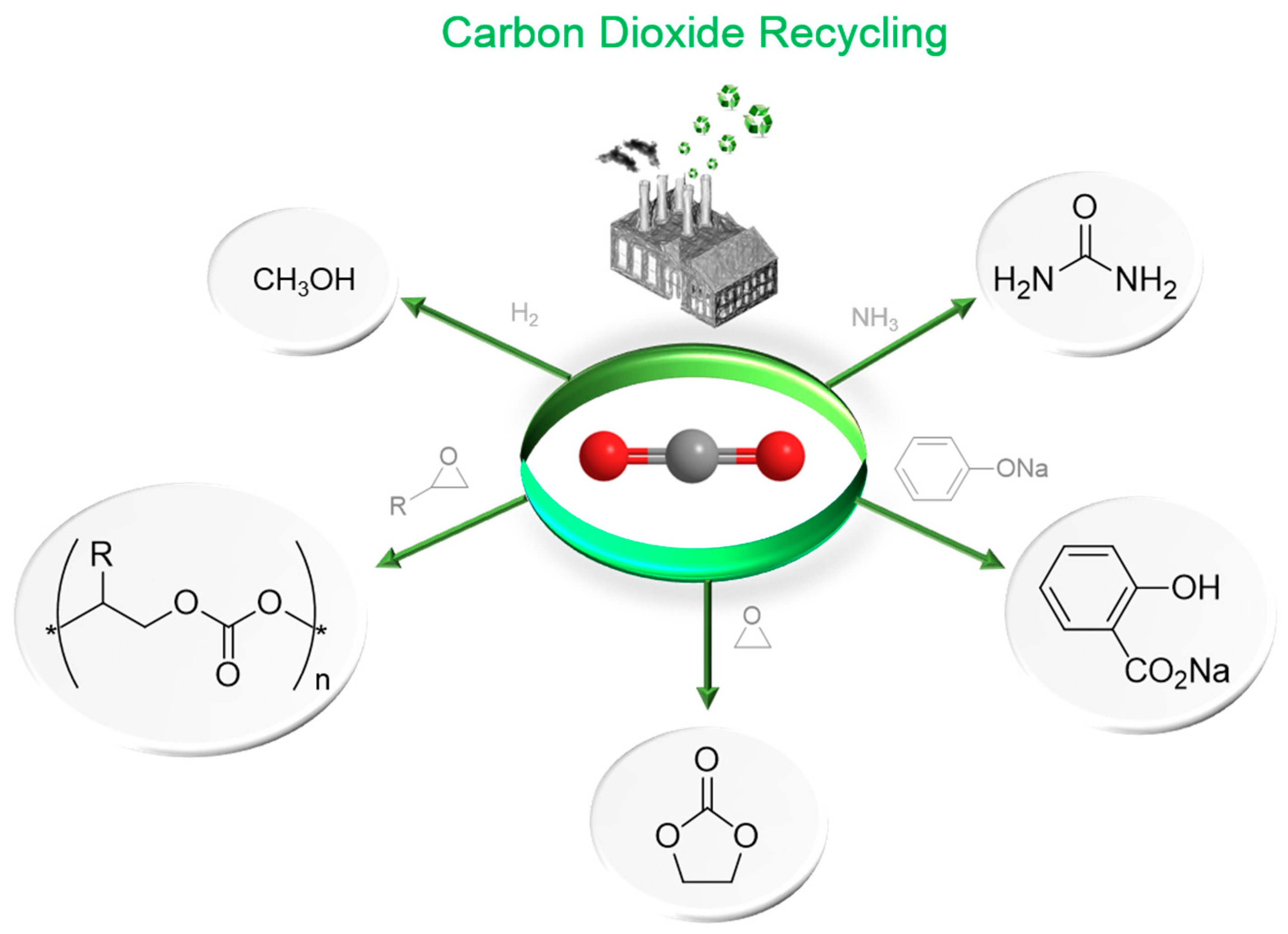
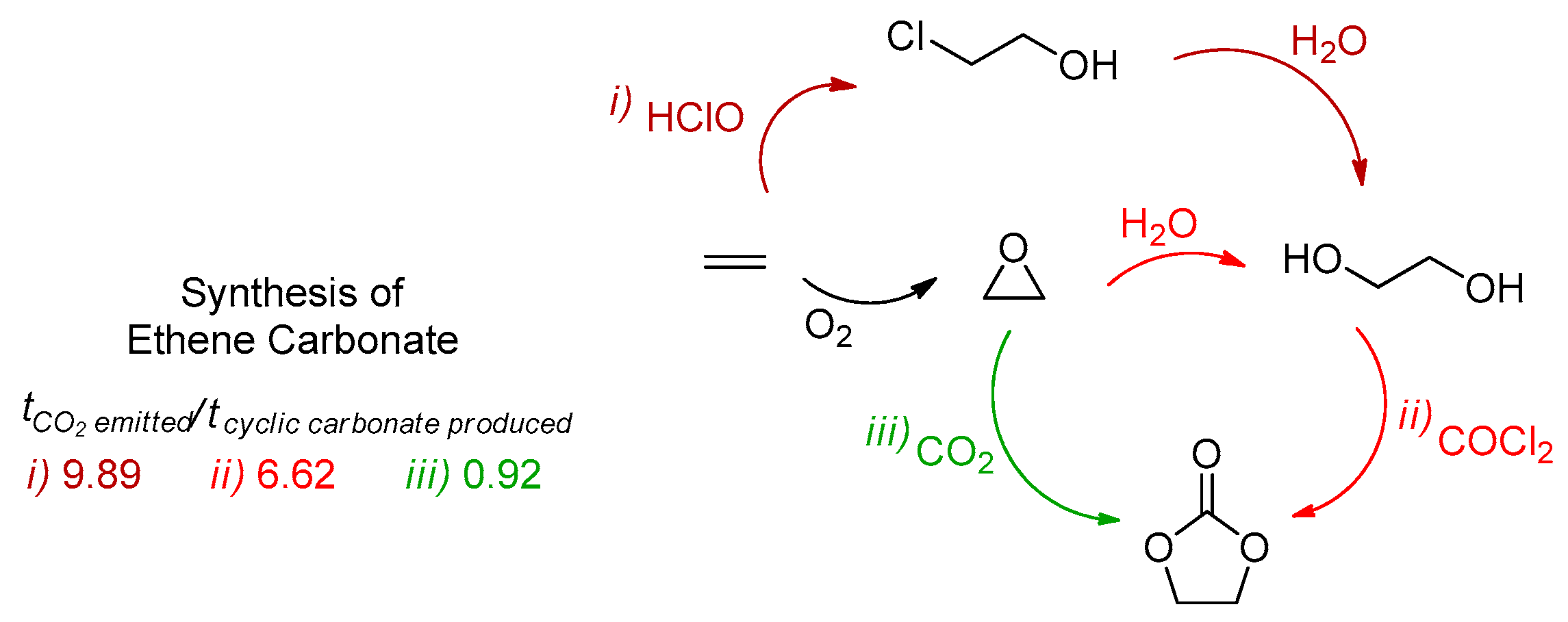
1.1. Carbon Dioxide: From Waste to Feedstock
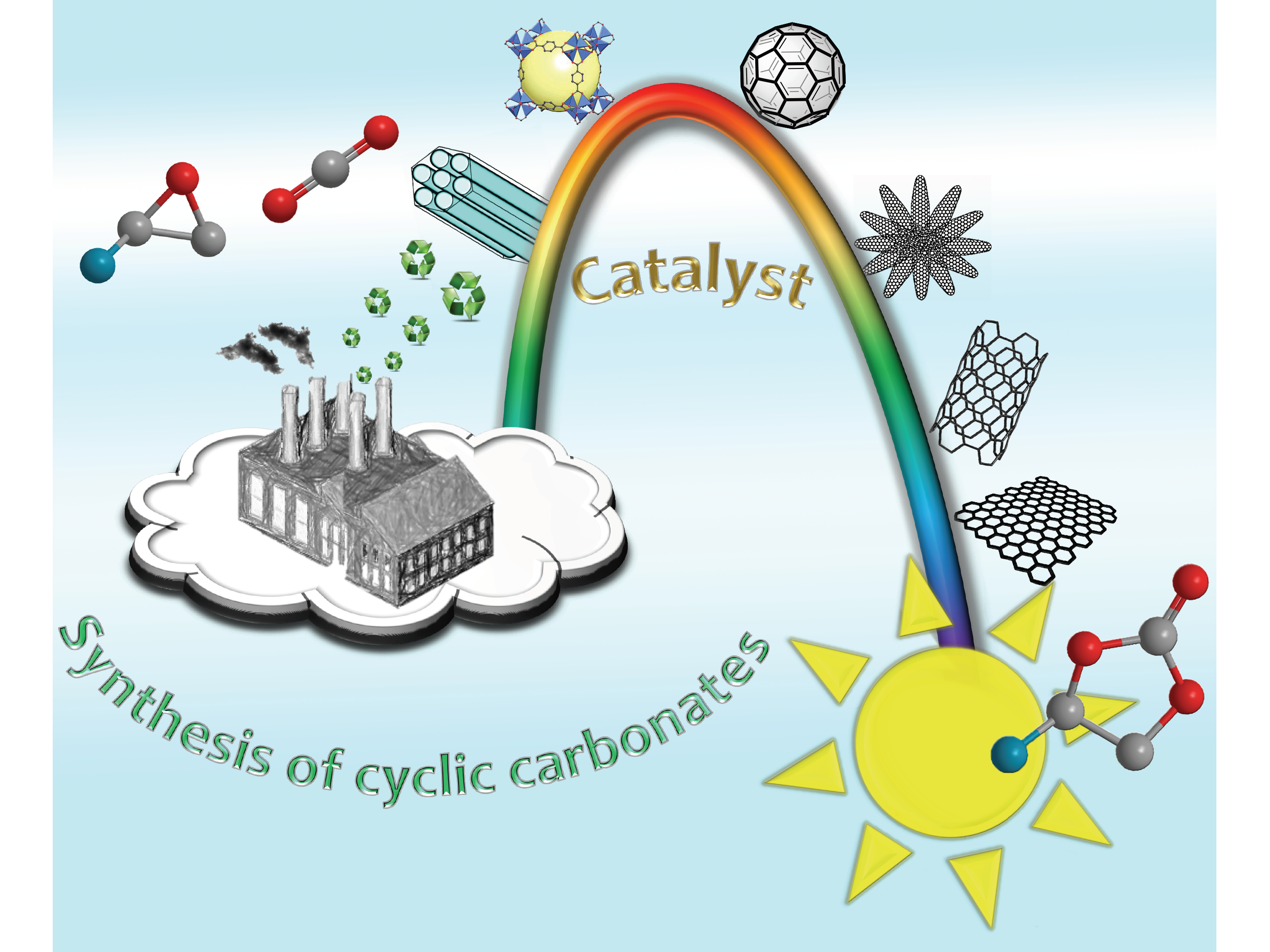
1.2. Carbon Dioxide Conversion into Cyclic Carbonates
1.3. Catalytic Systems for the Synthesis of Cyclic Carbonates
2. Hybrid Catalysts for the Synthesis of Cyclic Carbonates
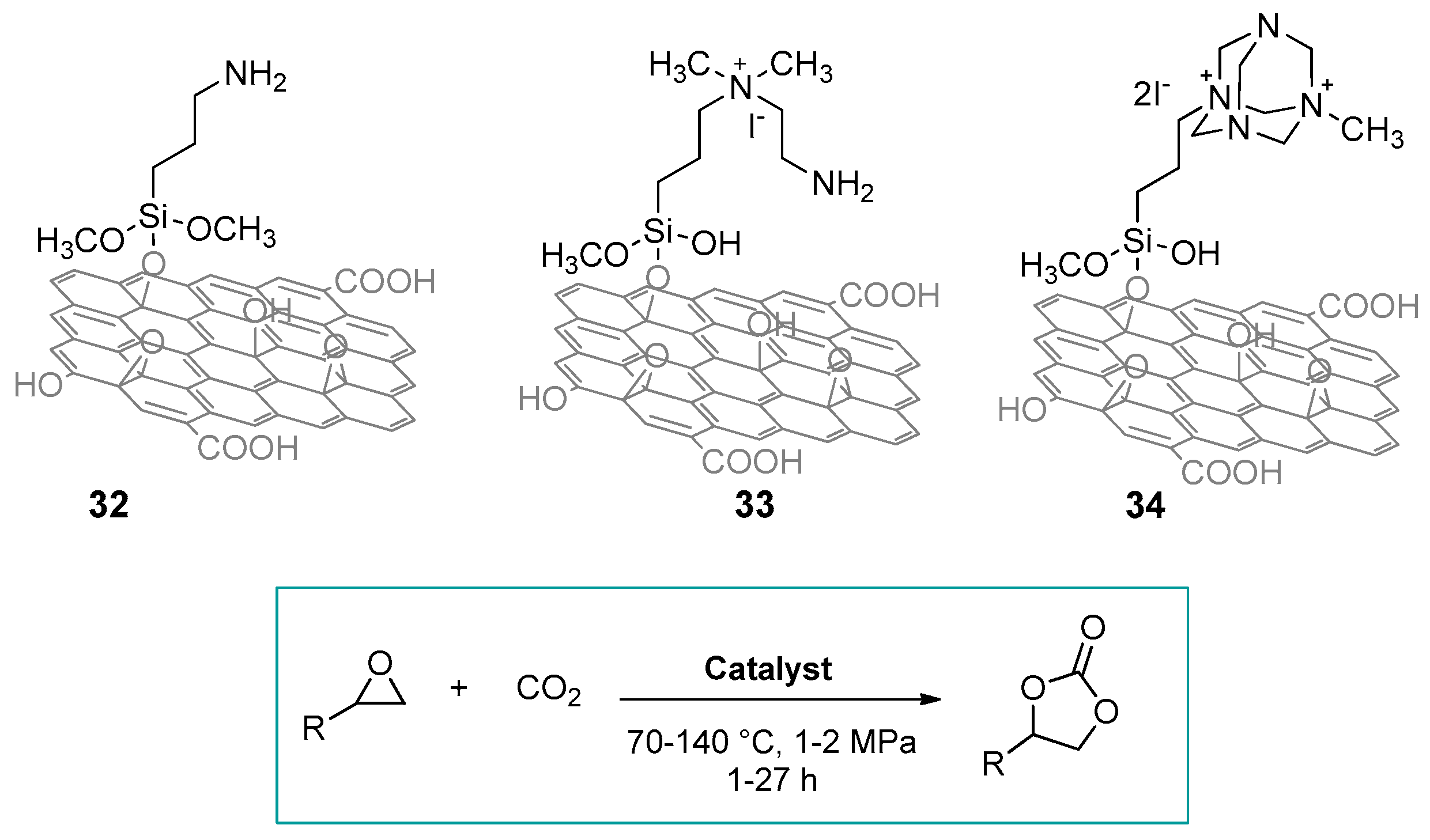
2.1. Silica-Based Hybrid Catalysts
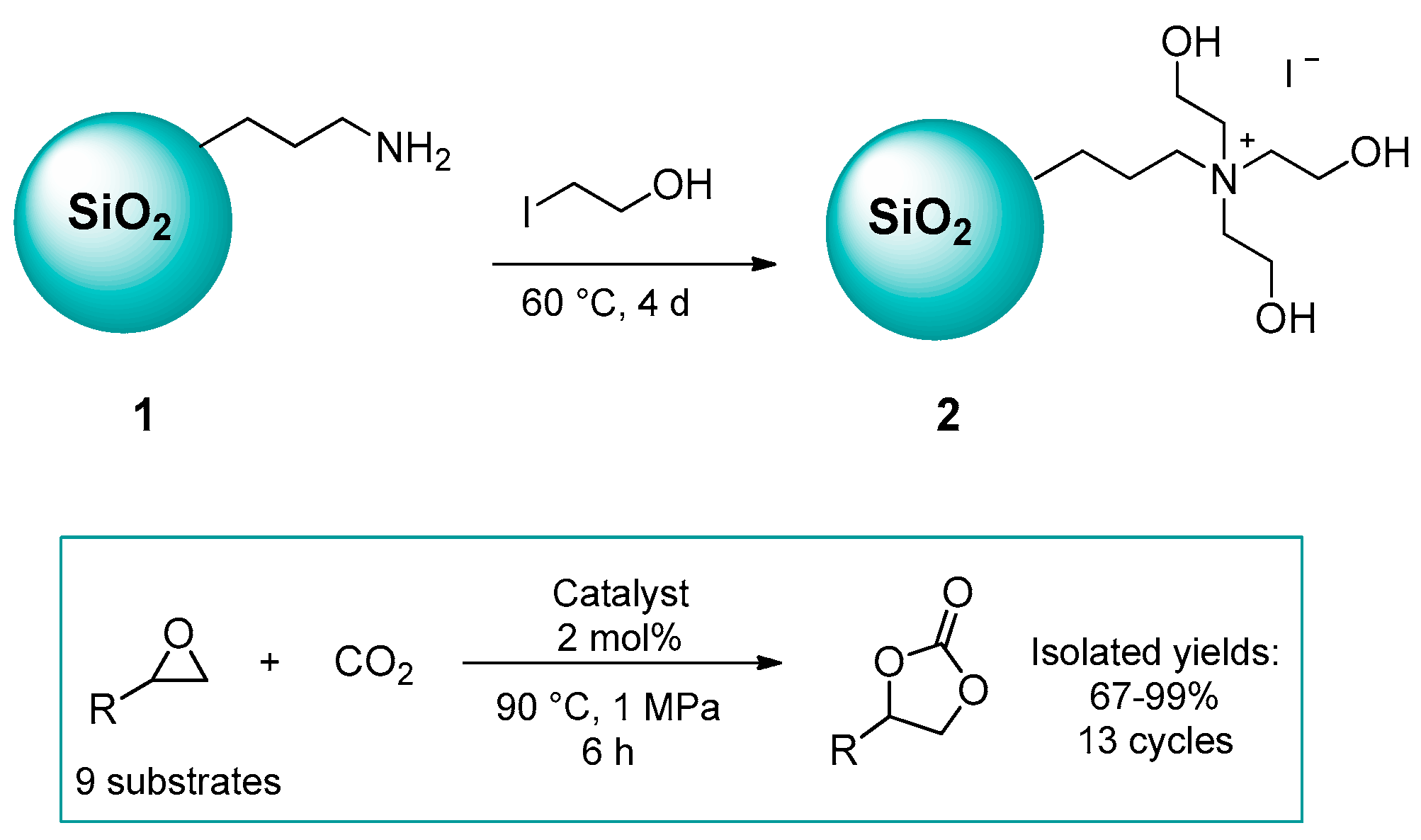
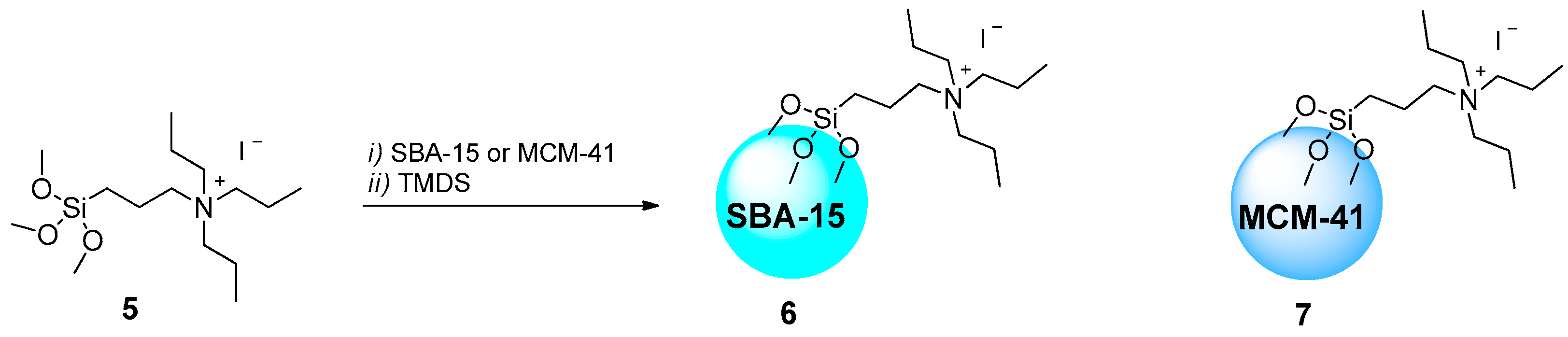
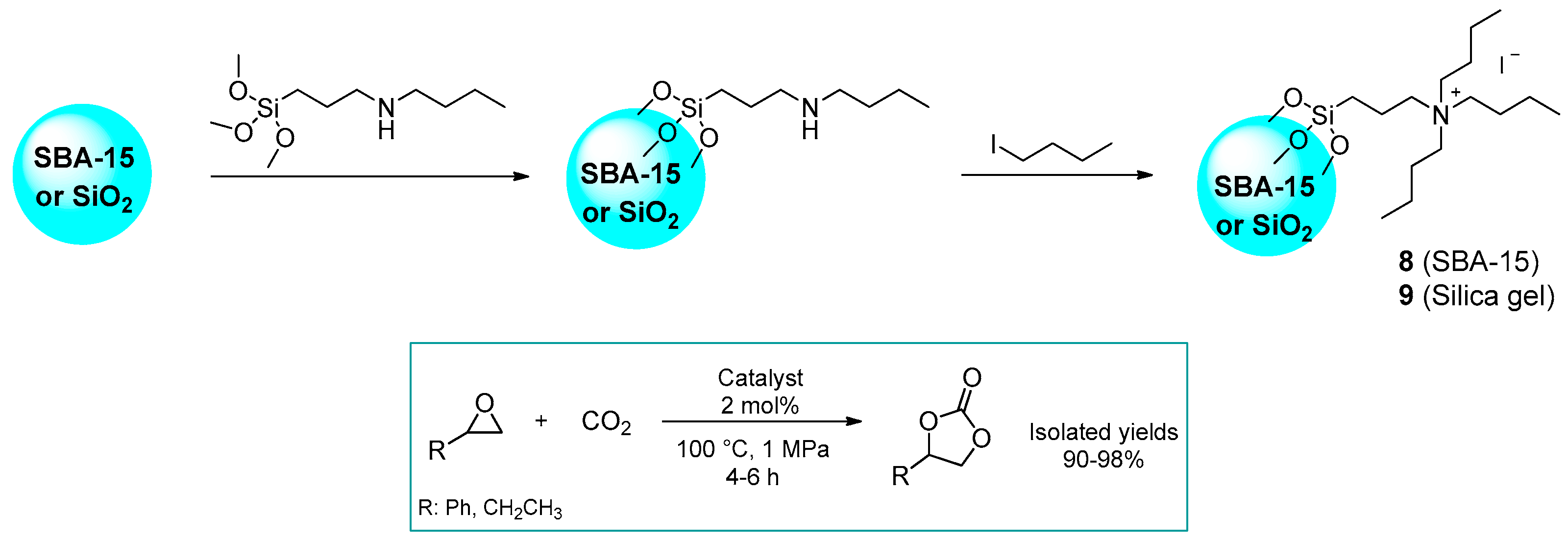
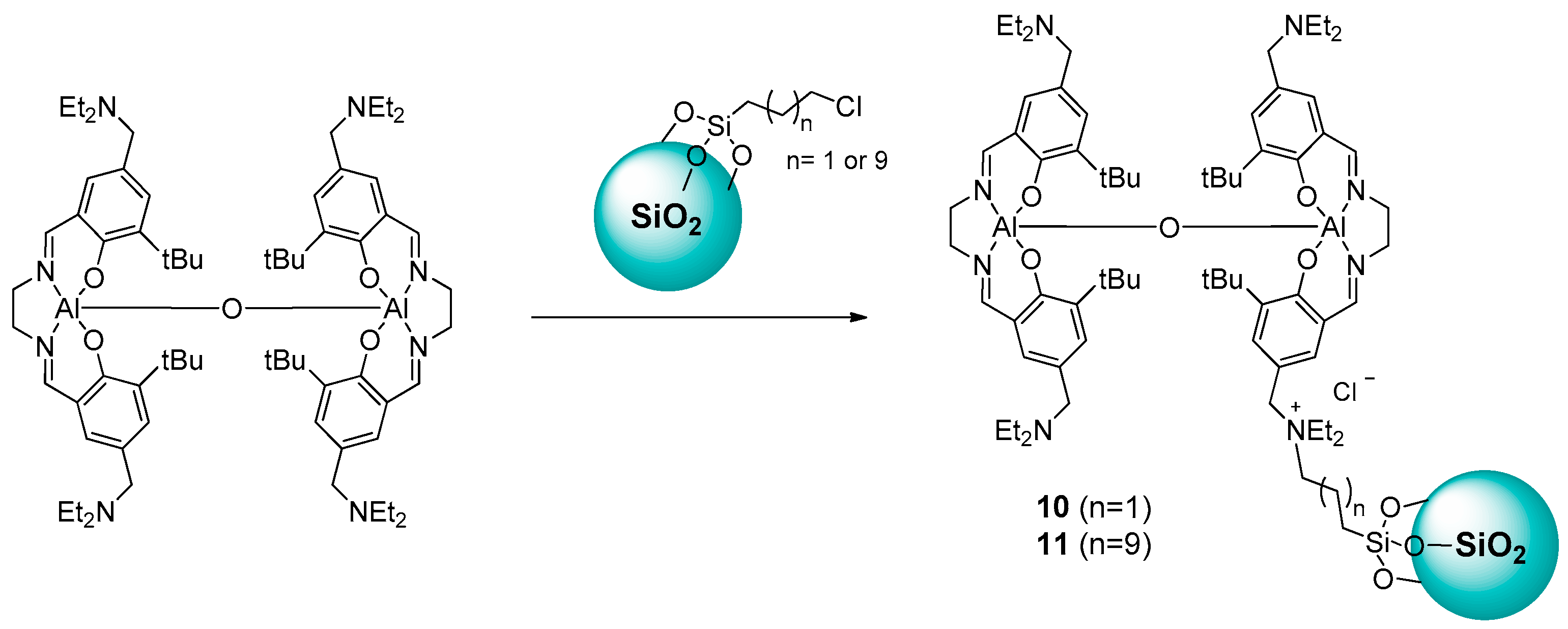
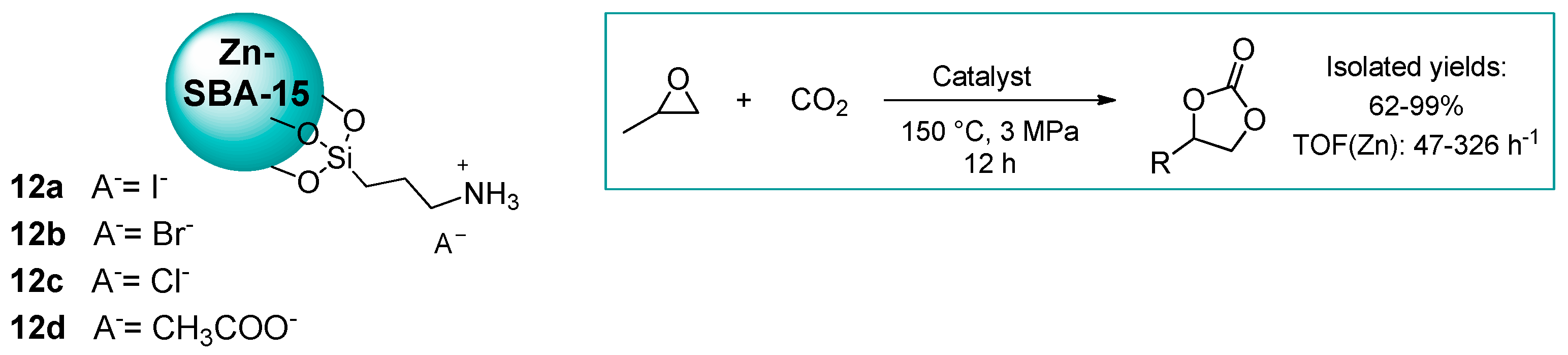
2.1.1. Ammonium-Functionalized Silica
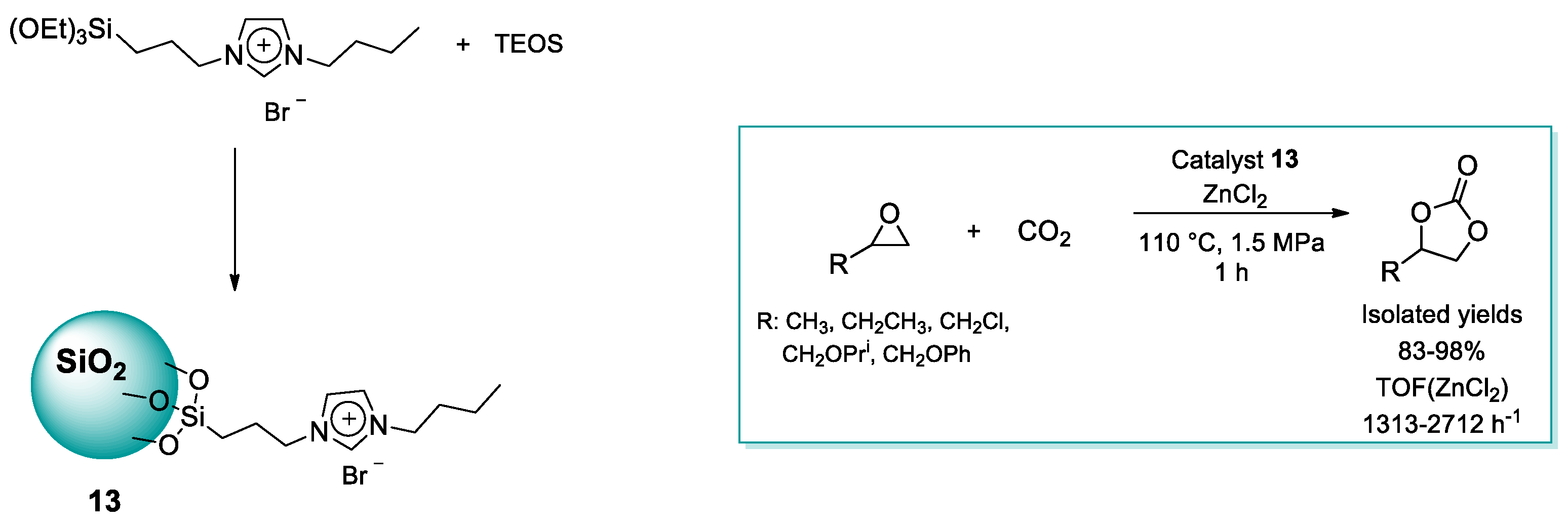
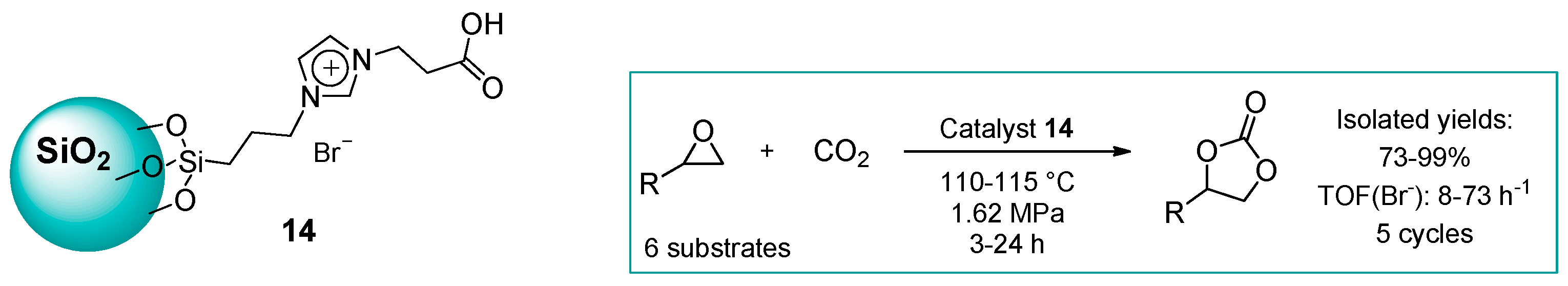
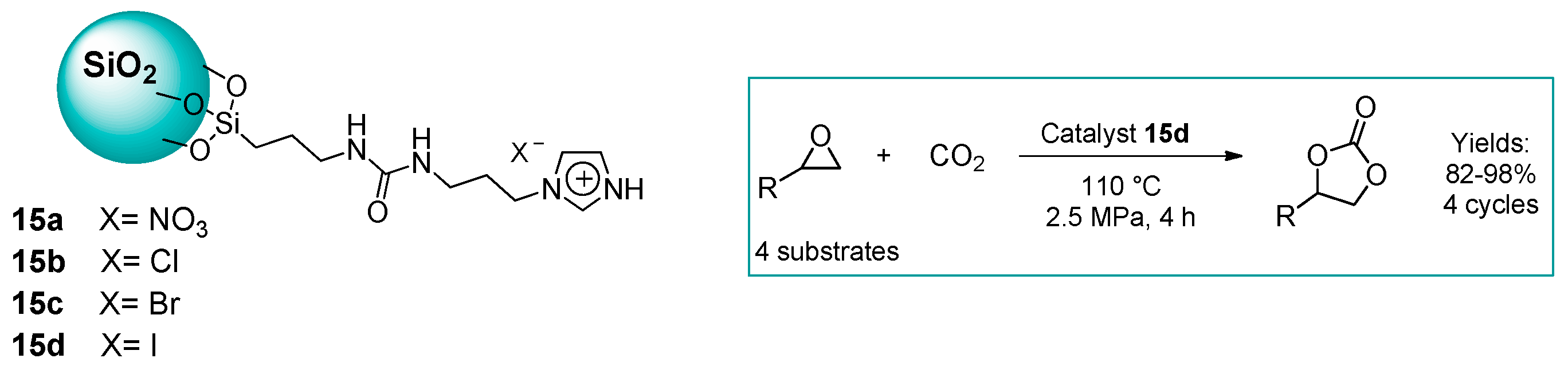
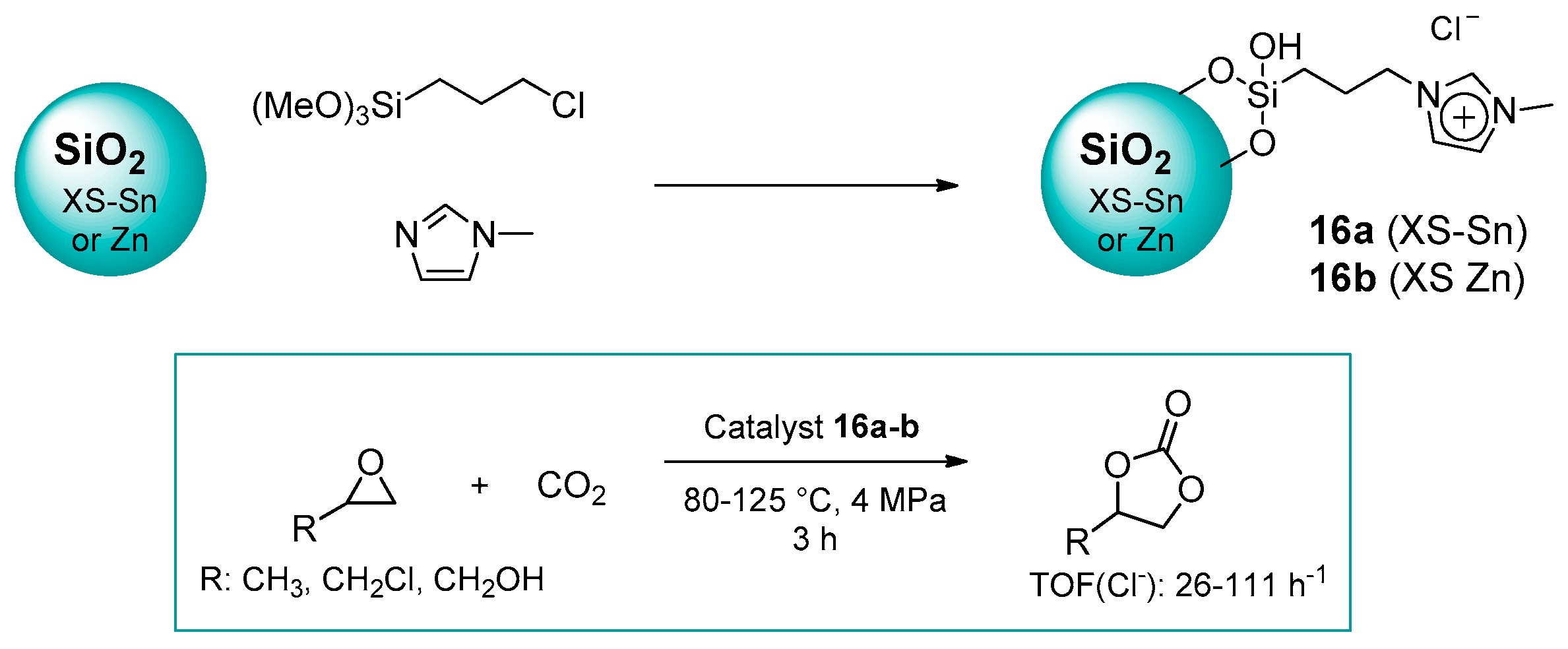
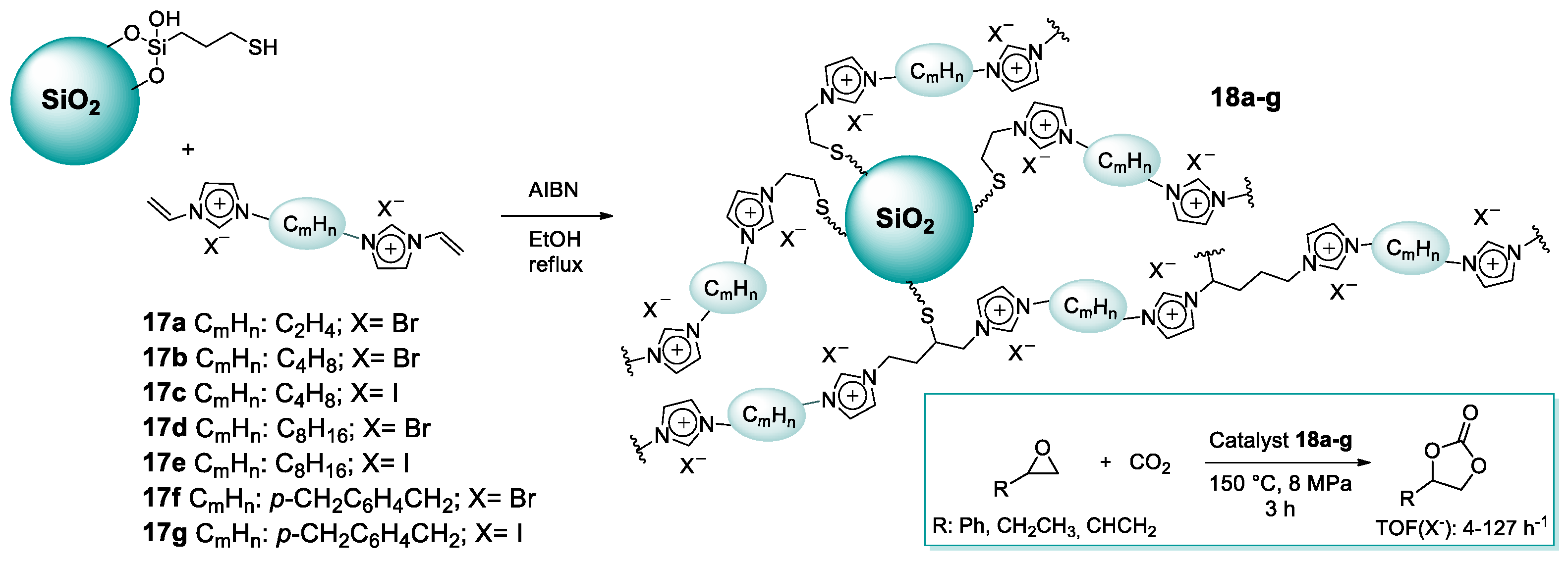
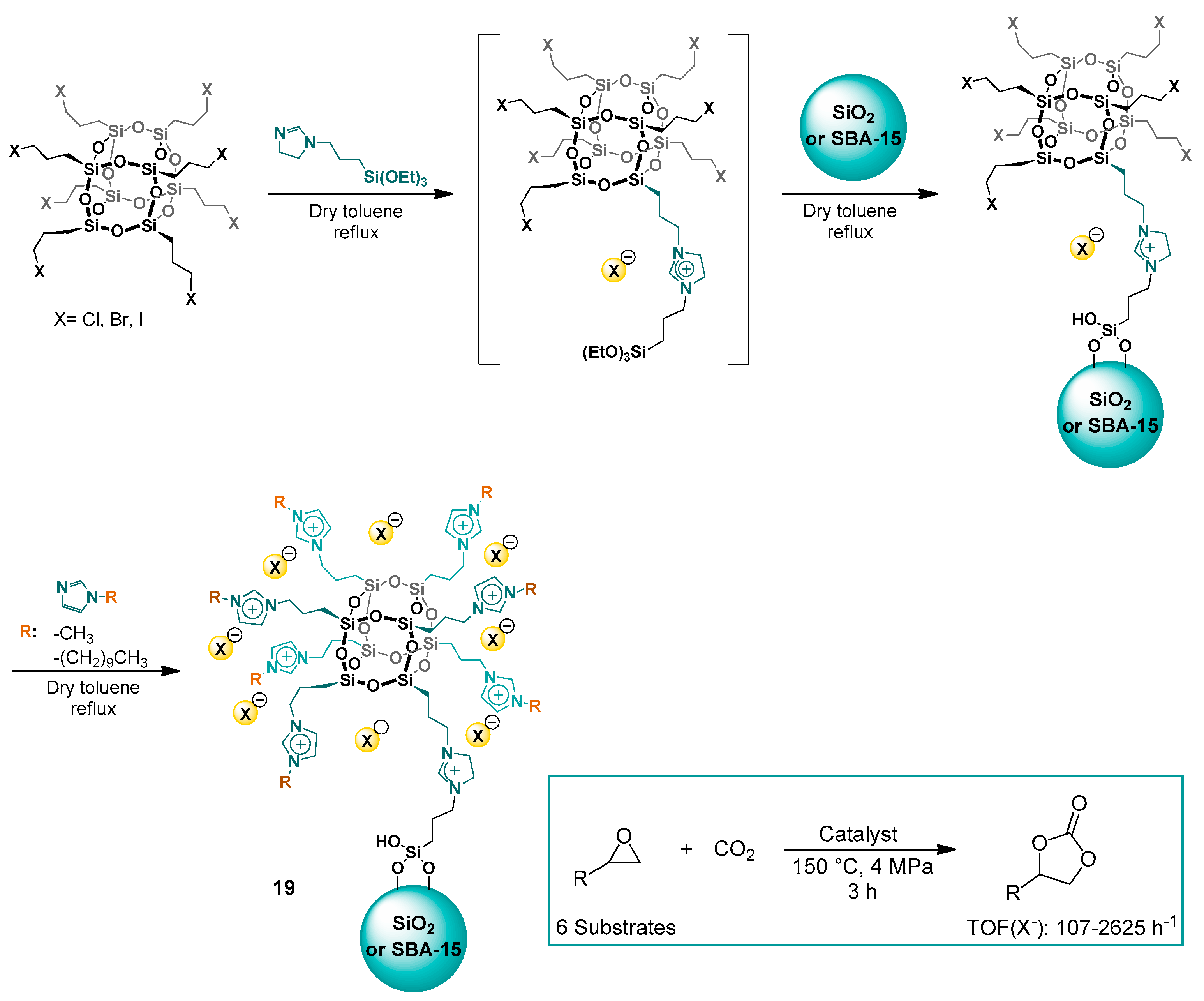
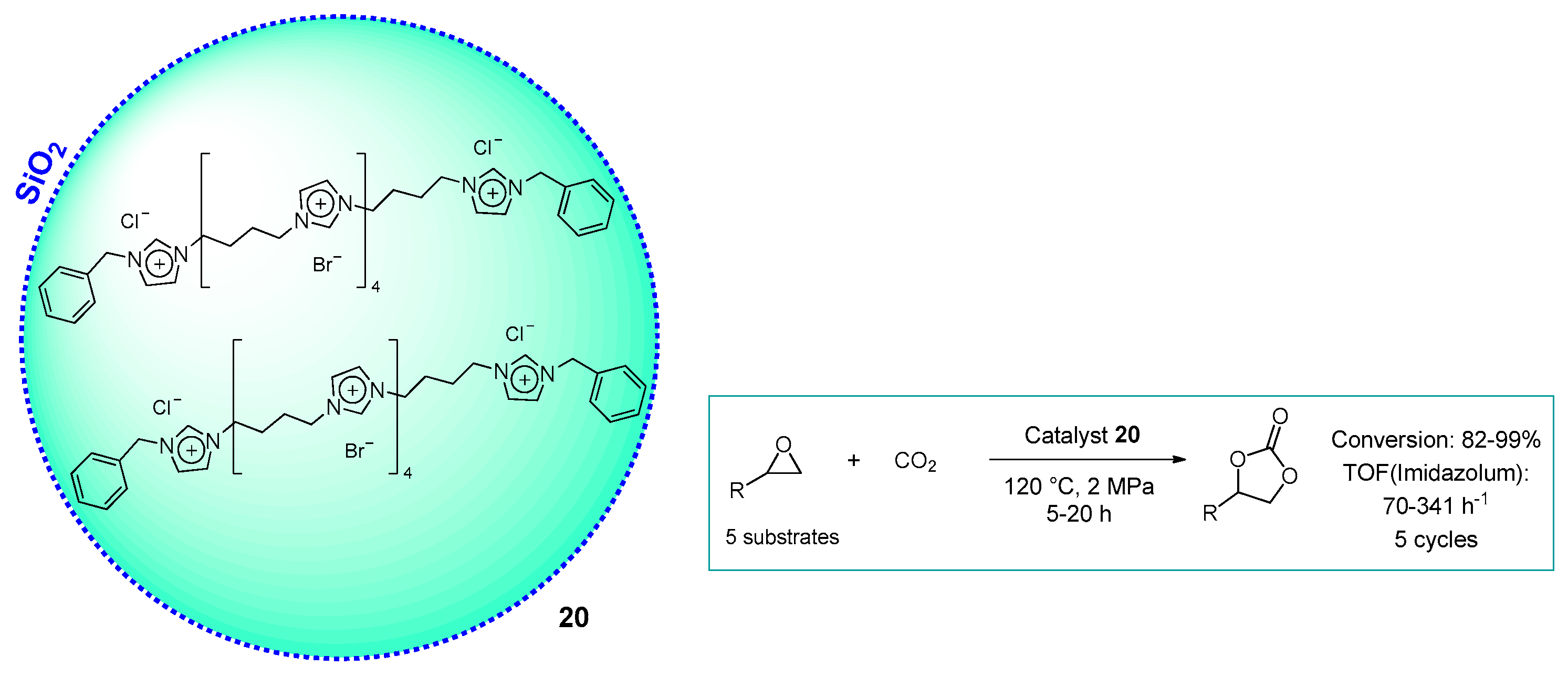
2.1.2. Imidazolium-Functionalized Silica
2.1.3. Phosphonium- and Pyridinium-Functionalized Silica
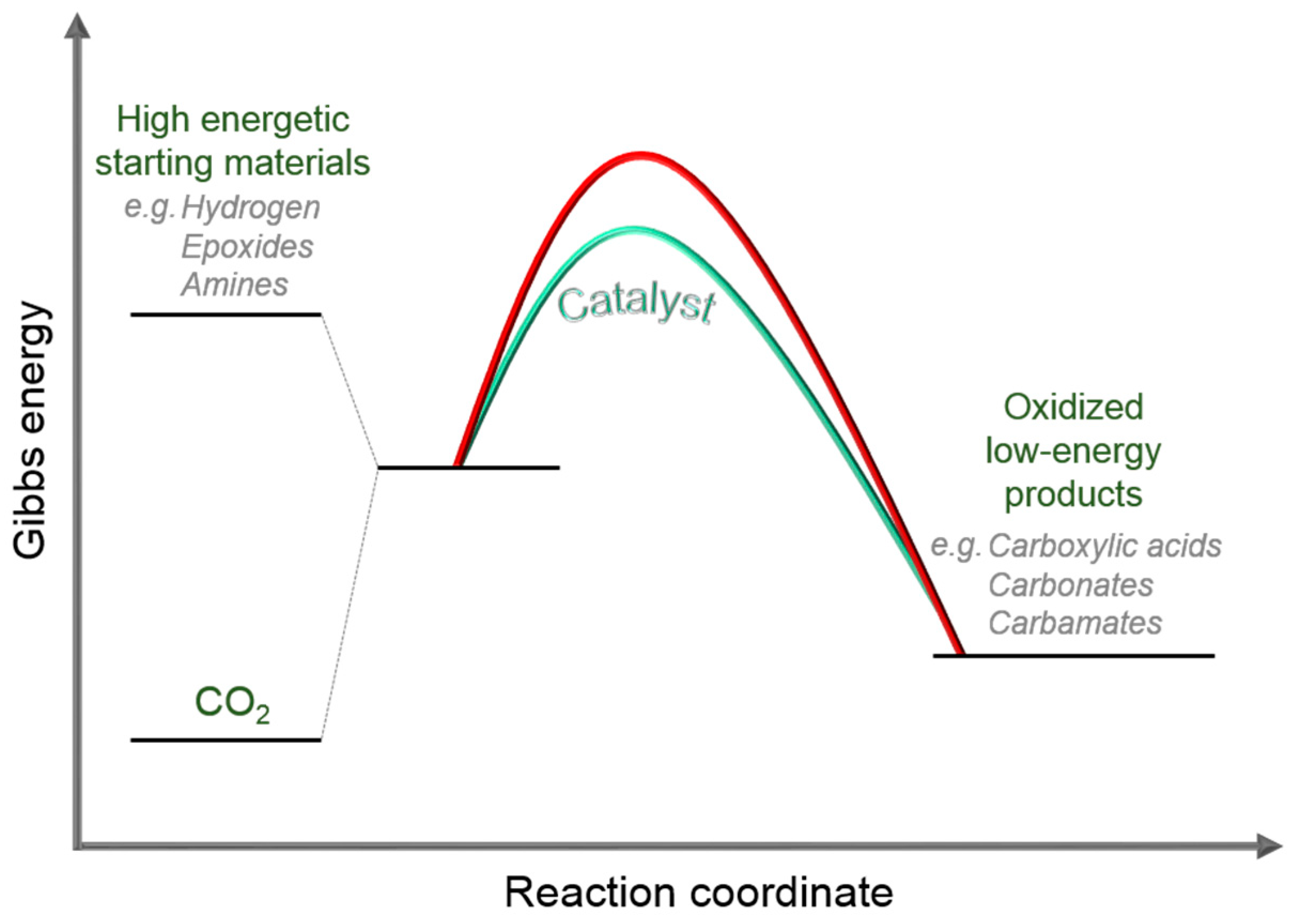
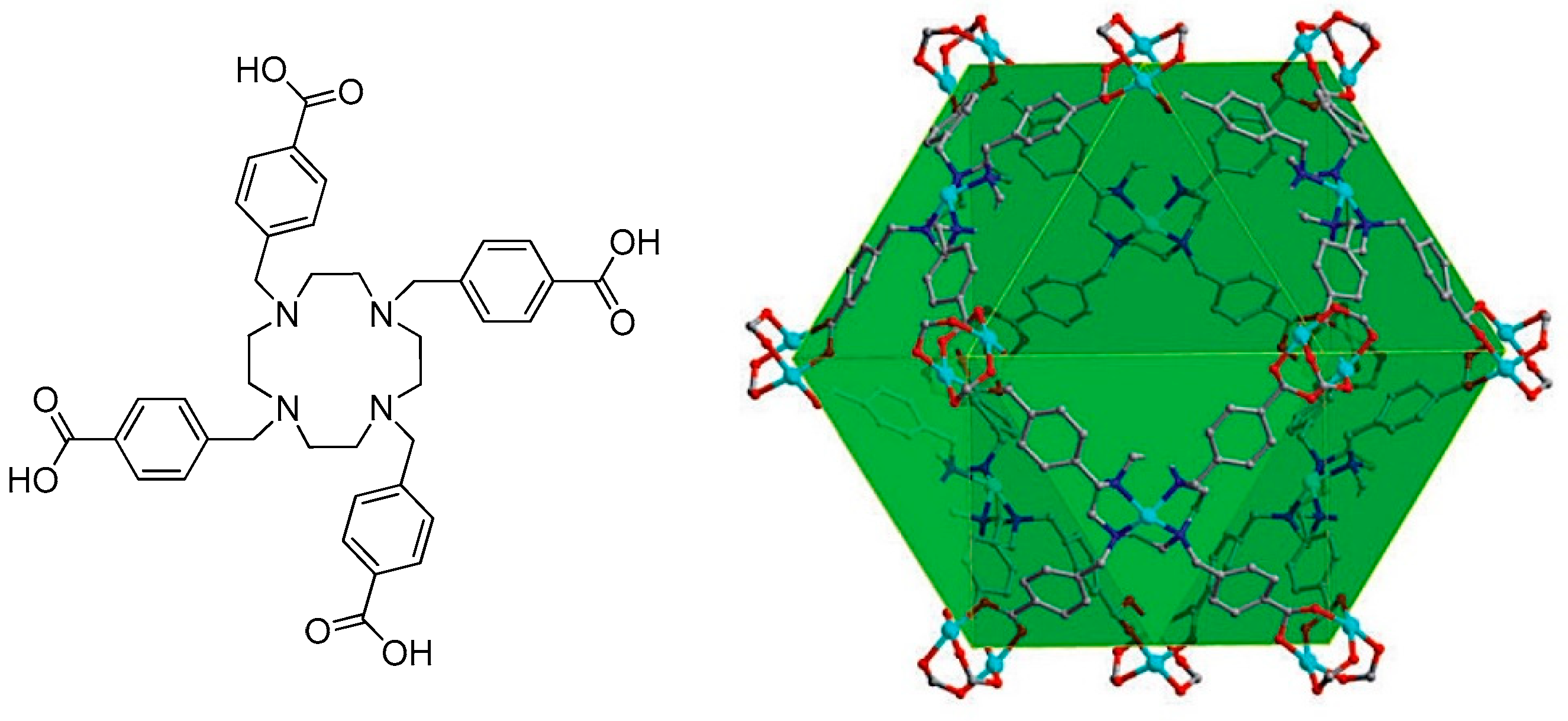
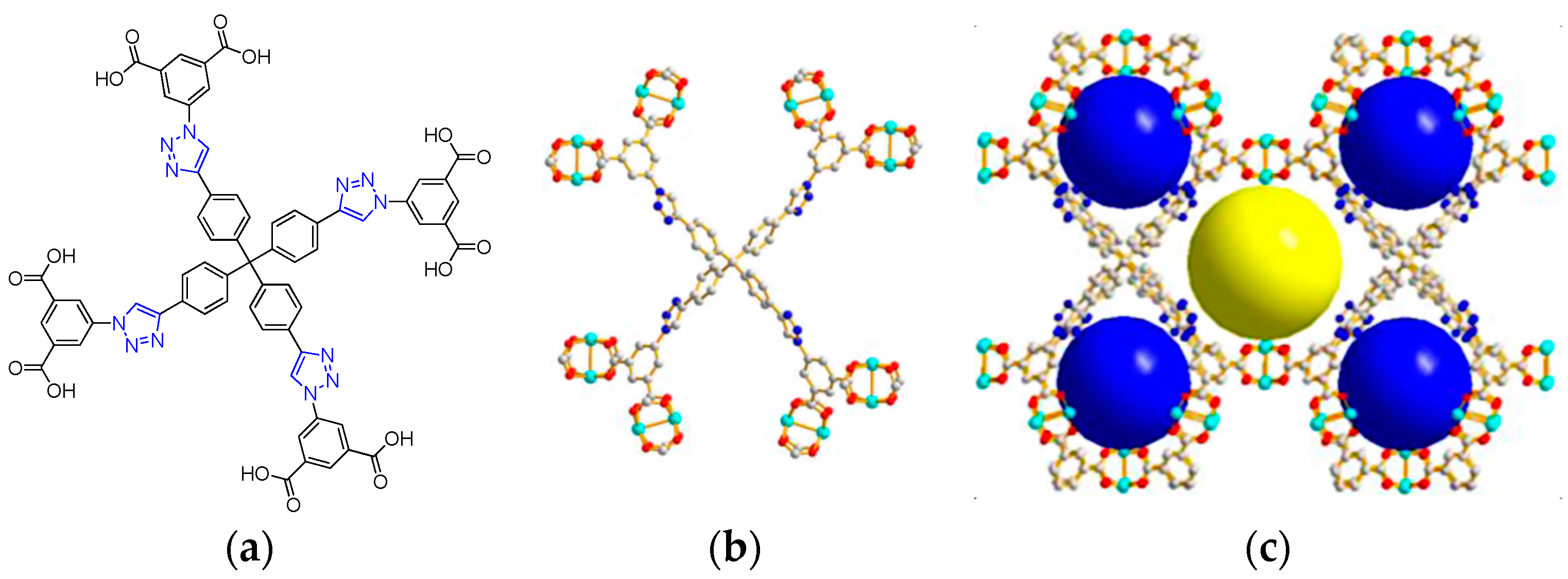
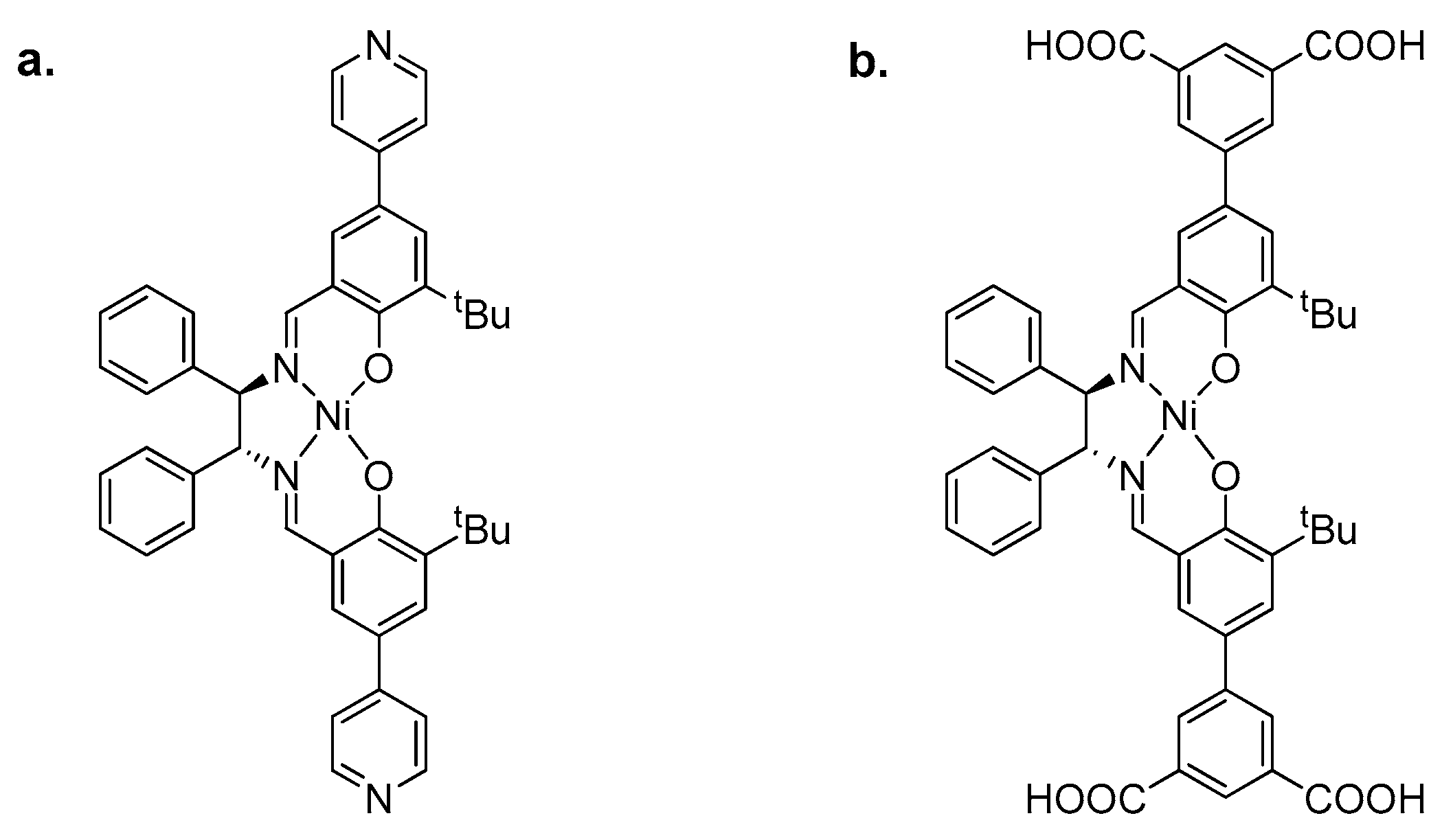
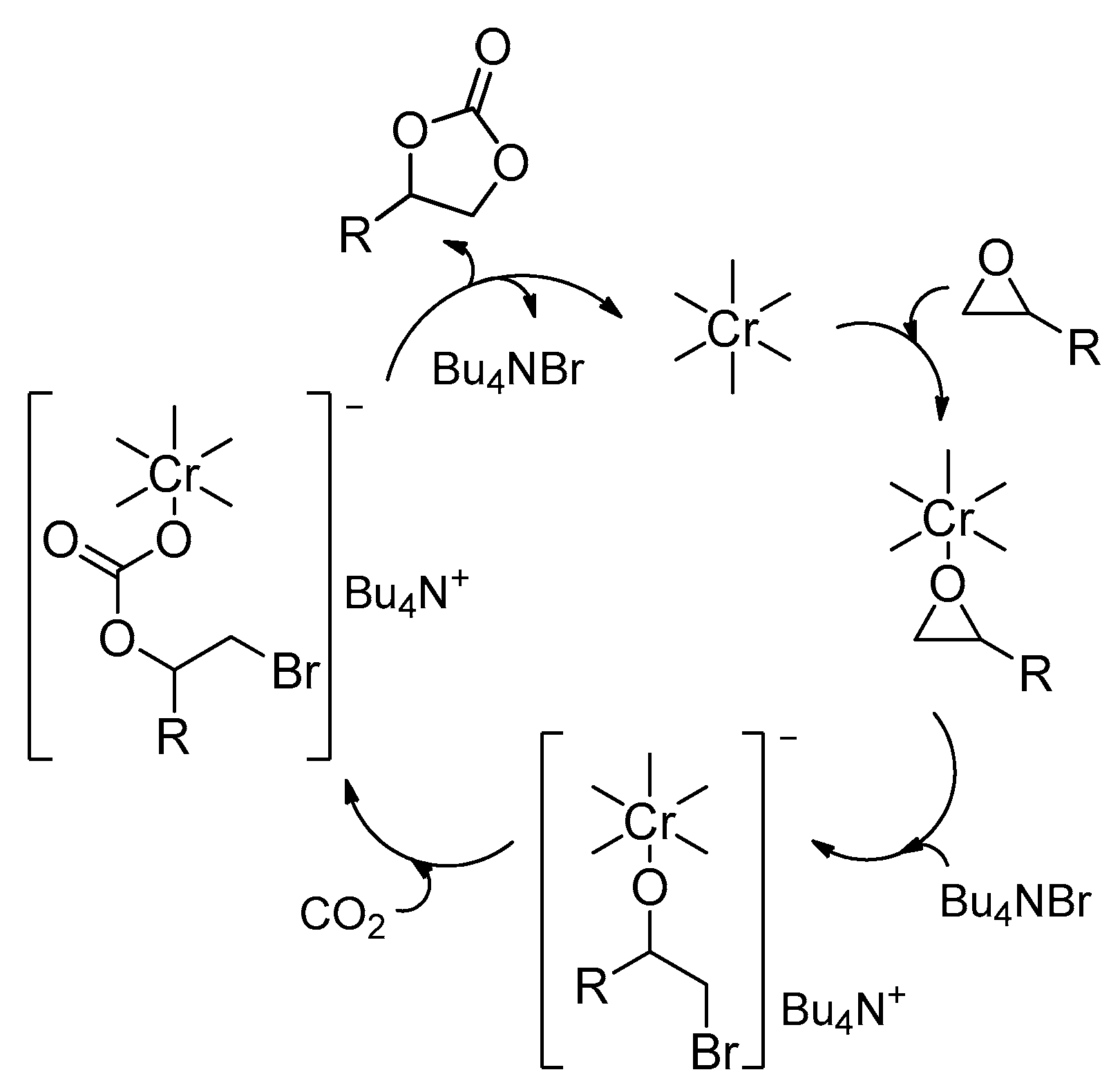
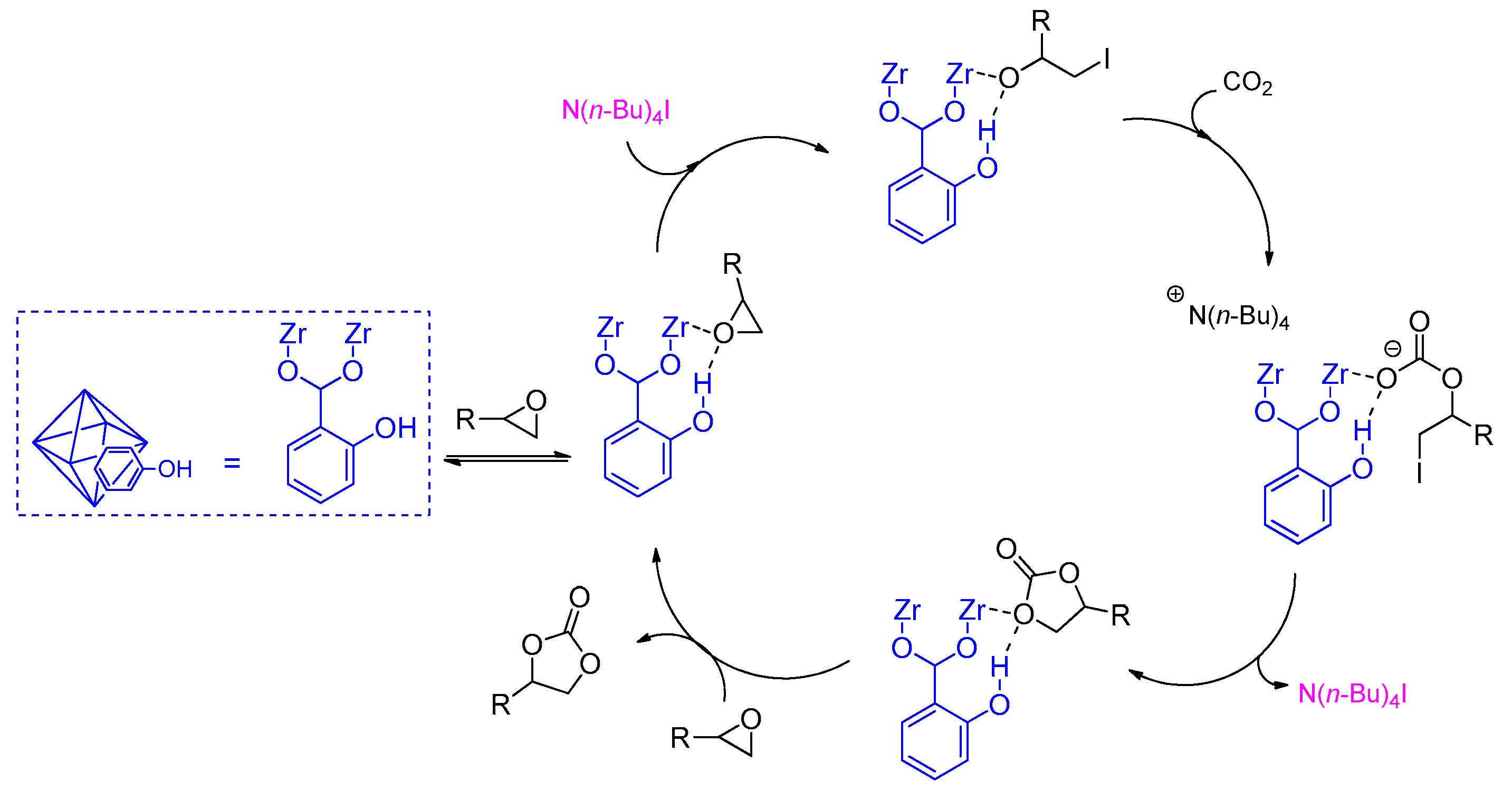
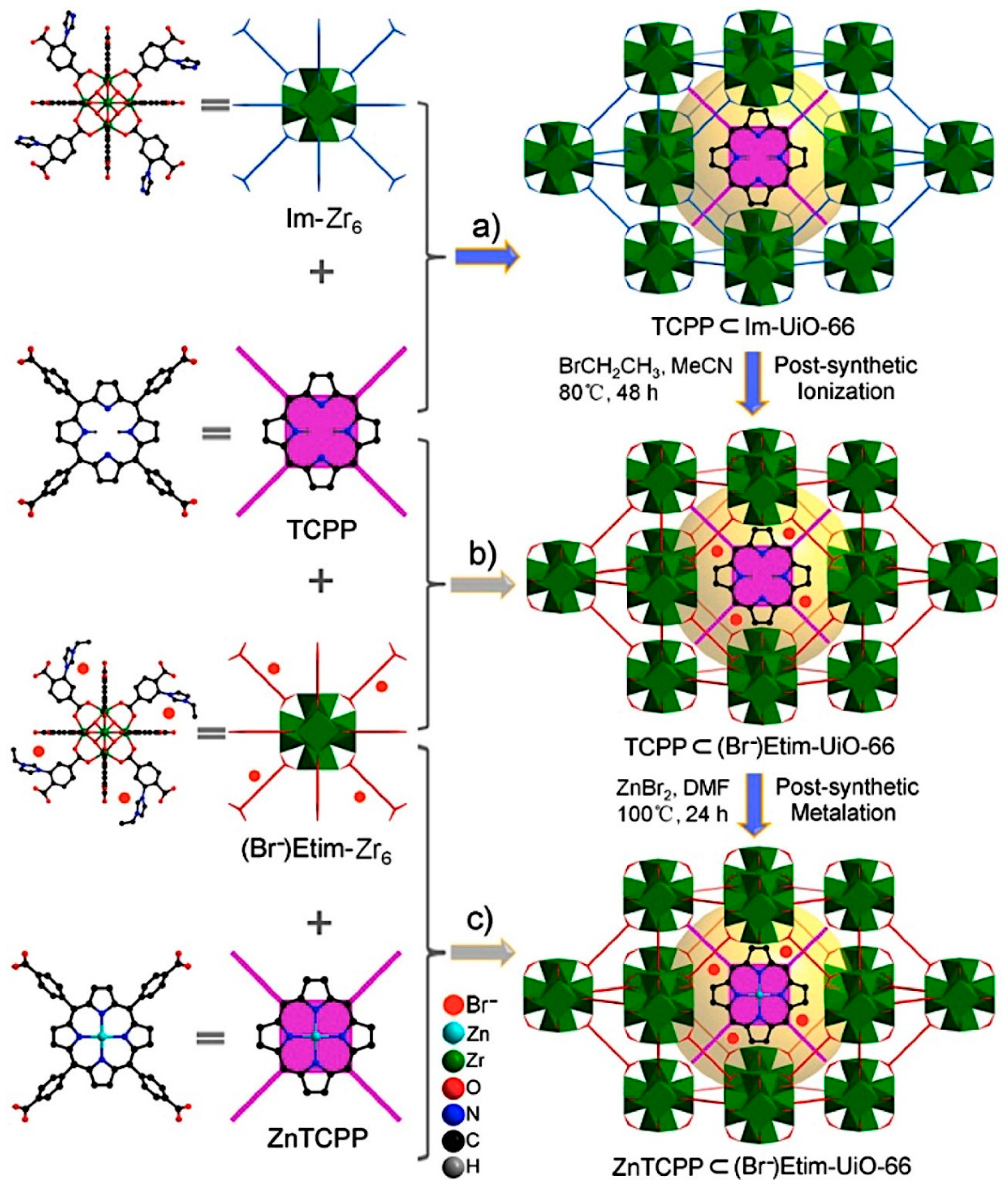
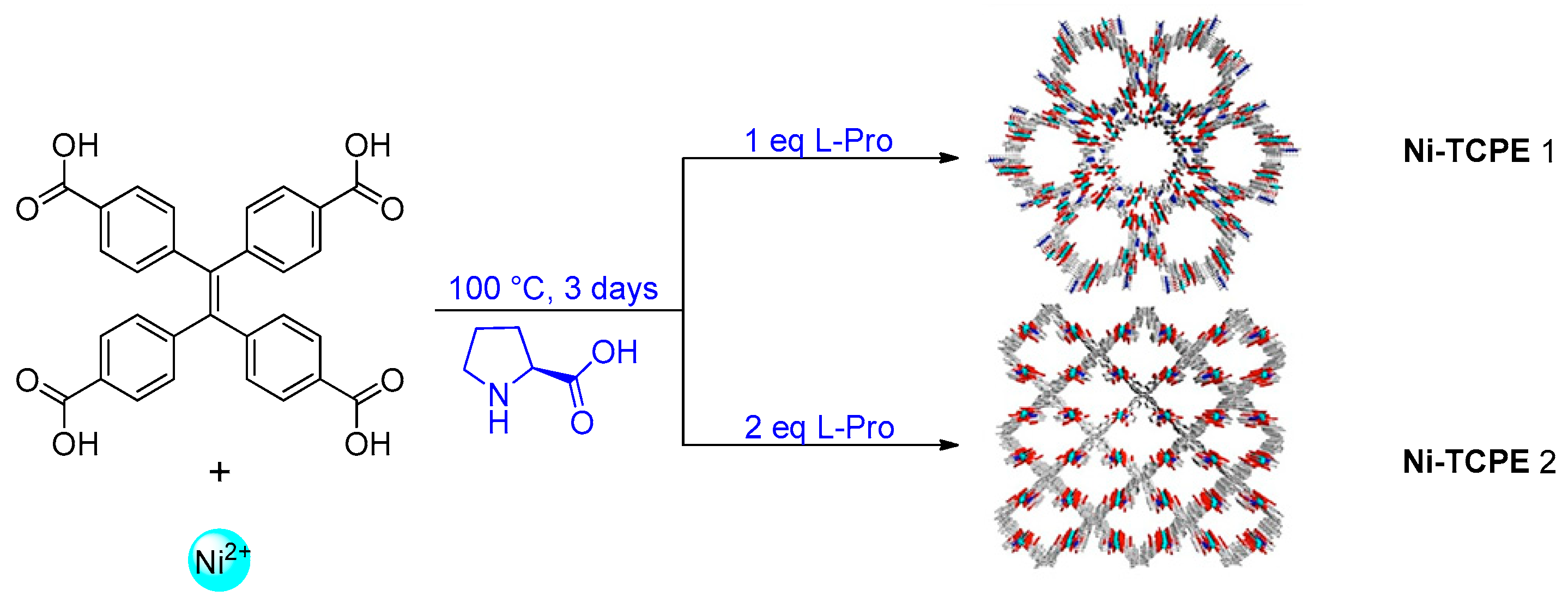
2.2. Metal–Organic Framework (MOF)-Based Catalysts
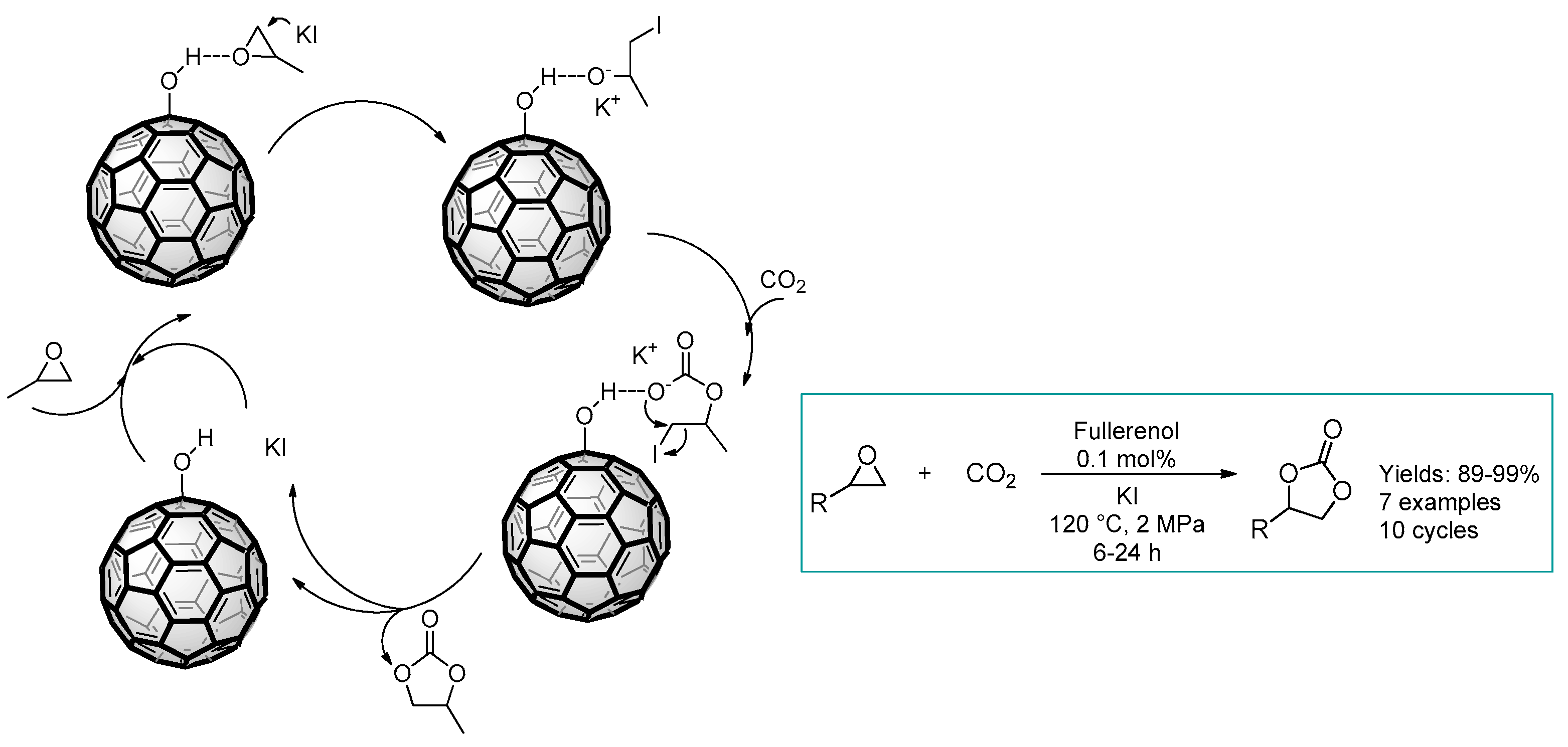
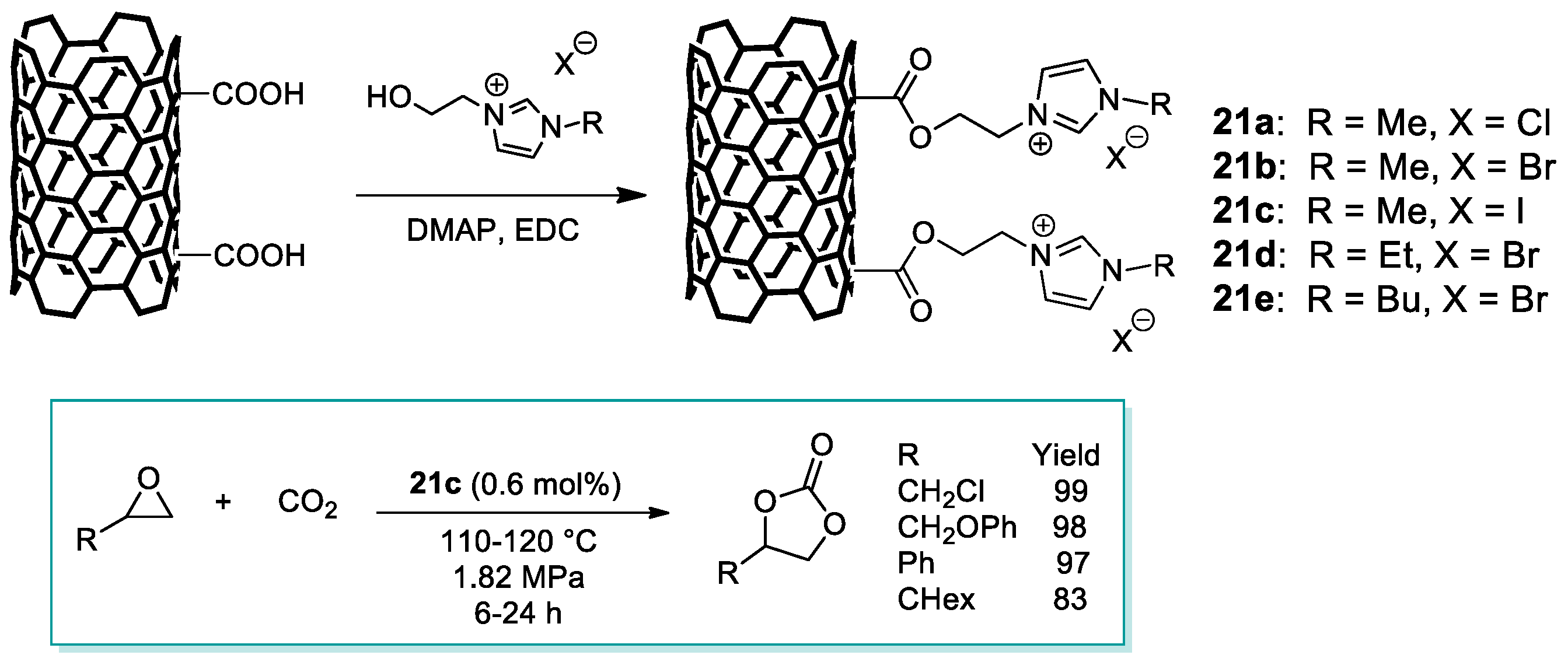
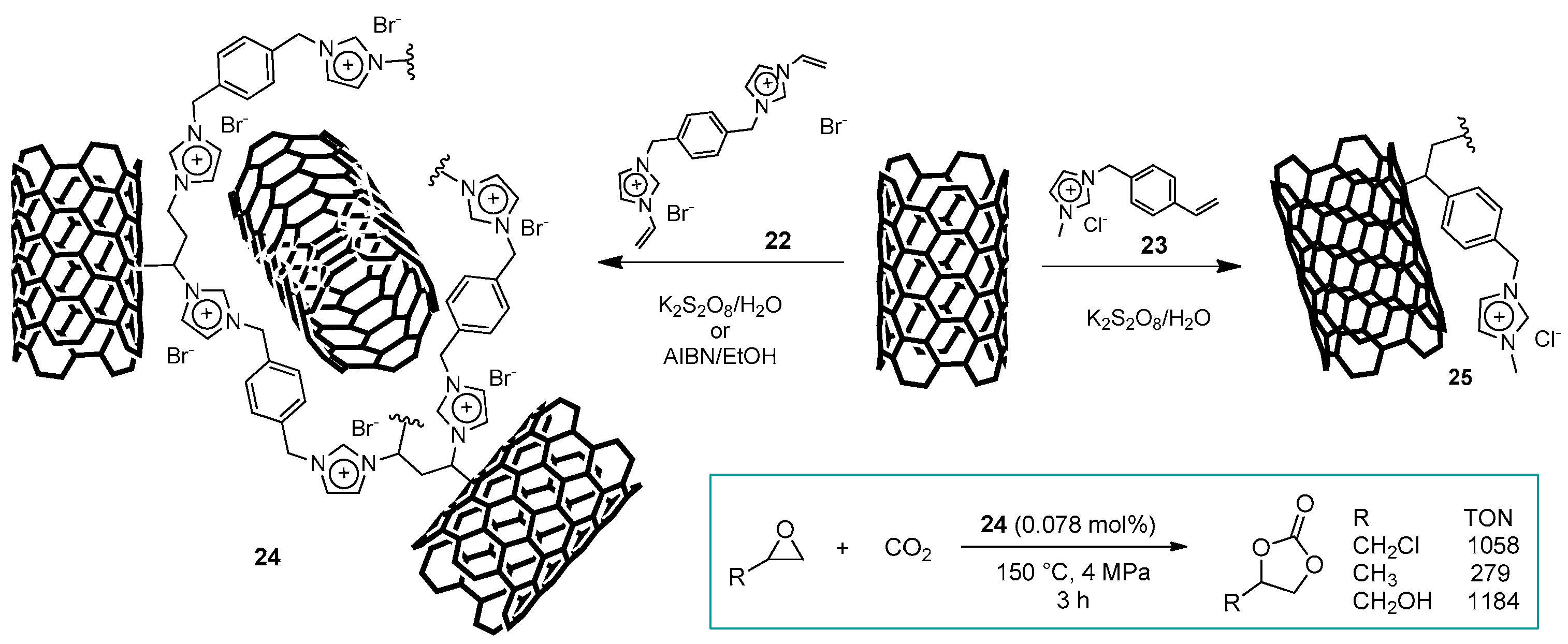
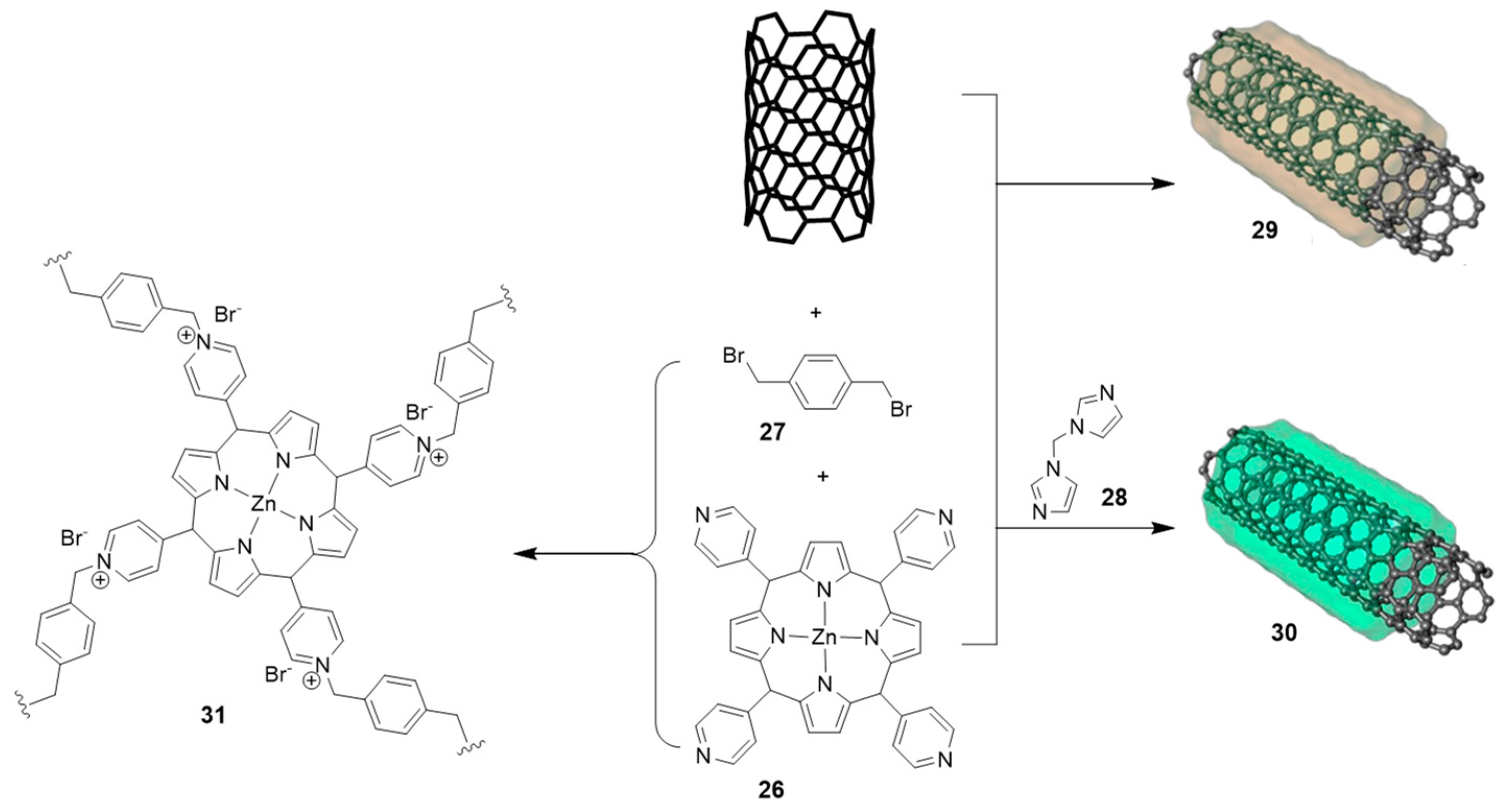
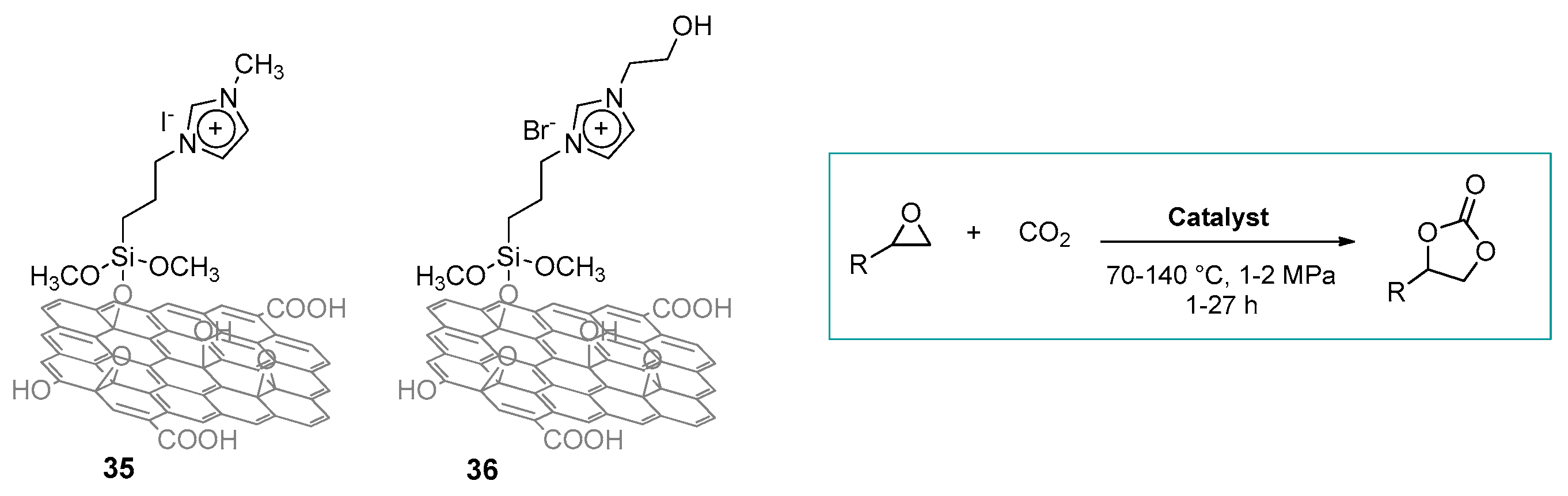
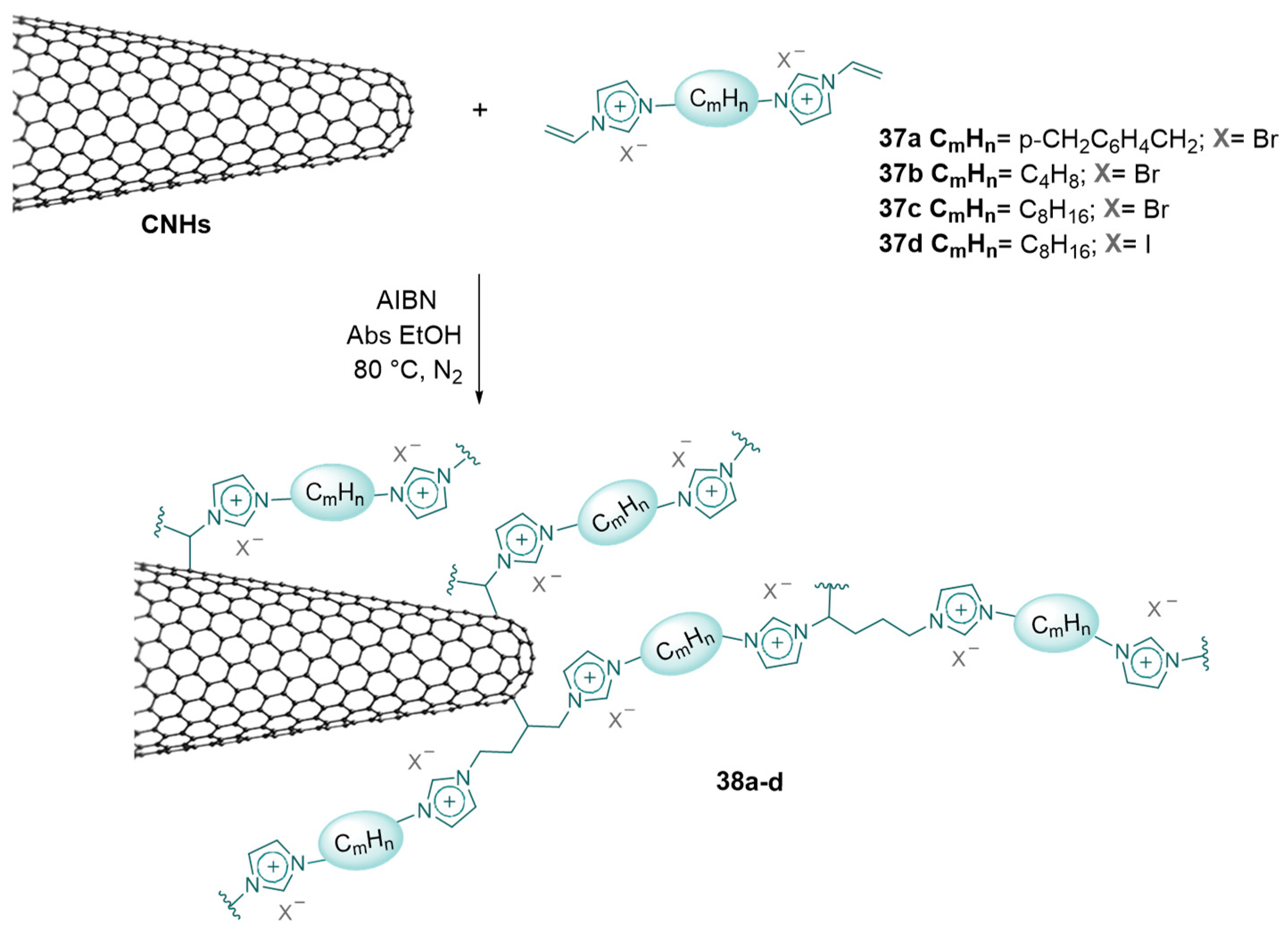
2.3. Nanocarbon-Based Catalysts for CO2 Conversion into Cyclic Carbonates
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahman, F.A.; Aziz, M.M.A.; Saidur, R.; Bakar, W.A.W.A.; Hainin, M.R.; Putrajaya, R.; Hassan, N.A. Pollution to solution: Capture and sequestration of carbon dioxide (CO2) and its utilization as a renewable energy source for a sustainable future. Renew. Sustain. Energy Rev. 2017, 71, 112–126. [Google Scholar] [CrossRef]
- Alper, E.; Yuksel Orhan, O. CO2 utilization: Developments in conversion processes. Petroleum 2017, 3, 109–126. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Maeda, C.; Miyazaki, Y.; Ema, T. Recent progress in catalytic conversions of carbon dioxide. Catal. Sci. Technol. 2014, 4, 1482–1497. [Google Scholar] [CrossRef]
- Quadrelli, E.A.; Centi, G.; Duplan, J.-L.; Perathoner, S. Carbon Dioxide Recycling: Emerging Large-Scale Technologies with Industrial Potential. ChemSusChem 2011, 4, 1194–1215. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Decortes, A.; Castilla, A.M.; Kleij, A.W. Salen-Complex-Mediated Formation of Cyclic Carbonates by Cycloaddition of CO2 to Epoxides. Angew. Chem. Int. Ed. 2010, 49, 9822–9837. [Google Scholar] [CrossRef]
- He, Q.; O’Brien, J.W.; Kitselman, K.A.; Tompkins, L.E.; Curtis, G.C.T.; Kerton, F.M. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride–metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- Comerford, J.W.; Ingram, I.D.V.; North, M.; Wu, X. Sustainable metal-based catalysts for the synthesis of cyclic carbonates containing five-membered rings. Green Chem. 2015, 17, 1966–1987. [Google Scholar] [CrossRef]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W.A.; Kühn, F.E. Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide by Using Organocatalysts. ChemSusChem 2015, 8, 2436–2454. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-fixation into cyclic and polymeric carbonates: Principles and applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef]
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 1312–1330. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pizzato, F.; Villuendas, P. Cyclic carbonates as sustainable solvents for proline-catalysed aldol reactions. Tetrahedron Asymmetry 2010, 21, 1262–1271. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Fan, L.; Kong, X.; Lu, Y. Progress in electrolytes for rechargeable Li-based batteries and beyond. Green Energy Environ. 2016, 1, 18–42. [Google Scholar] [CrossRef]
- Anastas, P.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Bhanage, B.M.; Fujita, S.-I.; Ikushima, Y.; Arai, M. Synthesis of dimethyl carbonate and glycols from carbon dioxide, epoxides, and methanol using heterogeneous basic metal oxide catalysts with high activity and selectivity. Appl. Catal. A 2001, 219, 259–266. [Google Scholar] [CrossRef]
- Nguyen, P.T.K.; Nguyen, H.T.D.; Nguyen, H.N.; Trickett, C.A.; Ton, Q.T.; Gutiérrez-Puebla, E.; Monge, M.A.; Cordova, K.E.; Gándara, F. New Metal–Organic Frameworks for Chemical Fixation of CO2. ACS Appl. Mater. Interfaces 2018, 10, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-Y.; Chen, Y.; Niu, Y.; Williams, K.; Cash, L.; Perez, P.J.; Wojtas, L.; Cai, J.; Chen, Y.-S.; Ma, S. Crystal Engineering of an nbo Topology Metal–Organic Framework for Chemical Fixation of CO2 under Ambient Conditions. Angew. Chem. Int. Ed. 2014, 53, 2615–2619. [Google Scholar] [CrossRef]
- Huh, S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts 2019, 9, 34. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Z.; Han, B.; Hu, S.; Li, W.; Xie, Y. Synthesis of cyclic carbonates from epoxides and CO2 catalyzed by potassium halide in the presence of β-cyclodextrin. Green Chem. 2008, 10, 1337–1341. [Google Scholar] [CrossRef]
- Vignesh Babu, H.; Muralidharan, K. Zn(ii), Cd(ii) and Cu(ii) complexes of 2,5-bis{N-(2,6-diisopropylphenyl)iminomethyl}pyrrole: Synthesis, structures and their high catalytic activity for efficient cyclic carbonate synthesis. Dalton Trans. 2013, 42, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Liu, B.; Gan, Q.; Li, H.; Darensbourg, D.J. Formation of Cyclic Carbonates from Carbon Dioxide and Epoxides Coupling Reactions Efficiently Catalyzed by Robust, Recyclable One-Component Aluminum-Salen Complexes. ACS Catal. 2012, 2, 2029–2035. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Lamb, K.J.; North, M. Cr(salophen) Complex Catalyzed Cyclic Carbonate Synthesis at Ambient Temperature and Pressure. ACS Catal. 2016, 6, 5012–5025. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, X. Lewis base-CO2 adducts as organocatalysts for CO2 transformation. Sci. China Chem. 2017, 60, 904–911. [Google Scholar] [CrossRef]
- Saptal, V.B.; Bhanage, B.M. Bifunctional Ionic Liquids Derived from Biorenewable Sources as Sustainable Catalysts for Fixation of Carbon Dioxide. ChemSusChem 2016, 10, 1145–1151. [Google Scholar] [CrossRef]
- Saptal, V.B.; Bhanage, B.M. Bifunctional Ionic Liquids for the Multitask Fixation of Carbon Dioxide into Valuable Chemicals. ChemCatChem 2015, 8, 244–250. [Google Scholar] [CrossRef]
- Luo, R.; Chen, Y.; He, Q.; Lin, X.; Xu, Q.; He, X.; Zhang, W.; Zhou, X.; Ji, H. Metallosalen-Based Ionic Porous Polymers as Bifunctional Catalysts for the Conversion of CO2 into Valuable Chemicals. ChemSusChem 2017, 10, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Hao, X.-J.; Wang, P.-P.; Li, J. Amino acid-based ionic liquids for CO2 conversion to form cyclic carbonate under solvent-free conditions. Mol. Catal. 2017, 433, 420–429. [Google Scholar] [CrossRef]
- Jutz, F.; Andanson, J.-M.; Baiker, A. Ionic Liquids and Dense Carbon Dioxide: A Beneficial Biphasic System for Catalysis. Chem. Rev. 2011, 111, 322–353. [Google Scholar] [CrossRef]
- Girard, A.-L.; Simon, N.; Zanatta, M.; Marmitt, S.; Gonçalves, P.; Dupont, J. Insights on recyclable catalytic system composed of task-specific ionic liquids for the chemical fixation of carbon dioxide. Green Chem. 2014, 16, 2815–2825. [Google Scholar] [CrossRef]
- Zhong, H.; Su, Y.; Chen, X.; Li, X.; Wang, R. Imidazolium- and Triazine-Based Porous Organic Polymers for Heterogeneous Catalytic Conversion of CO2 into Cyclic Carbonates. ChemSusChem 2017, 10, 4855–4863. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A Triazole-Containing Metal–Organic Framework as a Highly Effective and Substrate Size-Dependent Catalyst for CO2 Conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Vinu, A.; Hossain, K.Z.; Ariga, K. Recent Advances in Functionalization of Mesoporous Silica. J. Nanosci. Nanotechnol. 2005, 5, 347–371. [Google Scholar] [CrossRef] [PubMed]
- Brühwiler, D. Postsynthetic functionalization of mesoporous silica. Nanoscale 2010, 2, 887–892. [Google Scholar] [CrossRef]
- Kohrt, C.; Werner, T. Recyclable Bifunctional Polystyrene and Silica Gel-Supported Organocatalyst for the Coupling of CO2 with Epoxides. ChemSusChem 2015, 8, 2031–2034. [Google Scholar] [CrossRef] [PubMed]
- Hajipour, A.R.; Heidari, Y.; Kozehgary, G. Silica grafted ammonium salts based on DABCO as heterogeneous catalysts for cyclic carbonate synthesis from carbon dioxide and epoxides. RSC Adv. 2015, 5, 22373–22379. [Google Scholar] [CrossRef]
- Zakharova, M.V.; Kleitz, F.; Fontaine, F.-G. Carbon Dioxide Oversolubility in Nanoconfined Liquids for the Synthesis of Cyclic Carbonates. ChemCatChem 2017, 9, 1886–1890. [Google Scholar] [CrossRef]
- Kolle, J.M.; Sayari, A. Substrate dependence on the fixation of CO2 to cyclic carbonates over reusable porous hybrid solids. J. CO2 Util. 2018, 26, 564–574. [Google Scholar] [CrossRef]
- North, M.; Villuendas, P. Influence of Support and Linker Parameters on the Activity of Silica-Supported Catalysts for Cyclic Carbonate Synthesis. ChemCatChem 2012, 4, 789–794. [Google Scholar] [CrossRef]
- Liu, M.; Liu, B.; Liang, L.; Wang, F.; Shi, L.; Sun, J. Design of bifunctional NH3I-Zn/SBA-15 single-component heterogeneous catalyst for chemical fixation of carbon dioxide to cyclic carbonates. J. Mol. Catal. A Chem. 2016, 418–419, 78–85. [Google Scholar] [CrossRef]
- Xiao, L.-F.; Li, F.-W.; Peng, J.-J.; Xia, C.-G. Immobilized ionic liquid/zinc chloride: Heterogeneous catalyst for synthesis of cyclic carbonates from carbon dioxide and epoxides. J. Mol. Catal. A Chem. 2006, 253, 265–269. [Google Scholar] [CrossRef]
- Han, L.; Park, S.-W.; Park, D.-W. Silica grafted imidazolium-based ionic liquids: Efficient heterogeneous catalysts for chemical fixation of CO2 to a cyclic carbonate. Energy Environ. Sci. 2009, 2, 1286–1292. [Google Scholar] [CrossRef]
- Han, L.; Park, M.-S.; Choi, S.-J.; Kim, Y.-J.; Lee, S.-M.; Park, D.-W. Incorporation of Metal Ions into Silica-Grafted Imidazolium-Based Ionic Liquids to Efficiently Catalyze Cycloaddition Reactions of CO2 and Epoxides. Catal. Lett. 2012, 142, 259–266. [Google Scholar] [CrossRef]
- Han, L.; Choi, H.-J.; Choi, S.-J.; Liu, B.; Park, D.-W. Ionic liquids containing carboxyl acid moieties grafted onto silica: Synthesis and application as heterogeneous catalysts for cycloaddition reactions of epoxide and carbon dioxide. Green Chem. 2011, 13, 1023–1028. [Google Scholar] [CrossRef]
- Sadeghzadeh, S.M. A heteropolyacid-based ionic liquid immobilized onto fibrous nano-silica as an efficient catalyst for the synthesis of cyclic carbonate from carbon dioxide and epoxides. Green Chem. 2015, 17, 3059–3066. [Google Scholar] [CrossRef]
- Liu, M.; Lu, X.; Jiang, Y.; Sun, J.; Arai, M. Zwitterionic Imidazole-Urea Derivative Framework Bridged Mesoporous Hybrid Silica: A Highly Efficient Heterogeneous Nanocatalyst for Carbon Dioxide Conversion. ChemCatChem 2018, 10, 1860–1868. [Google Scholar] [CrossRef]
- Comès, A.; Collard, X.; Fusaro, L.; Atzori, L.; Cutrufello, M.G.; Aprile, C. Bi-functional heterogeneous catalysts for carbon dioxide conversion: Enhanced performances at low temperature. RSC Adv. 2018, 8, 25342–25350. [Google Scholar] [CrossRef]
- Aprile, C.; Giacalone, F.; Agrigento, P.; Liotta, L.F.; Martens, J.A.; Pescarmona, P.P.; Gruttadauria, M. Multilayered Supported Ionic Liquids as Catalysts for Chemical Fixation of Carbon Dioxide: A High-Throughput Study in Supercritical Conditions. ChemSusChem 2011, 4, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Agrigento, P.; Al-Amsyar, S.M.; Sorée, B.; Taherimehr, M.; Gruttadauria, M.; Aprile, C.; Pescarmona, P.P. Synthesis and high-throughput testing of multilayered supported ionic liquid catalysts for the conversion of CO2 and epoxides into cyclic carbonates. Catal. Sci. Technol. 2014, 4, 1598–1607. [Google Scholar] [CrossRef]
- Calabrese, C.; Liotta, L.F.; Giacalone, F.; Gruttadauria, M.; Aprile, C. Supported Polyhedral Oligomeric Silsesquioxane-Based (POSS) Materials as Highly Active Organocatalysts for the Conversion of CO2. ChemCatChem 2019, 11, 560–567. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, A.S.; Lee, J.-C.; Hong, S.M.; Hwang, S.S.; Koo, C.M. Multifunctional Mesoporous Ionic Gels and Scaffolds Derived from Polyhedral Oligomeric Silsesquioxanes. ACS Appl. Mater. Interfaces 2017, 9, 3616–3623. [Google Scholar] [CrossRef] [PubMed]
- Akbari, Z.; Ghiaci, M. Heterogenization of a Green Homogeneous Catalyst: Synthesis and Characterization of Imidazolium Ionene/Br–Cl–@SiO2 as an Efficient Catalyst for the Cycloaddition of CO2 with Epoxides. Ind. Eng. Chem. Res. 2017, 56, 9045–9053. [Google Scholar] [CrossRef]
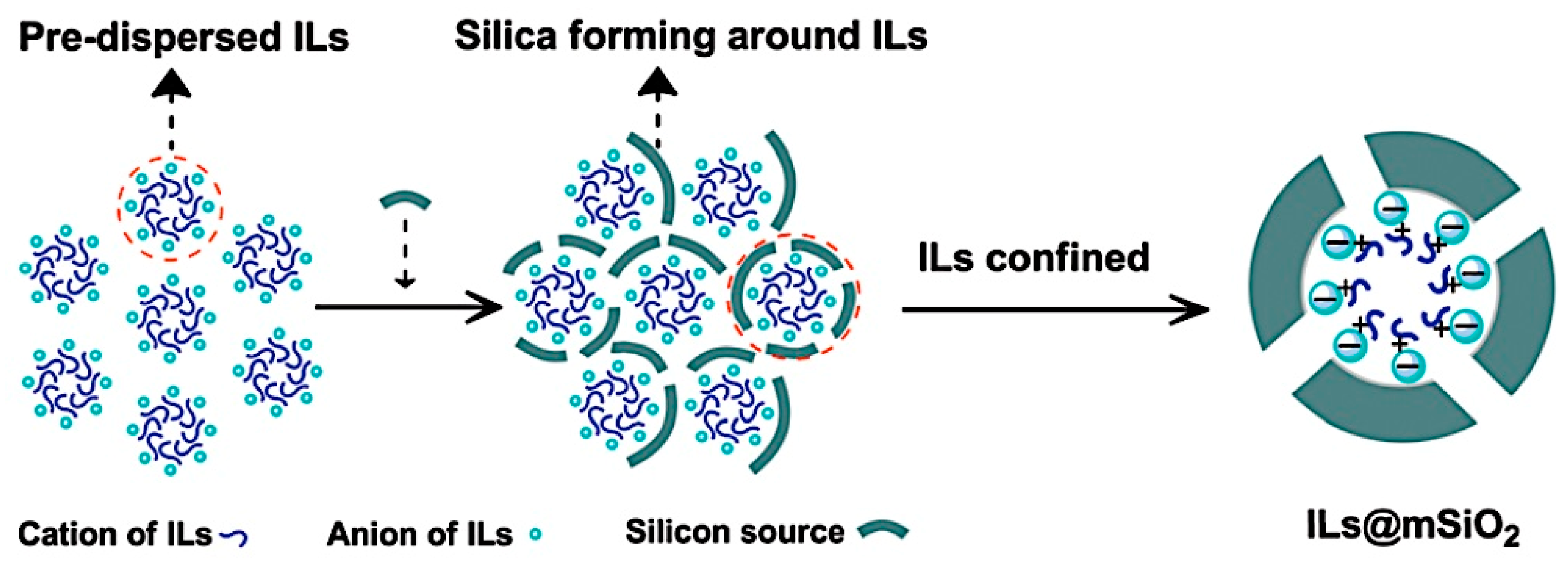
- Su, Q.; Qi, Y.; Yao, X.; Cheng, W.; Dong, L.; Chen, S.; Zhang, S. Ionic liquids tailored and confined by one-step assembly with mesoporous silica for boosting the catalytic conversion of CO2 into cyclic carbonates. Green Chem. 2018, 20, 3232–3241. [Google Scholar] [CrossRef]
- Takahashi, T.; Watahiki, T.; Kitazume, S.; Yasuda, H.; Sakakura, T. Synergistic hybrid catalyst for cyclic carbonate synthesis: Remarkable acceleration caused by immobilization of homogeneous catalyst on silica. Chem. Commun. 2006, 1664–1666. [Google Scholar] [CrossRef]
- Sakai, T.; Tsutsumi, Y.; Ema, T. Highly active and robust organic–inorganic hybrid catalyst for the synthesis of cyclic carbonates from carbon dioxide and epoxides. Green Chem. 2008, 10, 337–341. [Google Scholar] [CrossRef]
- Motokura, K.; Itagaki, S.; Iwasawa, Y.; Miyaji, A.; Baba, T. Silica-supported aminopyridinium halides for catalytic transformations of epoxides to cyclic carbonates under atmospheric pressure of carbon dioxide. Green Chem. 2009, 11, 1876–1880. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Tao, L.; Li, C.; Liu, L.; Chen, J.; Yang, Q. Cationic Zn-Porphyrin Immobilized in Mesoporous Silicas as Bifunctional Catalyst for CO2 Cycloaddition Reaction under Cocatalyst Free Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9237–9245. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Kirchon, A.; Feng, L.; Drake, H.F.; Joseph, E.A.; Zhou, H.-C. From fundamentals to applications: A toolbox for robust and multifunctional MOF materials. Chem. Soc. Rev. 2018, 47, 8611–8638. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Lu, Y.; Chen, K.; Zhao, Y.; Wang, P.; Sun, W.-Y. Metal–organic frameworks with catalytic centers: From synthesis to catalytic application. Coord. Chem. Rev. 2019, 378, 262–280. [Google Scholar] [CrossRef]
- Qiu, S.; Zhu, G. Molecular engineering for synthesizing novel structures of metal–organic frameworks with multifunctional properties. Coord. Chem. Rev. 2009, 253, 2891–2911. [Google Scholar] [CrossRef]
- Cui, W.-G.; Zhang, G.-Y.; Hu, T.-L.; Bu, X.-H. Metal-organic framework-based heterogeneous catalysts for the conversion of C1 chemistry: CO, CO2 and CH4. Coord. Chem. Rev. 2019, 387, 79–120. [Google Scholar] [CrossRef]
- Lei, Z.; Xue, Y.; Chen, W.; Qiu, W.; Zhang, Y.; Horike, S.; Tang, L. MOFs-Based Heterogeneous Catalysts: New Opportunities for Energy-Related CO2 Conversion. Adv. Energy Mater. 2018, 8, 1801587. [Google Scholar] [CrossRef]
- Alshammari, A.; Jiang, Z.; Cordova, K.E. Metal organic frameworks as emerging photocatalysts. In Semiconductor Photocatalysis: Materials, Mechanisms and Applications; Cao, W., Ed.; InTech: London, UK, 2016; pp. 302–341. [Google Scholar] [CrossRef]
- Noh, J.; Kim, D.; Lee, J.; Yoon, M.; Park, H.M.; Lee, M.K.; Kim, Y.; Kim, M. Three Component Controls in Pillared Metal-Organic Frameworks for Catalytic Carbon Dioxide Fixation. Catalysts 2018, 8, 565. [Google Scholar] [CrossRef]
- Zalomaeva, O.V.; Chibiryaev, A.M.; Kovalenko, K.A.; Kholdeeva, O.A.; Balzhinimaev, B.S.; Fedin, V.P. Cyclic carbonates synthesis from epoxides and CO2 over metal–organic framework Cr-MIL-101. J. Catal. 2013, 298, 179–185. [Google Scholar] [CrossRef]
- Kathalikkattil, A.C.; Roshan, R.; Tharun, J.; Babu, R.; Jeong, G.-S.; Kim, D.-W.; Cho, S.J.; Park, D.-W. A sustainable protocol for the facile synthesis of zinc-glutamate MOF: An efficient catalyst for room temperature CO2 fixation reactions under wet conditions. Chem. Commun. 2016, 52, 280–283. [Google Scholar] [CrossRef]
- Noh, J.; Kim, Y.; Park, H.; Lee, J.; Yoon, M.; Park, M.H.; Kim, Y.; Kim, M. Functional group effects on a metal-organic framework catalyst for CO2 cycloaddition. J. Ind. Eng. Chem. 2018, 64, 478–483. [Google Scholar] [CrossRef]
- Liang, J.; Xie, Y.-Q.; Wu, Q.; Wang, X.-Y.; Liu, T.-T.; Li, H.-F.; Huang, Y.-B.; Cao, R. Zinc Porphyrin/Imidazolium Integrated Multivariate Zirconium Metal–Organic Frameworks for Transformation of CO2 into Cyclic Carbonates. Inorg. Chem. 2018, 57, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, Y.; Qi, C.; Jiang, H. A chiral salen-based MOF catalytic material with high thermal, aqueous and chemical stabilities. Dalton Trans. 2017, 46, 7821–7832. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, J.; Ren, Y.; Jiang, H. A Ni(salen)-Based Metal–Organic Framework: Synthesis, Structure, and Catalytic Performance for CO2 Cycloaddition with Epoxides. Eur. J. Inorg. Chem. 2017, 2017, 4982–4989. [Google Scholar] [CrossRef]
- Tharun, J.; Bhin, K.-M.; Roshan, R.; Kim, D.W.; Kathalikkattil, A.C.; Babu, R.; Ahn, H.Y.; Won, Y.S.; Park, D.-W. Ionic liquid tethered post functionalized ZIF-90 framework for the cycloaddition of propylene oxide and CO2. Green Chem. 2016, 18, 2479–2487. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Dutta, S.; Hossain, M.S.A.; Shiddiky, M.J.A.; Tung, K.-L.; Shieh, F.-K.; Tsung, C.-K.; Wu, K.C.W.; Yamauchi, Y. Strategies for Improving the Functionality of Zeolitic Imidazolate Frameworks: Tailoring Nanoarchitectures for Functional Applications. Adv. Mater. 2017, 29, 1700213. [Google Scholar] [CrossRef]
- Bhin, K.M.; Tharun, J.; Roshan, K.R.; Kim, D.-W.; Chung, Y.; Park, D.-W. Catalytic performance of zeolitic imidazolate framework ZIF-95 for the solventless synthesis of cyclic carbonates from CO2 and epoxides. J. CO2 Util. 2017, 17, 112–118. [Google Scholar] [CrossRef]
- Zhou, Z.; He, C.; Xiu, J.; Yang, L.; Duan, C. Metal–Organic Polymers Containing Discrete Single-Walled Nanotube as a Heterogeneous Catalyst for the Cycloaddition of Carbon Dioxide to Epoxides. J. Am. Chem. Soc. 2015, 137, 15066–15069. [Google Scholar] [CrossRef] [PubMed]
- Su, D.S.; Perathoner, S.; Centi, G. Nanocarbons for the Development of Advanced Catalysts. Chem. Rev. 2013, 113, 5782–5816. [Google Scholar] [CrossRef]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef]
- Krueger, A. Carbon Materials and Nanotechnology; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- D’Souza, F.; Ito, O. Photosensitized electron transfer processes of nanocarbons applicable to solar cells. Chem. Soc. Rev. 2012, 41, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Su, D.S.; Schlögl, R. Nanostructured Carbon and Carbon Nanocomposites for Electrochemical Energy Storage Applications. ChemSusChem 2010, 3, 136–168. [Google Scholar] [CrossRef]
- Marchesan, S.; Prato, M. Nanomaterials for (Nano)medicine. ACS Med. Chem. Lett. 2013, 4, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Reina, G.; González-Domínguez, J.M.; Criado, A.; Vázquez, E.; Bianco, A.; Prato, M. Promises, facts and challenges for graphene in biomedical applications. Chem. Soc. Rev. 2017, 46, 4400–4416. [Google Scholar] [CrossRef] [PubMed]
- Martín, N. Carbon Nanoforms for Photovoltaics: Myth or Reality? Adv. Energy Mater. 2017, 7, 1601102. [Google Scholar] [CrossRef]
- Campisciano, V.; Gruttadauria, M.; Giacalone, F. Modified Nanocarbons for Catalysis. ChemCatChem 2019, 11, 90–133. [Google Scholar] [CrossRef]
- Schaetz, A.; Zeltner, M.; Stark, W.J. Carbon Modifications and Surfaces for Catalytic Organic Transformations. ACS Catal. 2012, 2, 1267–1284. [Google Scholar] [CrossRef]
- Sun, Y.-B.; Cao, C.-Y.; Yang, S.-L.; Huang, P.-P.; Wang, C.-R.; Song, W.-G. C60 fullerenol as an active and stable catalyst for the synthesis of cyclic carbonates from CO2 and epoxides. Chem. Commun. 2014, 50, 10307–10310. [Google Scholar] [CrossRef]
- Han, L.; Li, H.; Choi, S.-J.; Park, M.-S.; Lee, S.-M.; Kim, Y.-J.; Park, D.-W. Ionic liquids grafted on carbon nanotubes as highly efficient heterogeneous catalysts for the synthesis of cyclic carbonates. Appl. Catal. A 2012, 429–430, 67–72. [Google Scholar] [CrossRef]
- Baj, S.; Krawczyk, T.; Jasiak, K.; Siewniak, A.; Pawlyta, M. Catalytic coupling of epoxides and CO2 to cyclic carbonates by carbon nanotube-supported quaternary ammonium salts. Appl. Catal. A 2014, 488, 96–102. [Google Scholar] [CrossRef]
- Buaki-Sogó, M.; Vivian, A.; Bivona, L.A.; García, H.; Gruttadauria, M.; Aprile, C. Imidazolium functionalized carbon nanotubes for the synthesis of cyclic carbonates: Reducing the gap between homogeneous and heterogeneous catalysis. Catal. Sci. Technol. 2016, 6, 8418–8427. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Chen, J.; Yang, Q. Cationic Zn–Porphyrin Polymer Coated onto CNTs as a Cooperative Catalyst for the Synthesis of Cyclic Carbonates. ACS Appl. Mater. Interfaces 2018, 10, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, H.; Cao, F.; Ma, Y.; Qu, Y. Catalytic Behavior of Graphene Oxides for Converting CO2 into Cyclic Carbonates at One Atmospheric Pressure. ACS Sustain. Chem. Eng. 2018, 6, 4204–4211. [Google Scholar] [CrossRef]
- Saptal, V.B.; Sasaki, T.; Harada, K.; Nishio-Hamane, D.; Bhanage, B.M. Hybrid Amine-Functionalized Graphene Oxide as a Robust Bifunctional Catalyst for Atmospheric Pressure Fixation of Carbon Dioxide using Cyclic Carbonates. ChemSusChem 2016, 9, 644–650. [Google Scholar] [CrossRef]
- Lan, D.-H. Multi-functionalization of GO with multi-cationic ILs as high efficient metal-free catalyst for CO2 cycloaddition under mild conditions. Carbon 2018, 127, 245–254. [Google Scholar] [CrossRef]
- Lan, D.-H.; Chen, L.; Au, C.-T.; Yin, S.-F. One-pot synthesized multi-functional graphene oxide as a water-tolerant and efficient metal-free heterogeneous catalyst for cycloaddition reaction. Carbon 2015, 93, 22–31. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Wu, J.; Wu, H.; Zhang, W.-H.; Li, Y.-X. Graphene oxide immobilized with ionic liquids: Facile preparation and efficient catalysis for solvent-free cycloaddition of CO2 to propylene carbonate. RSC Adv. 2015, 5, 72361–72368. [Google Scholar] [CrossRef]
- Zhang, W.-H.; He, P.-P.; Wu, S.; Xu, J.; Li, Y.; Zhang, G.; Wei, X.-Y. Graphene oxide grafted hydroxyl-functionalized ionic liquid: A highly efficient catalyst for cycloaddition of CO2 with epoxides. Appl. Catal. A 2016, 509, 111–117. [Google Scholar] [CrossRef]
- Karousis, N.; Suarez-Martinez, I.; Ewels, C.P.; Tagmatarchis, N. Structure, Properties, Functionalization, and Applications of Carbon Nanohorns. Chem. Rev. 2016, 116, 4850–4883. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Liotta, L.F.; Carbonell, E.; Giacalone, F.; Gruttadauria, M.; Aprile, C. Imidazolium-Functionalized Carbon Nanohorns for the Conversion of Carbon Dioxide: Unprecedented Increase of Catalytic Activity after Recycling. ChemSusChem 2017, 10, 1202–1209. [Google Scholar] [CrossRef] [PubMed]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
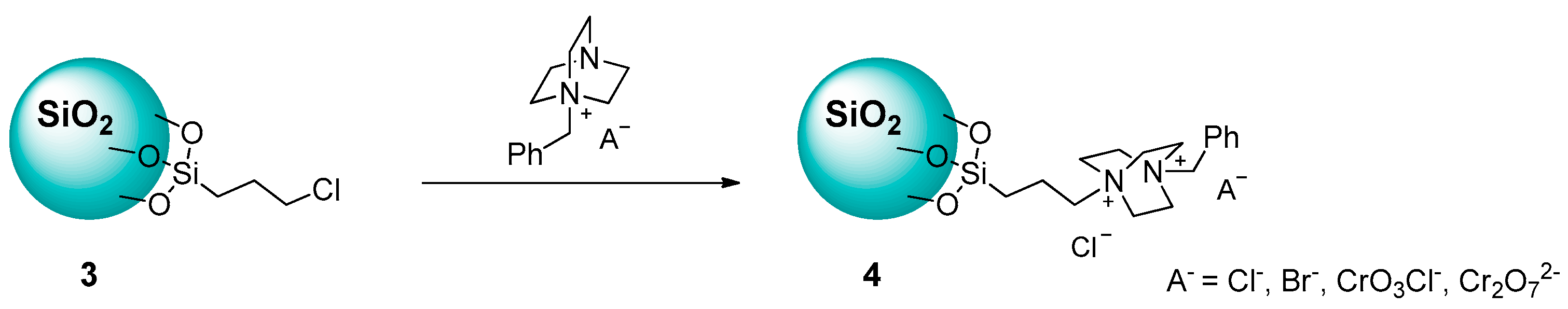{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Catalyst | Catalyst Loading | Conversion/Yields | Temperature | CO2 Pressure | Time | Reference |
|---|---|---|---|---|---|---|---|
| (mol%) | (%) | (°C) | (MPa) | (h) | |||
| 1 | 2 | 2 | 85 (Y) | 90 | 1.0 | 6 | [40] |
| 2 | 4 | 0.93 | 94 (Y) | 100 | 0.4 | 24 | [41] |
| 3 | 6 | 10 | 86 (Y) | r.t. | 0.1 | 24 | [42] |
| 4 | 7 | 10 | 99 (Y) | r.t. | 0.1 | 24 | [42] |
| 5 | 8 | 2 | 61 (Y) | 100 | 1.0 | 4 | [43] |
| 6 | 9 | 2 | 98 (Y) | 100 | 1.0 | 4 | [43] |
| 11 | 14 | 0.45 | 96 (Y) | 115 | 1.62 | 5 | [49] |
| 12 | 15d | 0.5 | 82 (Y) | 110 | 2.5 | 4 | [51] |
| 13 | 16b | 0.3 | 39 (C) | 125 | 4.0 | 3 | [52] |
| 14 | 18g | 0.43 | 99 (C) | 150 | 8.0 | 3 | [54] |
| 15 | 19 | 0.14 | 53 (C) | 150 | 4.0 | 3 | [55] |
| 17 | 20 | 0.25 | 98 (C) | 120 | 2.0 | 5 | [57] |
| 18 | ILs@mSiO2 | 0.26 | 89 (C) | 120 | 2.0 | 8 | [58] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrese, C.; Giacalone, F.; Aprile, C. Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates. Catalysts 2019, 9, 325. https://doi.org/10.3390/catal9040325
Calabrese C, Giacalone F, Aprile C. Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates. Catalysts. 2019; 9(4):325. https://doi.org/10.3390/catal9040325
Chicago/Turabian StyleCalabrese, Carla, Francesco Giacalone, and Carmela Aprile. 2019. "Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates" Catalysts 9, no. 4: 325. https://doi.org/10.3390/catal9040325
APA StyleCalabrese, C., Giacalone, F., & Aprile, C. (2019). Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates. Catalysts, 9(4), 325. https://doi.org/10.3390/catal9040325