Suzuki–Miyaura Coupling Using Monolithic Pd Reactors and Scaling-Up by Series Connection of the Reactors


Abstract
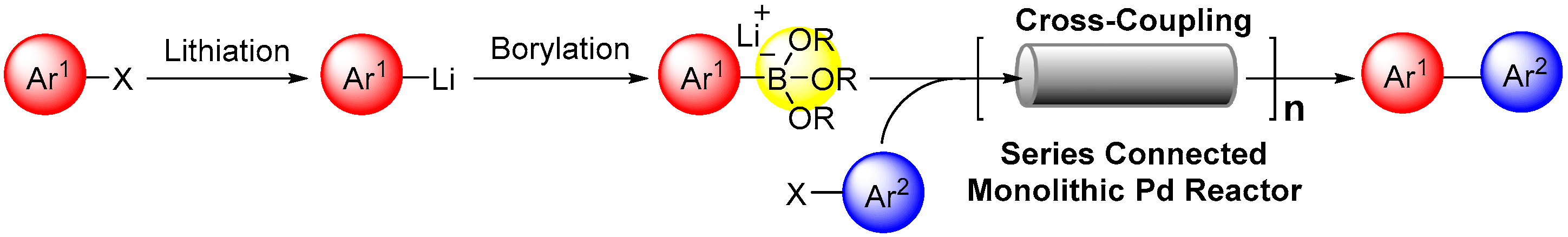
:1. Introduction
2. Results and Discussion
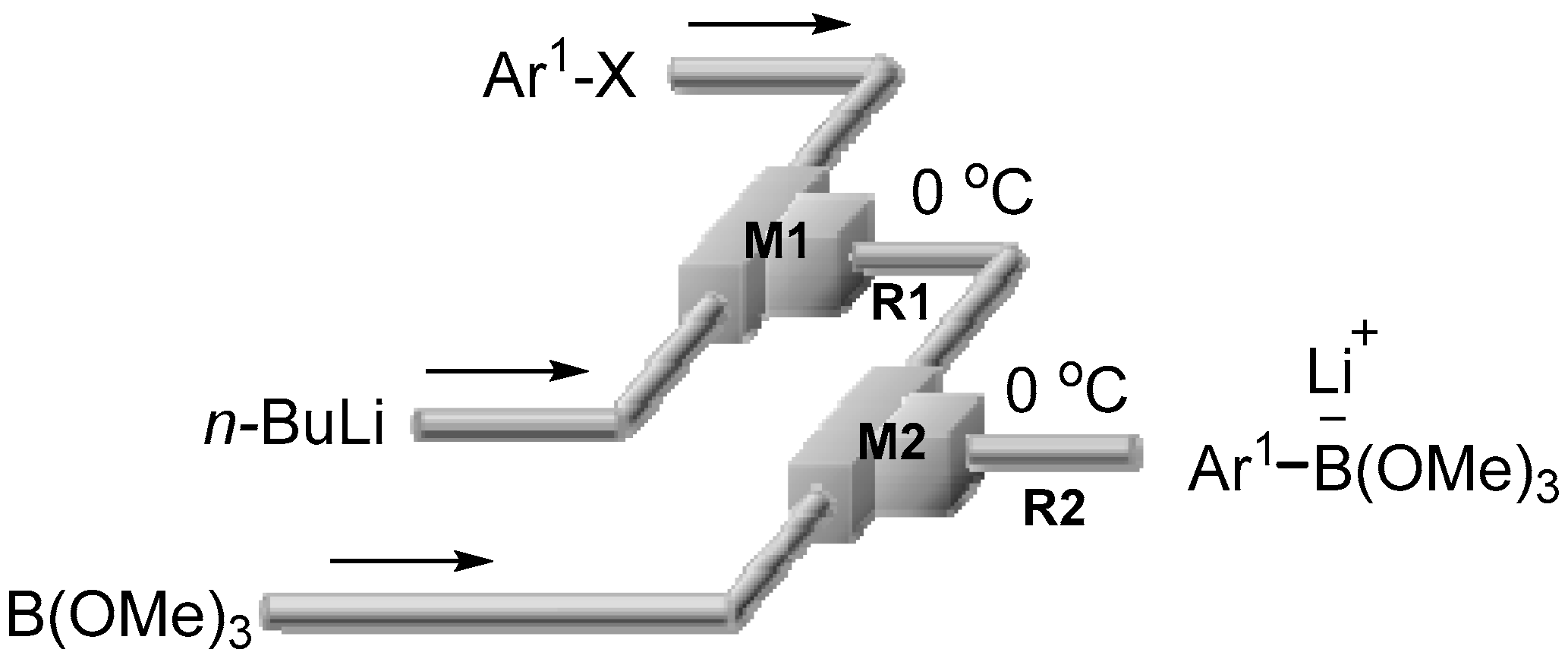
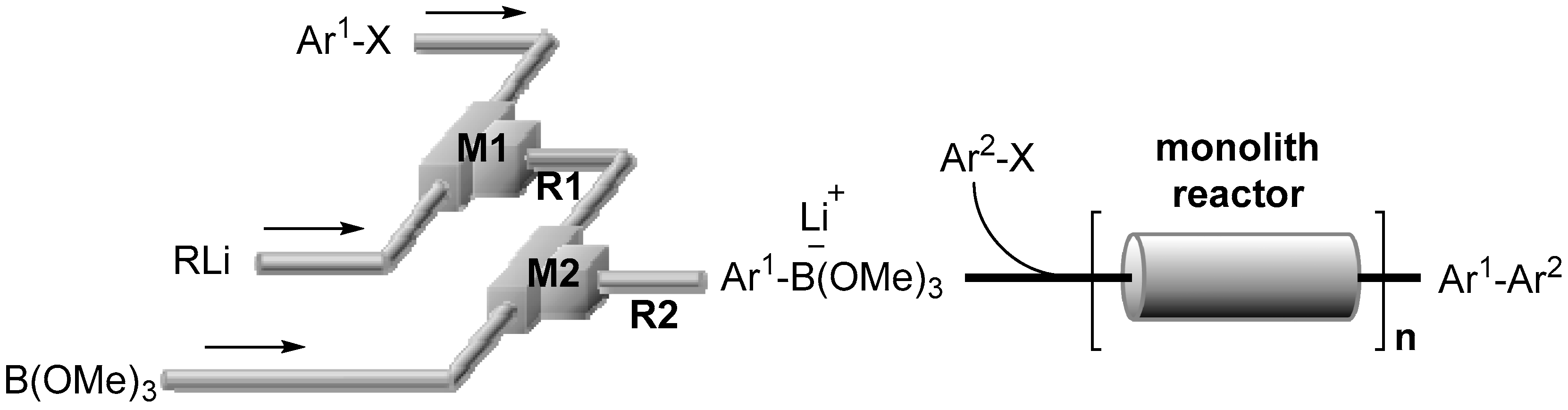
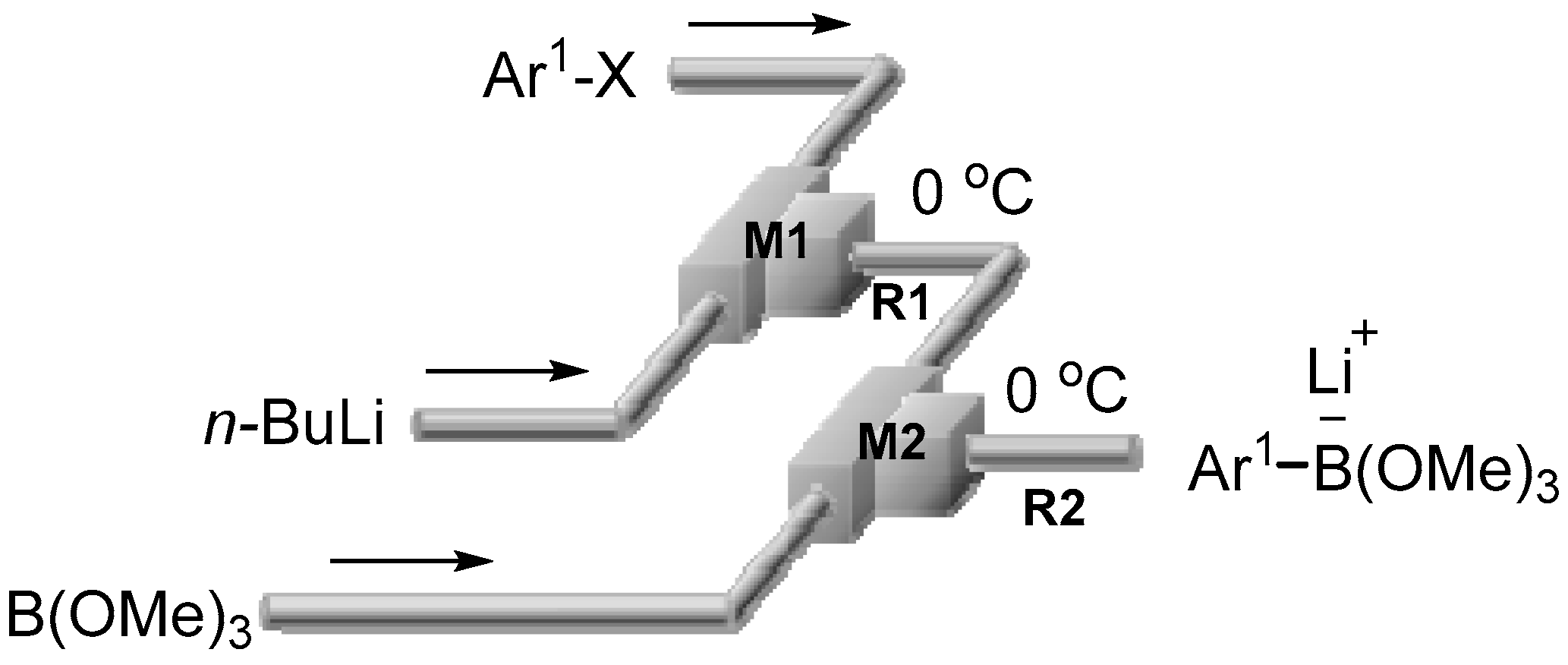
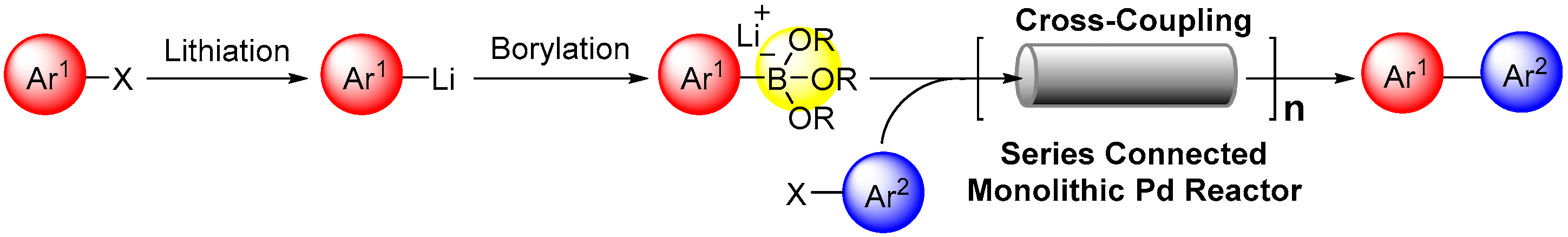
2.1. Preparation of Lithium Arylborates Using Flow Microreactors
2.2. Preparation of Polymer Monolith and Immobilization of Pd Catalyst
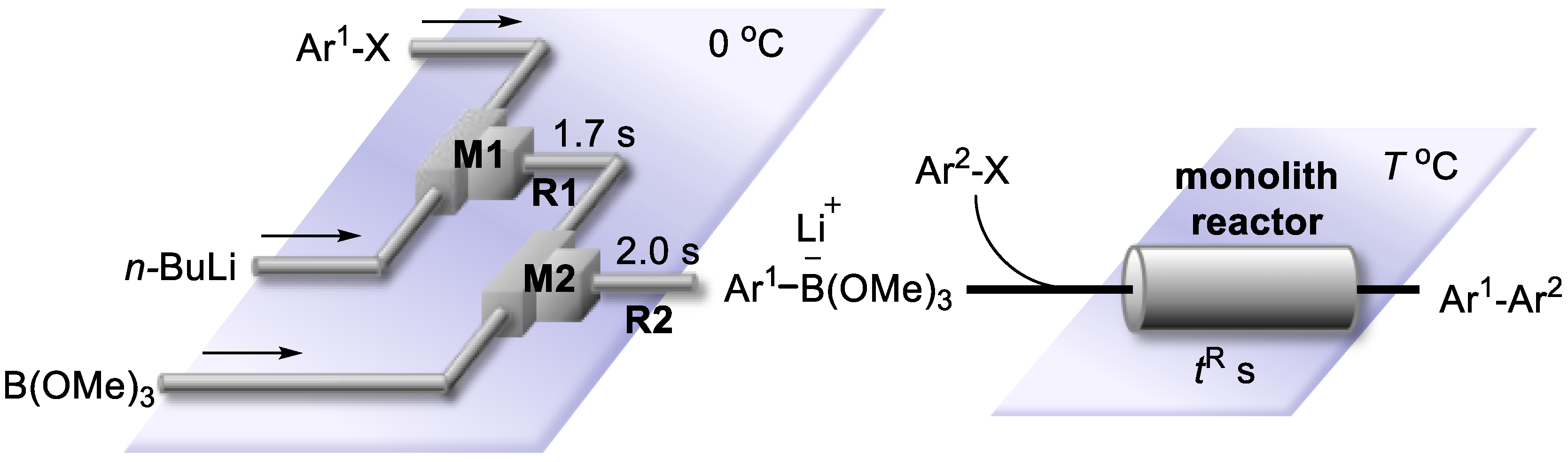
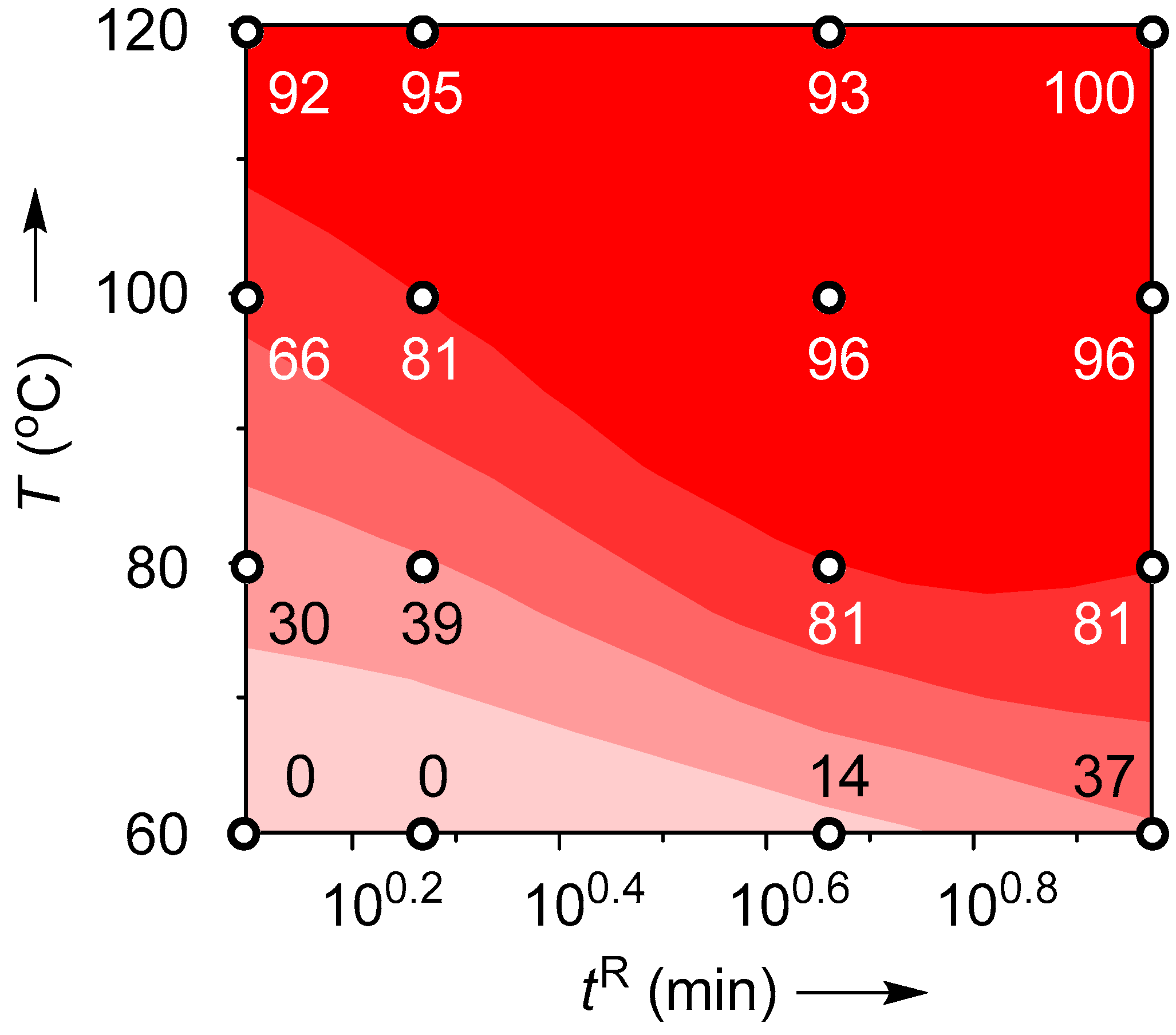
2.3. Suzuki–Miyaura Coupling Using the Pd Catalyst Supported by Polymer Monolith A
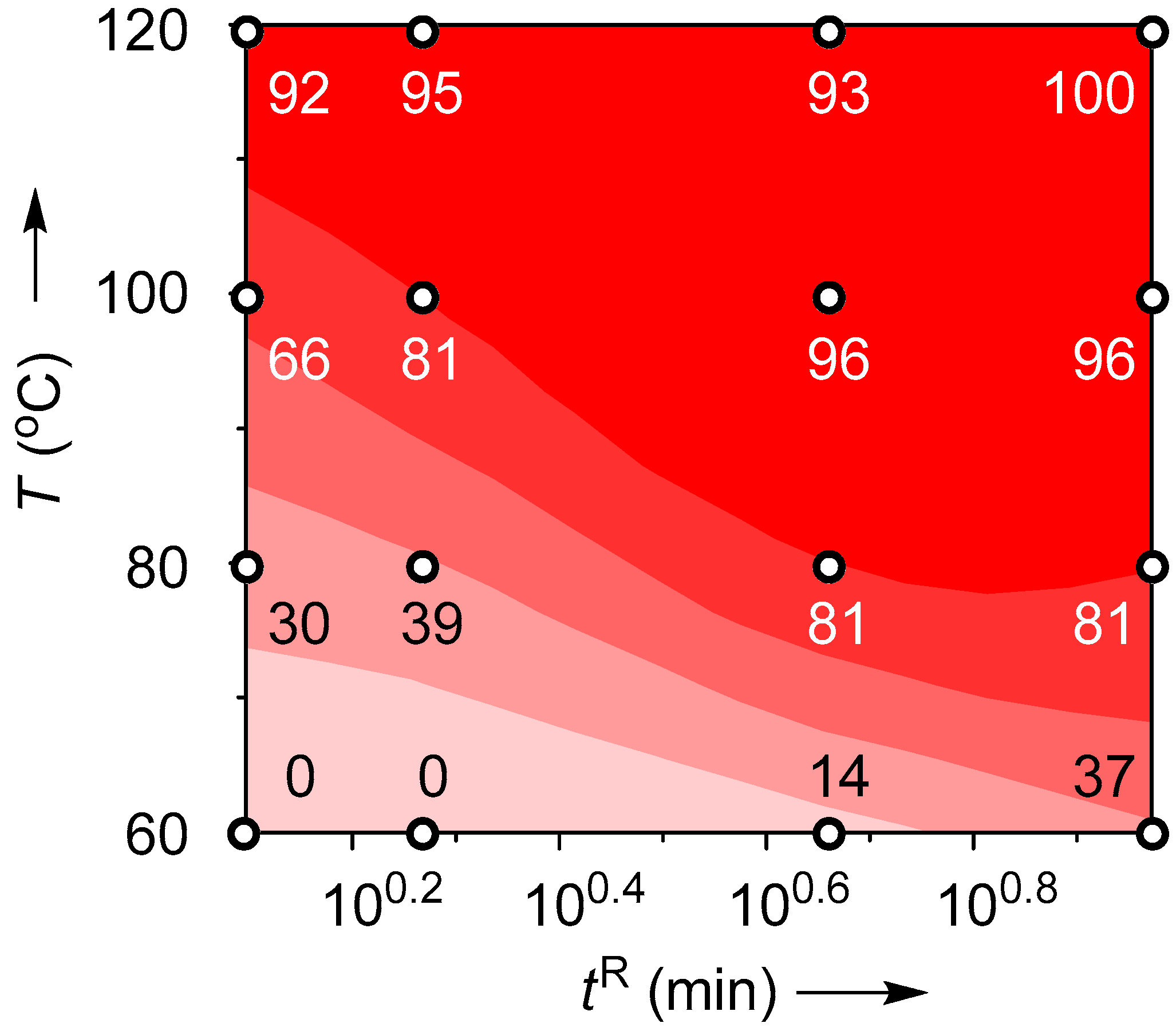
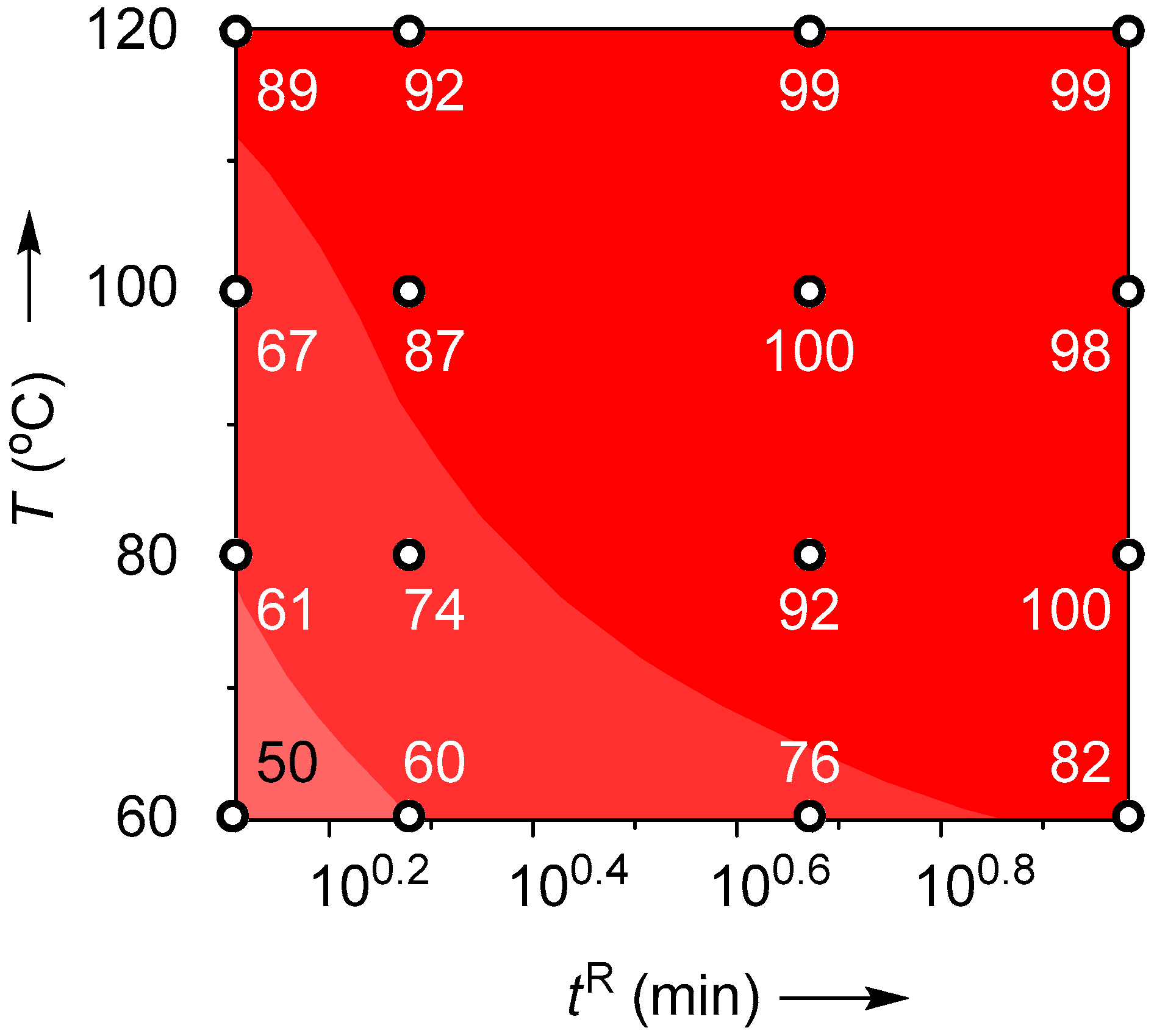
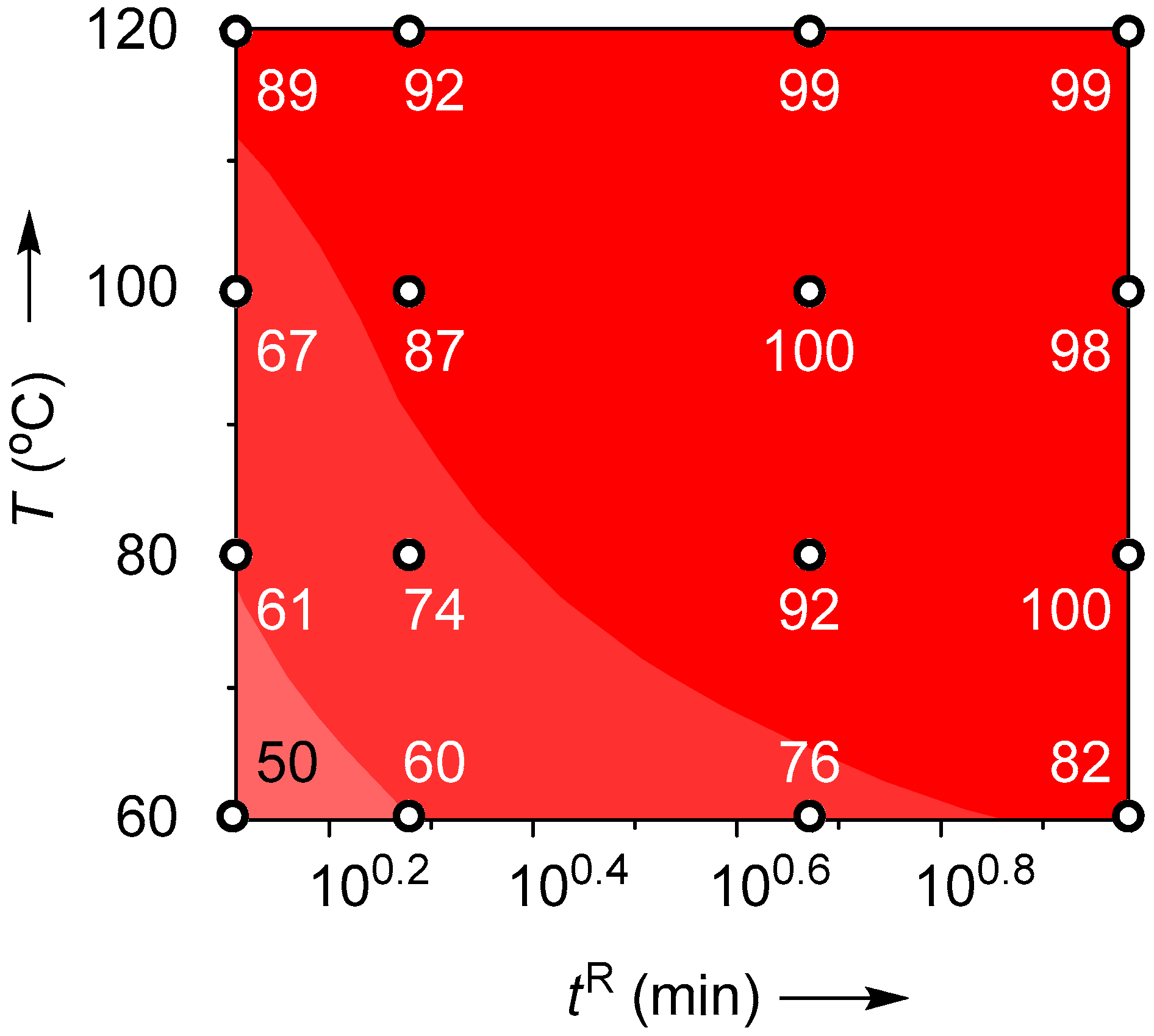
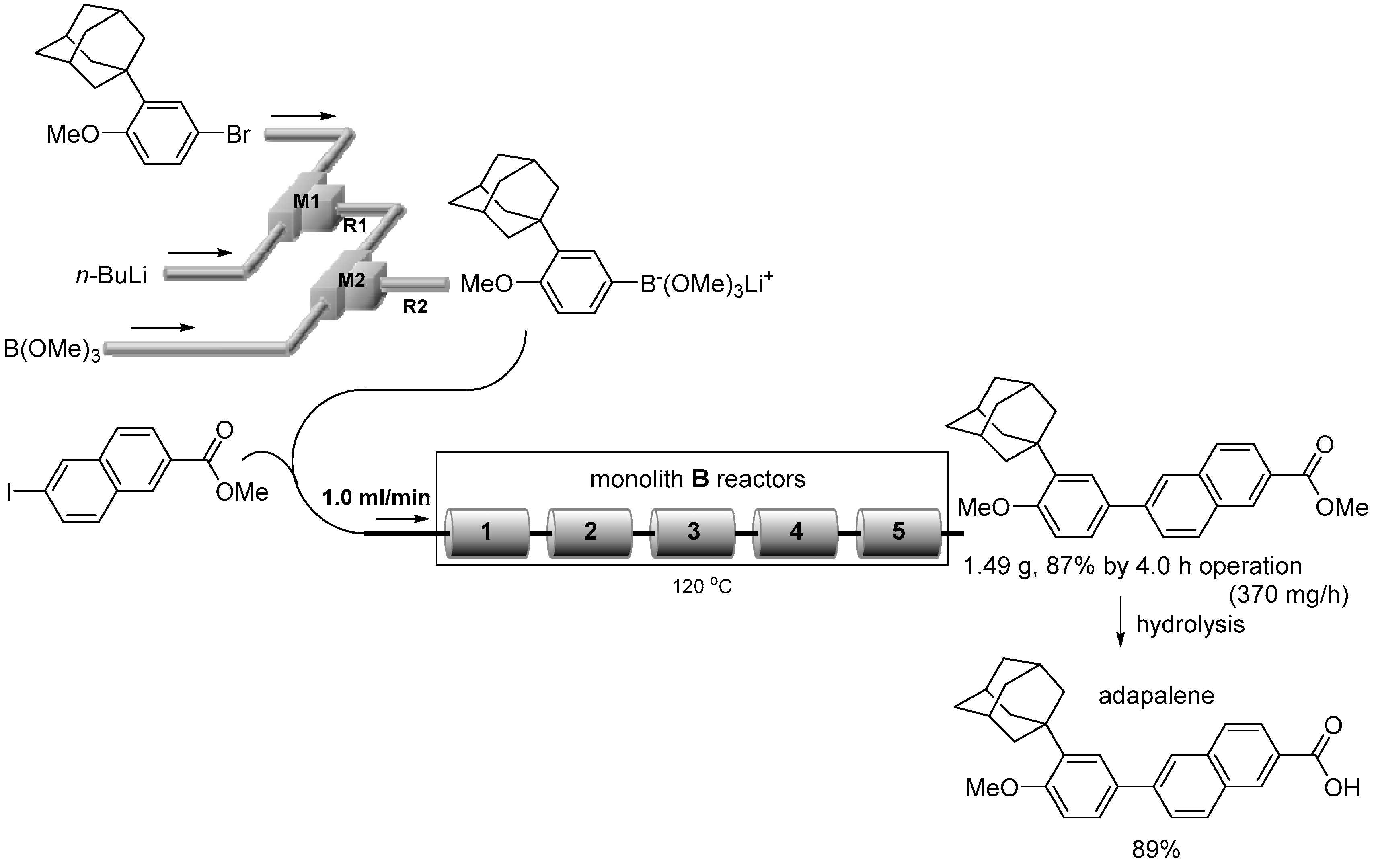
2.4. Suzuki–Miyaura Coupling Using the Pd Catalyst Supported by Monolith B
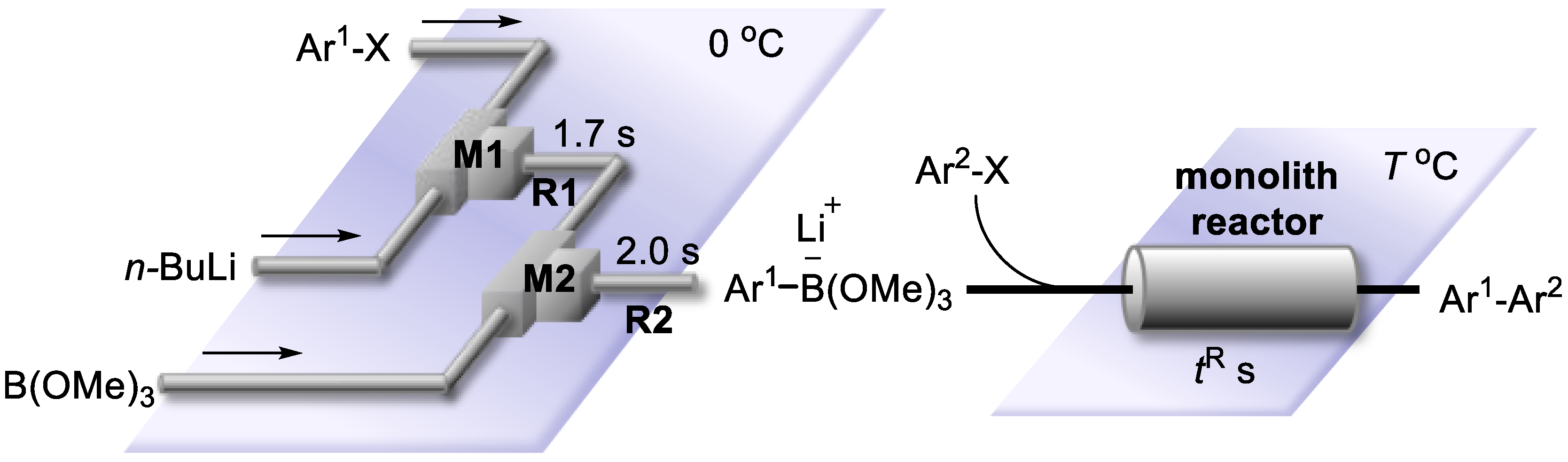
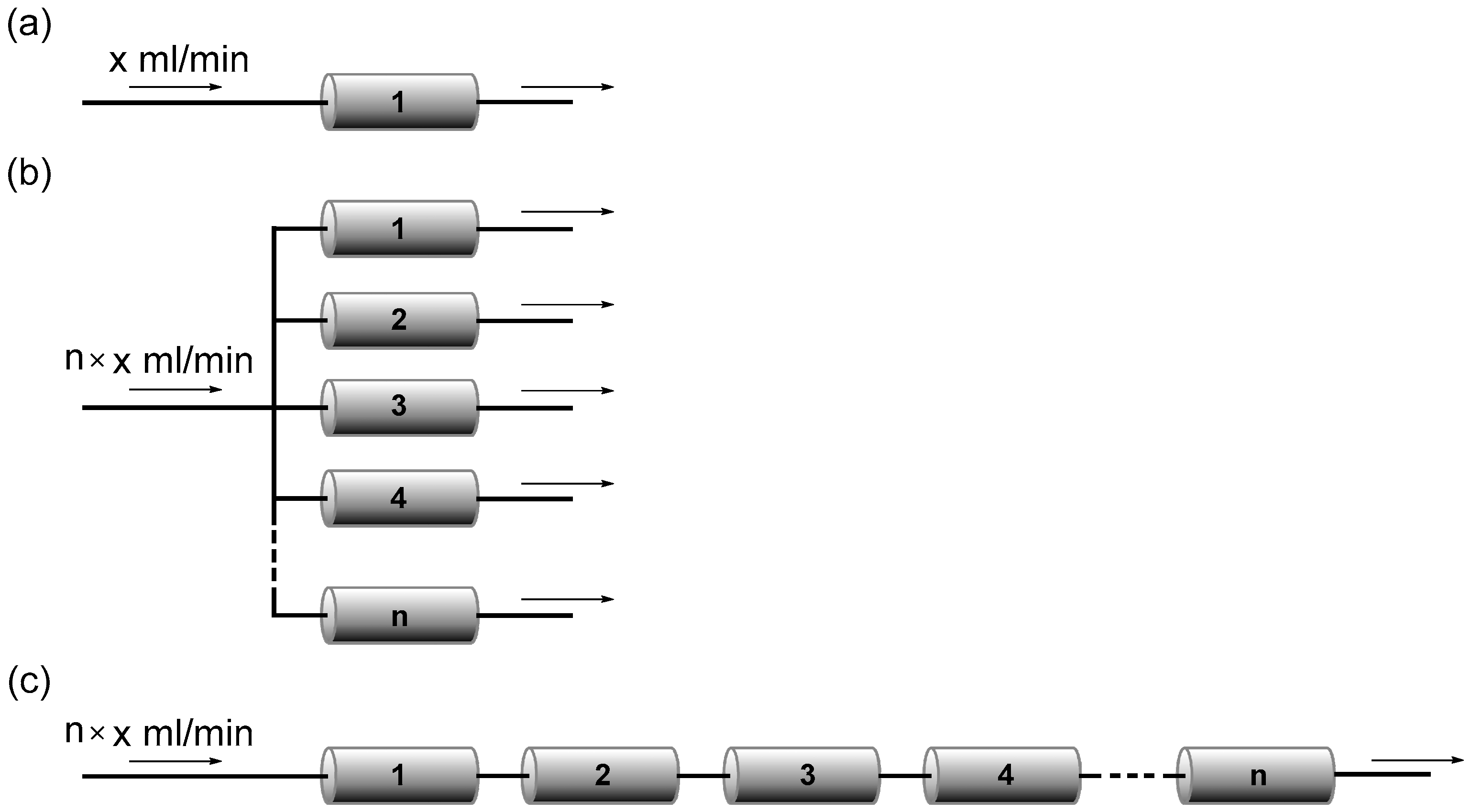
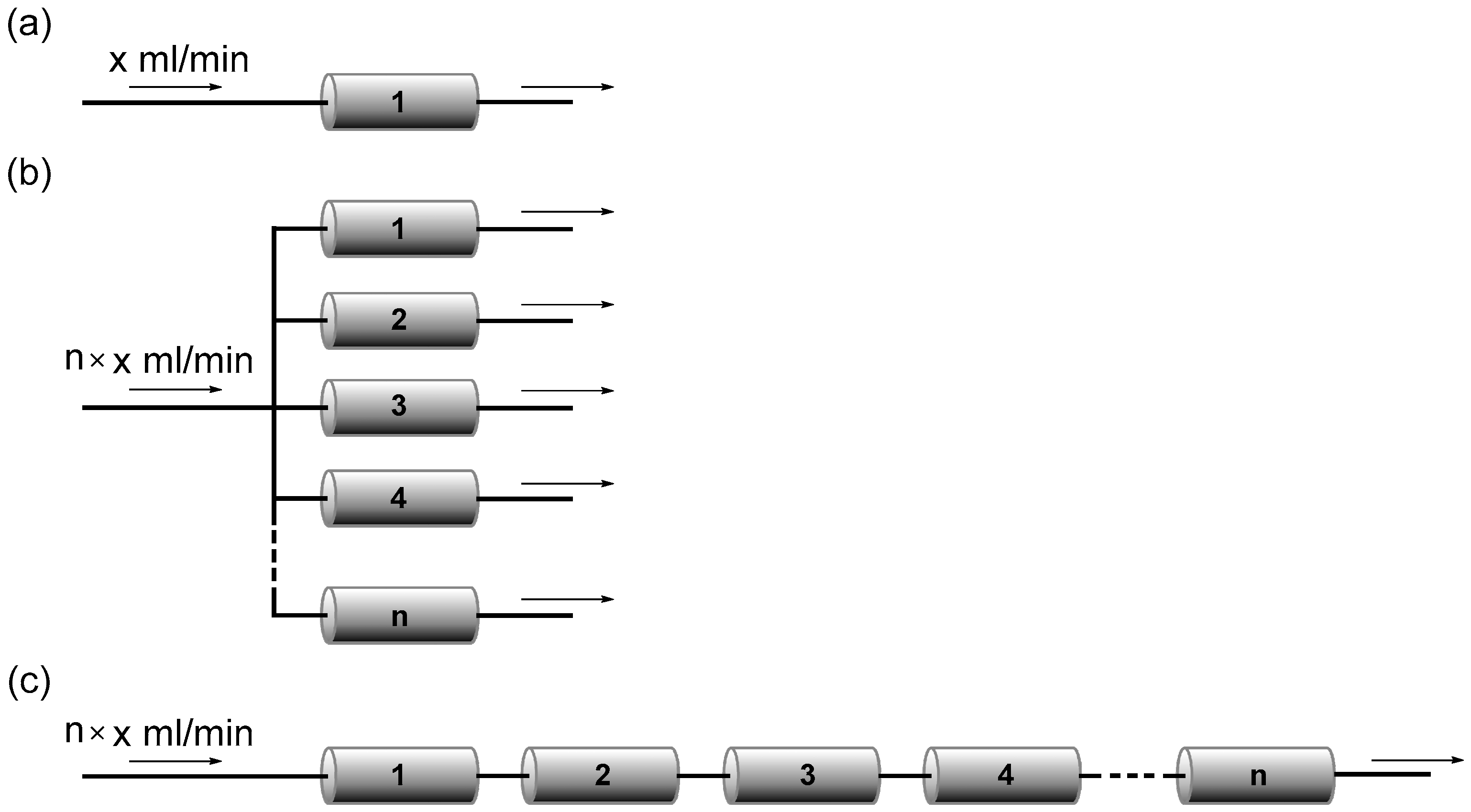
2.5. Series Connection
2.6. Recyclability of Monolithic Pd Catalysts
3. Materials and Methods
3.1. General
3.2. Preparation of the Pd Catalysts Supported by Monolith A and Monolith B
3.3. Cross-Coupling of Bromobenzene and p-Iodobenzonitrile by the Space Integation of Lithiation, Borylation, and Suzuki–Miyaura Coupling
3.4. Cross-Coupling of Aryl Bromides (Ar1–Br) and Aryl Halides (Ar2–X) by the Integration of Lithiation, Borylation and Suzuki–Miyaura Coupling in a Flow (Monolith A Reactor)
3.5. Cross-Coupling of Aryl Bromides (Ar1–Br) and Aryl Halides (Ar2–X) by the Integration of Lithiation, Borylation, and Suzuki–Miyaura Coupling Using a Single Monolith B Reactor or Three Monolith B Reactors Connected in Series
3.6. Cross-Coupling of Bromobenzene and p-Iodobenzonitrile by the Integration of Lithiation, Borylation, and Suzuki–Miyaura Coupling Using Multiple Monolith B Reactors Connected in Series
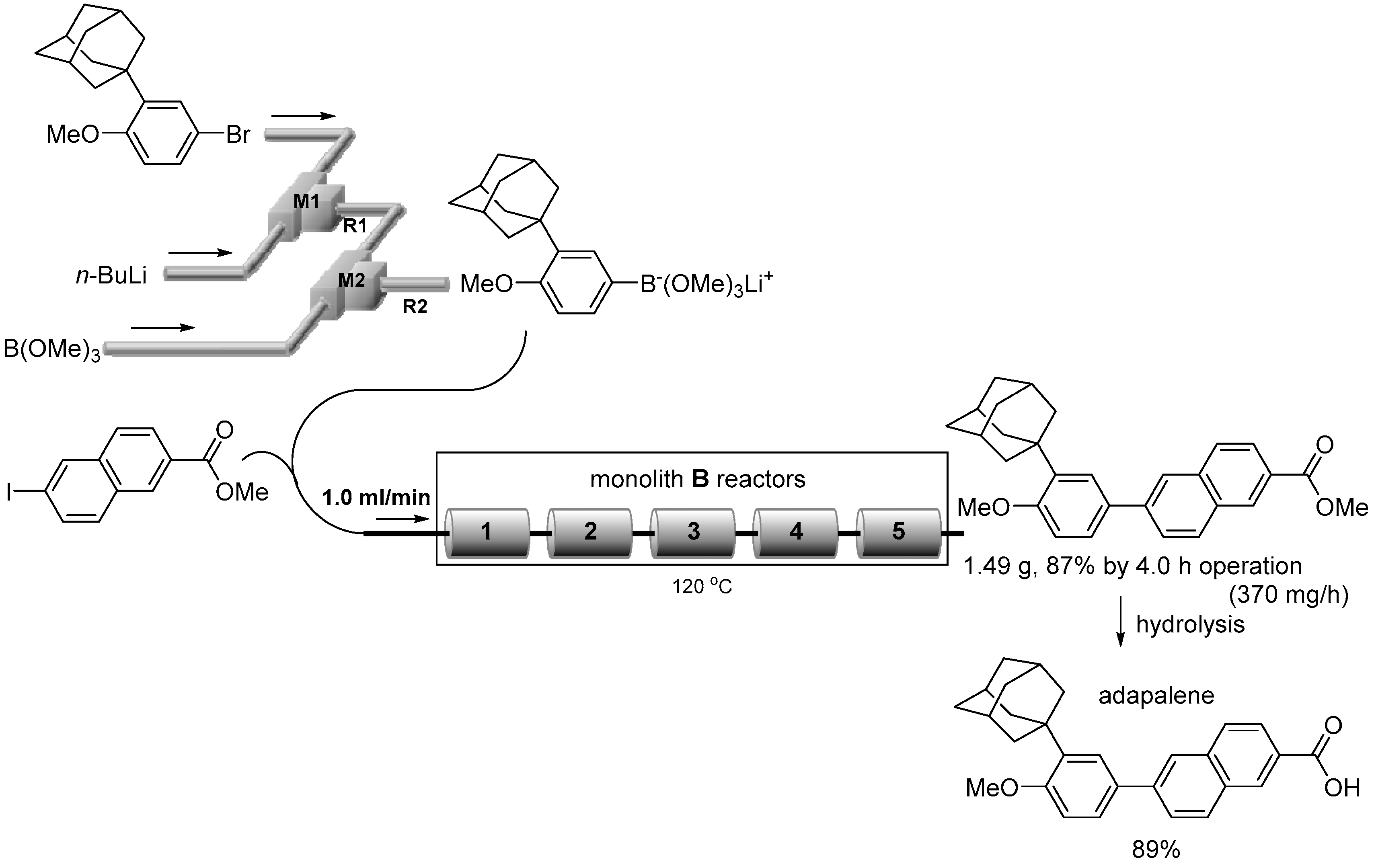
3.7. Synthesis of Adapalene
3.8. Recyclability of the Polymer Monolith A and Monolith B Reactors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Hessel, V.; Hardt, S.; Löwe, H. Chemical Micro Process Engineering; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Hessel, V.; Renken, A.; Schouten, J.C.; Yoshida, J. Micro Precess Engineering; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Wirth, T. Microreactors in Organic Chemistry and Catalysis; Wiley: New York, NY, USA, 2013. [Google Scholar]
- Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in microstructured reactors. Angew. Chem. Int. Ed. 2004, 43, 406–446. [Google Scholar] [CrossRef]
- Doku, G.N.; Verboom, W.; Reinhoudt, D.N.; Van den Berg, A. On-microchip multiphase chemistry—A review of microreactor design principles and reagent contacting modes. Tetrahedron 2005, 61, 2733–2742. [Google Scholar] [CrossRef]
- Yoshida, J.; Nagaki, A.; Iwasaki, T.; Suga, S. Enhancement of chemical selectivity by microreactors. Chem. Eng. Technol. 2005, 28, 259–266. [Google Scholar] [CrossRef]
- Watts, P.; Haswell, S.J. The application of micro reactors for organic synthesis. Chem. Soc. Rev. 2005, 34, 235–246. [Google Scholar] [CrossRef]
- Geyer, K.; Codée, J.D.C.; Seeberger, P.H. Microreactors as tools for synthetic Chemists-the chemists’ round-bottomed flask of the 21st century? Chem. Eur. J. 2006, 12, 8434–8442. [Google Scholar] [CrossRef]
- DeMello, A.J. Control and detection of chemical reactions in microfluidic systems. Nature 2006, 442, 394–402. [Google Scholar] [CrossRef]
- Song, H.; Chen, D.L.; Ismagilov, R.F. Reactions in droplets in microfluidic channels. Angew. Chem. Int. Ed. 2006, 45, 7336–7356. [Google Scholar] [CrossRef]
- Kobayashi, J.; Mori, Y.; Kobayashi, S. Multiphase organic synthesis in microchannel reactors. Chem. Asian J. 2006, 1, 22–35. [Google Scholar] [CrossRef]
- Brivio, M.; Verboom, W.; Reinhoudt, D.N. Miniaturized continuous flow reaction vessels: influence on chemical reactions. Lab Chip 2006, 6, 329–344. [Google Scholar] [CrossRef]
- Mason, B.P.; Price, K.E.; Steinbacher, J.L.; Bogdan, A.R.; McQuade, D.T. Greener approaches to organic synthesis using microreactor technology. Chem. Rev. 2007, 107, 2300–2318. [Google Scholar] [CrossRef]
- Ahmed-Omer, B.; Brandt, J.C.; Wirth, T. Advanced organic synthesis using microreactor technology. Org. Biomol. Chem. 2007, 5, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Watts, P.; Wiles, C. Recent advances in synthetic micro reaction technology. Chem. Commun. 2007, 443–467. [Google Scholar] [CrossRef]
- Fukuyama, T.; Rahman, M.T.; Sato, M.; Ryu, I. Adventures in inner space: microflow systems for practical organic synthesis. Synlett 2008, 151–163. [Google Scholar] [CrossRef]
- Hartman, R.L.; Jensen, K.F. Microchemical systems for continuous-flow synthesis. Lab Chip 2009, 9, 2495–2507. [Google Scholar] [CrossRef]
- McMullen, J.P.; Jensen, K.F. Integrated microreactors for reaction automation: new approaches to reaction development. Annu. Rev. Anal. Chem. 2010, 3, 19–42. [Google Scholar] [CrossRef]
- Yoshida, J.; Kim, H.; Nagaki, A. Green and sustainable chemical synthesis using flow microreactors. ChemSusChem 2011, 4, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Wiles, C.; Watts, P. Continuous flow reactors: a perspective. Green Chem. 2012, 14, 38–54. [Google Scholar] [CrossRef]
- Kirschning, A.; Kupracz, L.; Hartwig, J. New synthetic opportunities in miniaturized flow reactors with inductive heating. Chem. Lett. 2012, 41, 562–570. [Google Scholar] [CrossRef]
- Elvira, K.S.; iSolvas, X.C.; Wootton, R.C.R.; DeMello, A.J. The past, present and potential for microfluidic reactor technology in chemical synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef]
- Pastre, J.C.; Browne, D.L.; Ley, S.V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, I.R. The integration of flow reactors into synthetic organic chemistry. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. [Google Scholar] [CrossRef]
- Fukuyama, T.; Totoki, T.; Ryu, I. Carbonylation in microflow: close encounters of CO and reactive species. Green Chem. 2014, 16, 2042–2050. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow chemistry: Recent developments in the synthesis of pharmaceutical products. Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Gemoets, H.P.L.; Su, Y.; Shang, M.; Hessel, V.; Luque, R.; Noel, T. Liquid phase oxidation chemistry in continuous-flow microreactors. Chem. Soc. Rev. 2016, 45, 83–117. [Google Scholar] [CrossRef] [Green Version]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The hitchhiker’s guide to floe chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Cantillo, D.; Baghbanzadeh, M.; Kappe, C.O. In situ generated iron oxide nanocrystals as efficient and selective catalysts for the reduction of nitroarenes using a continuous flow method. Angew. Chem. Int. Ed. 2012, 51, 10190–10193. [Google Scholar] [CrossRef]
- Shu, W.; Buchwald, S.L. Enantioselective β-arylation of ketones enabled by lithiation/borylation/1,4-addition sequence under flow conditions. Angew. Chem. Int. Ed. 2012, 51, 5355–5358. [Google Scholar] [CrossRef]
- Lévesque, F.; Seeberger, P.H. Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem. Int. Ed. 2012, 51, 1706–1709. [Google Scholar] [CrossRef]
- Basavaraju, K.C.; Sharma, S.; Maurya, R.A.; Kim, D.P. Safe use of a toxic compound: Heterogeneous OsO4 catalysis in a nanobrush polymer microreactor. Angew. Chem. Int. Ed. 2013, 52, 6735–6738. [Google Scholar] [CrossRef]
- Brancour, C.; Fukuyama, T.; Mukai, Y.; Skrydstrup, T.; Ryu, I. Modernized low pressure carbonylation methods in batch and flow employing common acids as a CO source. Org. Lett. 2013, 15, 2794–2797. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.D.; Reiß, B.; Dai, C.; Stephenson, C.R.J. Batch to flow deoxygenation using visible light photoredox catalysis. Chem. Commun. 2013, 49, 4352–4354. [Google Scholar] [CrossRef]
- Battilocchio, C.; Hawkins, J.M.; Ley, S.V. A Mild and efficient flow procedure for the transfer hydrogenation of ketones and aldehydes using hydrous zirconia. Org. Lett. 2013, 15, 2278–2281. [Google Scholar] [CrossRef]
- Kleinke, A.S.; Jamison, T.F. Hydrogen-fee alkene reduction in continuous flow. Org. Lett. 2013, 15, 710–713. [Google Scholar] [CrossRef]
- Asano, K.; Uesugi, Y.; Yoshida, J. Pauson–Khand reactions in a photochemical flow microreactor. Org. Lett. 2013, 15, 2398–2401. [Google Scholar] [CrossRef] [PubMed]
- Guetzoyan, L.; Nikbin, N.; Baxendale, I.R.; Ley, S.V. Flow chemistry synthesis of zolpidem, alpidem and other GABAA agonists and their biological evaluation through the use of in-line frontal affinity chromatography. Chem. Sci. 2013, 4, 764–769. [Google Scholar] [CrossRef]
- Fuse, S.; Mifune, Y.; Takahashi, T. Efficient amide bond formation through a rapid and strong activation of carboxylic acids in a microflow reactor. Angew. Chem. Int. Ed. 2014, 53, 851–855. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Jamison, T.F. Continuous-flow synthesis of functionalized phenols by aerobic oxidation of Grignard reagents. Angew. Chem. Int. Ed. 2014, 53, 3353–3357. [Google Scholar] [CrossRef]
- Nagaki, A.; Takahashi, Y.; Yoshida, J. Extremely fast gas/liquid reactions in flow microreactors: Carboxylation of short-lived organolithiums. Chem. Eur. J. 2014, 20, 7931–7934. [Google Scholar] [CrossRef]
- Chen, M.; Ichikawa, S.; Buchwald, S.L. Rapid and efficient copper-catalyzed finkelstein reaction of (hetero)aromatics under continuous-flow conditions. Angew. Chem. Int. Ed. 2015, 54, 263–266. [Google Scholar] [CrossRef]
- Nagaki, A.; Takahashi, Y.; Yoshida, J. Generation and reaction of carbamoyl anions in flow: Applications in the three-component synthesis of functionalized α-ketoamides. Angew. Chem. Int. Ed. 2016, 55, 5327–5331. [Google Scholar] [CrossRef] [PubMed]
- Fuse, S.; Mifune, Y.; Nakamura, H.; Tanaka, H. Total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation. Nat. Commun. 2016, 7, 13491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Katcher, M.H.; Jamison, T.F. Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow. Nat. Chem. 2017, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Kim, H.; Yoshida, J. Aryllithium compounds bearing alkoxycarbonyl groups. Generation and reactions using a microflow system. Angew. Chem. Int. Ed. 2008, 47, 7833–7836. [Google Scholar] [CrossRef]
- Nagaki, A.; Kim, H.; Yoshida, J. Nitro-substituted aryl lithium compounds in microreactor synthesis: Switch between kinetic and thermodynamic control. Angew. Chem. Int. Ed. 2009, 48, 8063–8065. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Kim, H.; Moriwaki, Y.; Matsuo, C.; Yoshida, J. A flow microreactor system enables organolithium reactions without protecting alkoxycarbonyl groups. Chem. Eur. J. 2010, 16, 11167–11177. [Google Scholar] [CrossRef]
- Kim, H.; Nagaki, A.; Yoshida, J. A flow-microreactor approach to protecting-group-free synthesis using organolithium compounds. Nat. Commun. 2011, 2, 264. [Google Scholar] [CrossRef] [Green Version]
- Tomida, Y.; Nagaki, A.; Yoshida, J. Asymmetric carbolithiation of conjugated enynes: A flow microreactor enables the use of configurationally unstable intermediates before they epimerize. J. Am. Chem. Soc. 2011, 133, 3744–3747. [Google Scholar] [CrossRef]
- Nagaki, A.; Matsuo, C.; Kim, S.; Saito, K.; Miyazaki, A.; Yoshida, J. Lithiation of 1,2-dichloroethene in flow microreactors: Versatile synthesis of alkenes and alkynes by precise residence-time control. Angew. Chem. Int. Ed. 2012, 51, 3245–3248. [Google Scholar] [CrossRef]
- Nagaki, A.; Ichinari, D.; Yoshida, J. Three-Component coupling based on flash chemistry. Carbolithiation of benzyne with functionalized aryllithiums followed by reactions with electrophiles. J. Am. Chem. Soc. 2014, 136, 12245–12248. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Tsuchihashi, Y.; Haraki, S.; Yoshida, J. Benzyllithiums bearing aldehyde carbonyl groups. A flash chemistry approach. Org. Biomol. Chem. 2015, 13, 7140–7145. [Google Scholar] [CrossRef] [Green Version]
- Nagaki, A.; Imai, K.; Ishiuchi, S.; Yoshida, J. Reactions of difunctional electrophiles with functionalized aryllithium compounds: Remarkable chemoselectivity by flash chemistry. Angew. Chem. Int. Ed. 2015, 54, 1914–1918. [Google Scholar] [CrossRef]
- Nagaki, A.; Kim, S.; Miuchi, N.; Yamashita, H.; Hirose, K.; Yoshida, J. Switching between intermolecular and intramolecular reactions using flow microreactors: Lithiation of 2-bromo-2′-silylbiphenyls. Org. Chem. Front. 2016, 3, 1250–1253. [Google Scholar] [CrossRef]
- Yoshida, J.; Kim, H.; Nagaki, A. “Impossible” chemistries based on flow and micro. J. Flow. Chem. 2017, 7, 60–64. [Google Scholar] [CrossRef]
- Diederich, F.; Stang, P.J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Stanforth, S.P. Catalytic cross-coupling reactions in biaryl synthesis. Tetrahedron 1998, 54, 263–303. [Google Scholar] [CrossRef]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−aryl bond formation one century after the discovery of the ullmann reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-catalyzed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- Gildner, P.G.; Thomas, J. Colacot, T.J. Reactions of the 21st century: Two decades of innovative catalyst design for palladium-catalyzed cross-couplings. Organometallics 2015, 34, 5497–5508. [Google Scholar] [CrossRef]
- Roy, D.; Uozumi, Y. Recent advances in palladium-catalyzed cross-coupling reactions at ppm to ppb molar catalyst loadings. Adv. Synth. Catal. 2018, 360, 602–625. [Google Scholar] [CrossRef]
- Mennecke, K.; Sodolenko, W.; Kirschning, A. Carbon-carbon cross-coupling reactions under continuous flow conditions using poly(vinylpyridine) doped with palladium. Synthesis 2008, 1589–1599. [Google Scholar] [CrossRef]
- Muñoz, J.M.; Alcázar, J.; de la Hoz, A.; Díaz-Ortiz, A. Cross-coupling in flow using supported catalysts: Mild, clean, efficient and sustainable Suzuki–Miyaura coupling in a single pass. Adv. Synth. Catal. 2012, 354, 3456–3460. [Google Scholar] [CrossRef]
- Pavia, C.; Ballerini, E.; Bivona, L.A.; Giacalone, F.; Aprile, C.; Vaccaro, L.; Gruttadauria, M. Palladium supported on cross-linked imidazolium network on silica as highly sustainable catalysts for the Suzuki reaction under flow conditions. Adv. Synth. Catal. 2013, 355, 2007–2018. [Google Scholar] [CrossRef]
- Tanimu, A.; Jaenicke, S.; Alhooshani, K. Heterogeneous catalysis in continuous flow microreactors: A review of methods and applications. Chem. Eng. J. 2017, 327, 792–821. [Google Scholar] [CrossRef]
- Masuda, K.; Ichitsuka, T.; Koumura, N.; Sato, K.; Kobayashi, S. Flow fine synthesis with heterogeneous catalysts. Tetrahedron 2018, 74, 1705–1730. [Google Scholar] [CrossRef]
- Solodenko, W.; Wen, H.; Leue, S.; Stuhlmann, F.; Sourkouni-Argirusi, G.; Jas, G.; Schönfeld, H.; Kunz, U.; Kirschning, A. Development of a continuous-flow system for catalysis with palladium(0) particles. Eur. J. Org. Chem. 2004, 3601–3610. [Google Scholar] [CrossRef]
- Kunz, U.; Kirschning, A.; Wen, H.L.; Solodenko, W.; Cecilia, R.; Kappe, C.O.; Turek, T. Monolithic polymer/carrier materials: Versatile composites for fine chemical synthesis. Catal. Today 2005, 105, 318–324. [Google Scholar] [CrossRef]
- Michrowska, A.; Mennecke, K.; Kunz, U.; Kirschning, A.; Grela, K. A new concept for the noncovalent binding of a ruthenium-based olefin metathesis catalyst to polymeric phases: Preparation of a catalyst on raschig rings. J. Am. Chem. Soc. 2006, 128, 13261–13267. [Google Scholar] [CrossRef]
- Nikbin, N.; Ladlow, M.; Ley, S.V. Continuous flow ligand-free heck reactions using monolithic Pd [0] nanoparticles. Org. Process Res. Dev. 2007, 11, 458–462. [Google Scholar] [CrossRef]
- Mennecke, K.; Cecilia, R.; Glasnov, T.N.; Gruhl, S.; Vogt, C.; Feldhoff, A.; Vargas, M.A.L.; Kappe, C.O.; Kunz, U.; Kirschning, A. Palladium(0) nanoparticles on glass-polymer composite materials as recyclable catalysts: A comparison study on their use in batch and continuous flow processes. Adv. Synth. Catal. 2008, 350, 717–730. [Google Scholar] [CrossRef]
- Kundu, D.; Patra, A.K.; Sakamoto, J.; Uyama, H. A palladium-loaded mesoporous polymer monolith as reusable heterogeneous catalyst for cross-coupling reactions. React. Funct. Polym. 2014, 79, 8–13. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Immobilized transition metals as catalysts for cross-couplings in continuous flow—A critical assessment of the reaction mechanism and metal leaching. ChemCatChem 2014, 6, 3286–3305. [Google Scholar] [CrossRef]
- Munirathinam, R.; Huskens, J.; Verboom, W. Supported catalysis in continuous-flow microreactors. Adv. Synth. Catal. 2015, 357, 1093–1123. [Google Scholar] [CrossRef]
- Ramarao, C.; Ley, S.V.; Smith, S.C.; Shirley, I.M.; DeAlmeida, N. Encapsulation of palladium in polyurea microcapsules. Chem. Commun. 2002, 1132–1133. [Google Scholar] [CrossRef]
- Lee, C.K.Y.; Holmes, A.B.; Ley, S.V.; McConvey, I.F.; Al-Duri, B.; Leeke, G.A.; Santos, R.C.D.; Seville, J.P.K. Efficient batch and continuous flow Suzuki cross-coupling reactions under mild conditions, catalysed by polyurea-encapsulated palladium (II) acetate and tetra-n-butylammonium salts. Chem. Commun. 2005, 2175–2177. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Griffiths-Jones, C.M.; Ley, S.V.; Tranmer, G.K. Microwave-assisted Suzuki coupling reactions with an encapsulated palladium catalyst for batch and continuous-flow transformations. Chem. Eur. J. 2006, 12, 4407–4416. [Google Scholar] [CrossRef]
- Phan, N.T.S.; Brown, D.H.; Styring, P. A facile method for catalyst immobilisation on silica: nickel-catalysed Kumada reactions in mini-continuous flow and batch reactors. Green Chem. 2004, 6, 526–532. [Google Scholar] [CrossRef]
- Al-Hashimi, M.; Qazi, A.; Sullivan, A.C.; Wilson, J.R.H. Dithio palladium modified silicas—New heterogeneous catalysts for Suzuki cross-coupling reactions. J. Mol. Catal. A 2007, 278, 160–164. [Google Scholar] [CrossRef]
- Lim, J.; Riduan, S.N.; Lee, S.S.; Ying, J.Y. Siliceous mesocellular foam-supported aza(bisoxazoline)-copper catalysts. Adv. Synth. Catal. 2008, 350, 1295–1308. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Len, C.; Fihri, A. Silica-supported palladium: Sustainable catalysts for cross-coupling reactions. Coord. Chem. Rev. 2009, 253, 2599–2626. [Google Scholar] [CrossRef]
- Ceylan, S.; Friese, C.; Lammel, C.; Mazac, K.; Kirschning, A. Inductive heating for organic synthesis by using functionalized magnetic nanoparticles inside microreactors. Angew. Chem. Int. Ed. 2008, 47, 8950–8953. [Google Scholar] [CrossRef] [PubMed]
- Laska, U.; Frost, C.G.; Price, G.J.; Plucinski, P.K. Easy-separable magnetic nanoparticle-supported Pd catalysts: Kinetics, stability and catalyst re-use. J. Catal. 2009, 268, 318–328. [Google Scholar] [CrossRef]
- Houlding, T.K.; Gao, P.; Degirmenci, V.; Tchabanenko, K.; Rebrov, E.V. Mechanochemical synthesis of TiO2/NiFe2O4 magnetic catalysts for operation under RF field. Mater. Sci. Eng. B 2015, 193, 175–180. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Kashinath, D. Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar] [CrossRef] [Green Version]
- Maluenda, I.; Navarro, O. Recent developments in the Suzuki-Miyaura reaction: 2010–2014. Molecules 2015, 20, 7528–7557. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ward, T.R. Recent advances in the palladium catalyzed Suzuki–Miyaura cross-coupling reaction in water. Catal. Lett. 2016, 146, 820–840. [Google Scholar] [CrossRef]
- Ishiyama, T.; Itoh, Y.; Kitano, T.; Miyaura, N. Synthesis of arylboronates via the palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett. 1997, 38, 3447–3450. [Google Scholar] [CrossRef] [Green Version]
- Giroux, A.; Han, Y.; Prasit, P. One pot biaryl synthesis via in situ boronate formation. Tetrahedron Lett. 1997, 38, 3841–3844. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Chemistry of Group 13 element-transition metal linkage—The platinum- and palladium-catalyzed reactions of (alkoxo)diborons. J. Organomet. Chem. 2000, 611, 392–402. [Google Scholar] [CrossRef]
- Willis, D.M.; Strongin, R.M. Palladium-catalyzed borylation of aryldiazonium tetrafluoroborate salts. A new synthesis of arylboronic esters. Tetrahedron Lett. 2000, 41, 8683–8686. [Google Scholar] [CrossRef]
- Todd, M.H.; Abell, C. Novel chemical tagging method for combinatorial synthesis utilizing suzuki chemistry and fourier transform ion cyclotron resonance mass spectrometry. J. Comb. Chem. 2001, 3, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Seidel, G. Microwave-assisted synthesis of pinacol boronates from aryl chlorides catalyzed by a palladium/imidazolium salt system. Org. Lett. 2002, 4, 541–543. [Google Scholar] [CrossRef]
- Billingsley, K.L.; Barder, T.E.; Buchwald, S.L. Palladium-catalyzed borylation of aryl chlorides: Scope, applications, and computational studies. Angew. Chem. Int. Ed. 2007, 46, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Jiang, Y.; Qiu, D.; Zhang, Y.; Wang, J. Direct conversion of arylamines to pinacol boronates: A metal-free borylation process. Angew. Chem. Int. Ed. 2010, 49, 1846–1849. [Google Scholar] [CrossRef] [PubMed]
- Maddaford, S.P.; Keay, B.A. Scope and limitations of the palladium-catalyzed cross-coupling reaction of in situ generated organoboranes with aryl and vinyl halides. J. Org. Chem. 1994, 59, 6501–6503. [Google Scholar] [CrossRef]
- Andersen, N.G.; Maddaford, S.P.; Keay, B.A. A modified in situ Suzuki cross-coupling of haloarenes for the preparation of C2-symmetric biaryls. J. Org. Chem. 1996, 61, 9556–9559. [Google Scholar] [CrossRef]
- Brown, S.D.; Armstrong, R.W. Synthesis of tetrasubstituted ethylenes on solid support via resin capture. J. Am. Chem. Soc. 1996, 118, 6331–6332. [Google Scholar] [CrossRef]
- Carbonnelle, A.C.; Zhu, J. A novel synthesis of biaryl-containing macrocycles by a domino Miyaura arylboronate formation: Intramolecular Suzuki reaction. Org. Lett. 2000, 2, 3477–3480. [Google Scholar] [CrossRef]
- Li, W.; Nelson, D.P.; Jensen, M.S.; Hoerrner, R.S.; Cai, D.; Larsen, R.D.; Reider, P.J. An improved protocol for the preparation of 3-pyridyl- and some arylboronic acids. J. Org. Chem. 2002, 67, 5394–5397. [Google Scholar] [CrossRef]
- Zhu, L.; Duquette, J.; Zhang, M. An improved preparation of arylboronates: application in one-pot Suzuki biaryl synthesis. J. Org. Chem. 2003, 68, 3729–3732. [Google Scholar] [CrossRef]
- Shu, W.; Pellegatti, L.; Oberli, M.A.; Buchwald, S.L. Continuous-flow synthesis of biaryls enabled by multistep solid-handling in a lithiation/borylation/Suzuki–Miyaura cross-coupling sequence. Angew. Chem. Int. Ed. 2011, 50, 10665–10669. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J. Flash Chemistry. Fast Organic Synthesis in Microsystems; Wiley-Blackwell: Chichester, UK, 2008. [Google Scholar]
- Yoshida, J. Flash chemistry using electrochemical method and microsystems. Chem. Commun. 2005, 4509–4516. [Google Scholar] [CrossRef]
- Yoshida, J.; Nagaki, A.; Yamada, T. Flash chemistry: fast chemical synthesis by using microreactors. Chem. Eur. J. 2008, 14, 7450–7459. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J. Flash chemistry: flow microreactor synthesis based on high-resolution reaction time control. Chem. Rec. 2010, 10, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Takahashi, Y.; Nagaki, A. Flash chemistry: flow chemistry that cannot be done in batch. Chem. Commun. 2013, 49, 9896–9904. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Konno, N.; Watabe, Y.; Ichitsuka, T.; Nagaki, A.; Yoshida, J.; Ichikawa, J. Flash generation and borylation of 1-(trifluoromethyl)vinyllithium toward synthesis of α-(trifluoromethyl)styrenes. J. Fluorine Chem. 2018, 207, 72–76. [Google Scholar] [CrossRef]
- Nagaki, A.; Moriwaki, Y.; Yoshida, J. Flow synthesis of arylboronic esters bearing electrophilic functional groups and space integration with Suzuki–Miyaura coupling without intentionally added base. Chem. Commun. 2012, 48, 11211–11213. [Google Scholar] [CrossRef] [PubMed]
- Comer, E.; Organ, M.G. A microcapillary system for simultaneous, parallel microwave-assisted synthesis. Chem. Eur. J. 2005, 11, 7223–7227. [Google Scholar] [CrossRef] [PubMed]
- Gömann, A.; Deverell, J.A.; Munting, K.F.; Jones, R.C.; Rodemann, T.; Canty, A.J.; Smith, J.A.; Guijt, R.M. Palladium-mediated organic synthesis using porous polymer monolith formed in situ as a continuous catalyst support structure for application in microfluidic devices. Tetrahedron 2009, 65, 1450–1454. [Google Scholar] [CrossRef]
- He, P.; Haswell, S.J.; Fletcher, P.D.I.; Kelly, S.M.; Mansfield, A. Scaling up of continuous-flow, microwave-assisted, organic reactions by varying the size of Pd-functionalized catalytic monoliths. Beilstein J. Org. Chem. 2011, 7, 1150–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, W.R.; Plucinski, P.; Frost, C.G. Robust and reusable supported palladium catalysts for cross-coupling reactions in flow. Catal. Sci. Technol. 2014, 4, 948–954. [Google Scholar] [CrossRef]
- Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R.P.; Mandoli, A. A chiral organocatalytic polymer-based monolithic reactor. Green Chem. 2014, 16, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Jumde, R.P.; Marelli, M.; Scotii, N.; Mandoli, A.; Psaro, R.; Evangelisti, C. Ultrafine palladium nanoparticles immobilized into poly(4-vinylpyridine)-based porous monolith for continuous-flow Mizoroki–Heck reaction. J. Mol. Catal. A 2016, 414, 55–61. [Google Scholar] [CrossRef]
- Suga, S.; Yamada, D.; Yoshida, J. Cationic three-component coupling involving an optically active enamine derivative. From time integration to space integration of reactions. Chem. Lett. 2010, 39, 404–406. [Google Scholar] [CrossRef]
- Nagaki, A.; Kenmoku, A.; Moriwaki, Y.; Hayashi, A.; Yoshida, J. Cross-coupling in a flow microreactor. Space integration of lithiation and Murahashi coupling. Angew. Chem. Int. Ed. 2010, 49, 7543–7547. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Saito, K.; Nokami, T.; Nagaki, A. Space integration of reactions: An approach to increase capability of organic synthesis. Synlett 2011, 1189–1194. [Google Scholar] [CrossRef]
- Nagaki, A.; Hirose, K.; Moriwaki, Y.; Mitamura, K.; Matsukawa, K.; Ishizuka, N.; Yoshida, J. Integration of borylation of aryllithiums and Suzuki–Miyaura coupling using monolithic Pd catalyst. Catal. Sci. Technol. 2016, 6, 4690–4694. [Google Scholar] [CrossRef]
- Nagaki, A.; Kim, H.; Usutani, H.; Matsuo, C.; Yoshida, J. Generation and reaction of cyano-substituted aryllithium compounds using microreactors. Org. Biomol. Chem. 2010, 8, 1212–1217. [Google Scholar] [CrossRef]
- Nagaki, A.; Takabayashi, N.; Tomida, Y.; Yoshida, J. Synthesis of unsymmetrical biaryls by means of mono-selective reaction of polyhaloarenes using integrated microflow system. Org. Lett. 2008, 10, 3937–3940. [Google Scholar] [CrossRef]
- Hessel, V.; Hardt, S.; Loewe, H. Chemical Micro Process Engineering: Fundamentals, Modelling and Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Löwe, H.; Ziogas, A. Numbering-up of micro devices: a first liquid-flow splitting unit. Chem. Eng. J. 2004, 101, 421–429. [Google Scholar]
- Anderson, N.G. Using continuous processes to increase production. Org. Process Res. Dev. 2012, 16, 852–869. [Google Scholar] [CrossRef]
- Nagaki, A.; Hirose, K.; Tonomura, O.; Taniguchi, S.; Taga, T.; Hasebe, S.; Ishizuka, N.; Yoshida, J. Design of a numbering-up system of monolithic microreactors and its application to synthesis of a key intermediate of valsartan. Org. Process Res. Dev. 2016, 20, 687–691. [Google Scholar] [CrossRef]
- Su, Y.; Kuijpers, K.; Hessel, V.; Noël, T. A convenient numbering-up strategy for the scale-up of gas–liquid photoredox catalysis in flow. React. Chem. Eng. 2016, 1, 73–81. [Google Scholar] [CrossRef]
- Commenge, J.M.; Falk, L.; Corriou, J.P.; Matlosz, M. Optimal design for flow uniformity in microchannel reactors. AIChE J. 2002, 48, 345–358. [Google Scholar] [CrossRef]
- Tonomura, O.; Tominari, T.; Kano, M.; Hasebe, S. Operation policy for micro chemical plants with external numbering-up structure. Chem. Eng. J. 2008, 135, S131–S137. [Google Scholar] [CrossRef] [Green Version]
- Amador, C.; Gavriilidis, A.; Angeli, P. Flow distribution in different microreactor scale-out geometries and the effect of manufacturing tolerances and channel blockage. Chem. Eng. J. 2004, 101, 379–390. [Google Scholar] [CrossRef]
- Wang, J. Theory of flow distribution in manifolds. Chem. Eng. J. 2011, 168, 1331–1345. [Google Scholar] [CrossRef]
- Won, J.W.; Park, K.M.; Choi, S.J.; Chang, P. Serial connection of packed-bed reactors with different reaction temperatures: enhanced operational stability for enzymatically interesterified trans-free lipid production. Eur. Food Res. Technol. 2012, 235, 647–657. [Google Scholar] [CrossRef]
- Irvine, M.W.; Costa, B.M.; Dlaboga, D.; Culley, G.R.; Hulse, R.; Scholefield, C.L.; Atlason, P.; Fang, G.; Eaves, R.; Morley, R.; et al. Piperazine-2,3-dicarboxylic acid derivatives as dual antagonists of NMDA and GluK1-containing kainate receptors. J. Med. Chem. 2012, 55, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Grossman, O.; Gelman, D. Novel trans-spanned palladium complexes as efficient catalysts in mild and amine-free cyanation of aryl bromides under air. Org. Lett. 2006, 8, 1189–1191. [Google Scholar] [CrossRef]
- Ackermann, L.; Gschrei, C.J.; Althammera, A.; Riederer, M. Cross-coupling reactions of aryl and vinyl chlorides catalyzed by a palladium complex derived from an air-stable H-phosphonate. Chem. Commun. 2006, 1419–1421. [Google Scholar] [CrossRef]
- Bandari, R.; Höche, T.; Prager, A.; Dirnberger, K.; Buchmeiser, M.R. Ring-opening metathesis polymerization based pore-size-selective functionalization of glycidyl methacrylate based monolithic media: access to size-stable nanoparticles for ligand-free metal catalysis. Chem. Eur. J. 2010, 16, 4650–4658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G. Efficient protocol for the phosphine-free Suzuki-Miyaura reaction catalyzed by palladium on carbon at room temperature. Synthesis 2005, 537–542. [Google Scholar] [CrossRef]
- Kuhl, N.; Hopkinson, M.N.; Glorius, F. Selective rhodium(III)-catalyzed cross-dehydrogenative coupling of furan and thiophene derivatives. Angew. Chem. Int. Ed. 2012, 51, 8230–8234. [Google Scholar] [CrossRef] [PubMed]
- Cahiez, G.; Chaboche, C.; Mahuteau-Betzer, F.; Ahr, M. Iron-catalyzed homo-coupling of simple and functionalized arylmagnesium reagents. Org. Lett. 2005, 7, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Yi, J.; Deng, X.; Fu, Y.; Liu, L. Cu-catalyzed carbon-heteroatom coupling reactions under mild conditions promoted by resin-bound organic ionic bases. J. Org. Chem. 2011, 76, 800–810. [Google Scholar] [CrossRef]
- Amatore, M.; Gosmini, C. Efficient cobalt-catalyzed formation of unsymmetrical biaryl compounds and its application in the synthesis of a sartan intermediate. Angew. Chem. Int. Ed. 2008, 47, 2089–2092. [Google Scholar] [CrossRef]
- Schiek, M.; Al-Shamerya, K.; Lützen, A. Synthesis of symmetrically and unsymmetrically para-functionalized p-ouaterphenylenes. Synthesis 2007, 613–621. [Google Scholar] [CrossRef]
- Nising, C.F.; Schmid, U.K.; Nieger, M.; Bräse, S. A new protocol for the one-pot synthesis of symmetrical biaryls. J. Org. Chem. 2004, 69, 6830–6833. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. Synthesis of heteroaryl compounds through cross-coupling reaction of aryl bromides or benzyl halides with thienyl and pyridyl aluminum reagents. J. Org. Chem. 2014, 79, 230–239. [Google Scholar] [CrossRef]
- Izquierdo, F.; Corpet, M.; Nolan, S.P. The Suzuki–Miyaura reaction performed using a palladium–N-heterocyclic carbene catalyst and a weak inorganic base. Eur. J. Org. Chem. 2015, 1920–1924. [Google Scholar] [CrossRef]
- Kristensen, J.; Lysén, M.; Vedsø, P.; Begtrup, M. Synthesis of ortho substituted arylboronic esters by in situ trapping of unstable lithio intermediates. Org. Lett. 2001, 3, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiang, J. A high yield and pilot-scale process for the preparation of adapalene. Org. Process Res. Dev. 2006, 10, 285–288. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ar1-X | Conditions of Lithiation and Borylation | Ar2-X | Product | Yield (%) a | ||
|---|---|---|---|---|---|---|
| tR1 (s) | Temperature (°C) | Condition (a) c | Condition (b) d | |||
 | 1.7 | 0 |  | 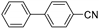 | 96 | 100 |
| 1.7 | 0 | 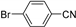 | 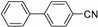 | 0 | 3 | |
| 1.7 | 0 | 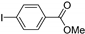 |  | 76 | 87 | |
| 1.7 | 0 | 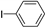 | 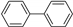 | 6 | 41 | |
| 1.7 | 0 |  | 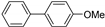 | 1 | 29 | |
| 1.7 | 0 | 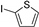 | 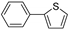 | 86 | 92 | |
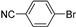 | 0.059 | 0 | 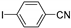 | 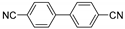 | 68 | 91 |
| 0.059 | 0 | 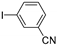 | 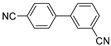 | 17 | 83 | |
| 0.059 | 0 | 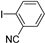 | 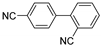 | 12 | 91 | |
| 0.059 | 0 |  | 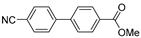 | 34 | 84 | |
 | 0.059 | 0 | 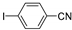 |  | 63 | 97 |
| 0.059 | 0 |  | 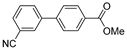 | 15 | 87 | |
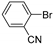 | 0.059 | 24 |  | 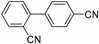 | 63 | 98 |
| 0.059 | 24 | 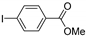 |  | 2 | 52 | |
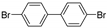 | 0.059 | 0 | 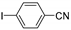 | 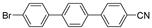 | 0 | 54 b |
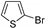 | 1.7 | 0 | 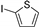 |  | 83 | 94 |
| 1.7 | 0 |  | 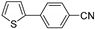 | 78 | 87 | |
| 1.7 | 0 | 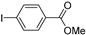 | 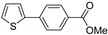 | 71 | 86 | |
| Reactor | n (Number of Reactors) | Flow Rate (mL/min) | Residence Time (min) | Pressure Drop (MPa) | Yield (%) | Productivity (mg/h) |
|---|---|---|---|---|---|---|
| A | 1 | 0.2 | 9.4 | 0.9 | 96 | 52 |
| A | 1 | 0.4 | 4.7 | 1.7 | 96 | 100 |
| A | 1 | 1.0 | 1.9 | 3.9 | 81 | 220 |
| A | 1 | 1.5 | 1.3 | 5.9 | 66 | 270 |
| B | 1 | 0.2 | 9.6 | 0.3 | 98 | 53 |
| B | 1 | 0.4 | 4.8 | 0.4 | 100 | 110 |
| B | 1 | 1.0 | 1.9 | 0.7 | 87 | 230 |
| B | 1 | 1.5 | 1.3 | 0.9 | 67 | 270 |
| B | 3 | 0.6 | 9.6 | 1.4 | 98 | 160 |
| B | 3 | 1.2 | 4.8 | 3.0 | 97 | 310 |
| B | 3 | 3.0 | 1.9 | 6.8 | 86 | 690 |
| B | 3 | 4.5 | 1.3 | 9.0 | 55 | 670 |
| B | 5 | 1.0 | 9.6 | 3.3 | 100 | 270 |
| B | 5 | 2.0 | 4.8 | 6.6 | 99 | 530 |
| B | 5 | 5.0 | 1.9 | 14 | 75 | 1000 |
| B | 5 | 7.5 | 1.3 | 19 | 52 | 1000 |
| Ar1-X | Conditions of Lithiation and Borylation | Ar2-X | Product | Yield (%) a | ||
|---|---|---|---|---|---|---|
| tR1 (s) | Temperature (°C) | n = 1 | n = 3 | |||
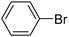 | 1.7 | 0 |  | 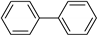 | 62 | 87 |
| 1.7 | 0 | 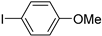 | 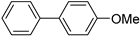 | 41 | 74 | |
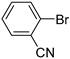 | 0.059 | 24 | 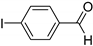 | 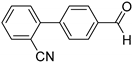 | 26 | 65 |
| 0.059 | 24 | 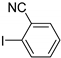 |  | 63 | 94 | |
| 0.059 | 24 | 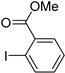 | 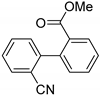 | 43 | 82 | |
| Cycle Number | Yield (%) a | |
|---|---|---|
| Monolith A | Monolith B | |
| 1 | 97 | 97 |
| 2 | 96 | 100 |
| 3 | 96 | 96 |
| 4 | 100 | 97 |
| 5 | 97 | 100 |
| 6 | 94 | 97 |
| 7 | 93 | 93 |
| 8 | 95 | 96 |
| 9 | 98 | 96 |
| 10 | 99 | 99 |
| 11 | 99 | 98 |
| 12 | 99 | 99 |
| 13 | 97 | 99 |
| 14 | 99 | 99 |
| 15 | 95 | 98 |
| T (°C) | Flow Rate (mL/min) | tR (min) | Yield (%) |
|---|---|---|---|
| 60 | 0.2 | 9.4 | 37 |
| 0.4 | 4.7 | 14 | |
| 1.0 | 1.9 | 0 | |
| 1.5 | 1.3 | 0 | |
| 80 | 0.2 | 9.4 | 81 |
| 0.4 | 4.7 | 81 | |
| 1.0 | 1.9 | 39 | |
| 1.5 | 1.3 | 30 | |
| 100 | 0.2 | 9.4 | 96 |
| 0.4 | 4.7 | 96 | |
| 1.0 | 1.9 | 81 | |
| 1.5 | 1.3 | 66 | |
| 120 | 0.2 | 9.4 | 100 |
| 0.4 | 4.7 | 95 | |
| 1.0 | 1.9 | 93 | |
| 1.5 | 1.3 | 96 |
| T (°C) | Flow Rate (mL/min) | tR (min) | Yield (%) |
|---|---|---|---|
| 60 | 0.2 | 9.6 | 50 |
| 0.4 | 4.8 | 60 | |
| 1.0 | 1.9 | 76 | |
| 1.5 | 1.3 | 82 | |
| 80 | 0.2 | 9.6 | 61 |
| 0.4 | 4.8 | 74 | |
| 1.0 | 1.9 | 92 | |
| 1.5 | 1.3 | 100 | |
| 100 | 0.2 | 9.6 | 67 |
| 0.4 | 4.8 | 87 | |
| 1.0 | 1.9 | 100 | |
| 1.5 | 1.3 | 98 | |
| 120 | 0.2 | 9.6 | 89 |
| 0.4 | 4.8 | 92 | |
| 1.0 | 1.9 | 99 | |
| 1.5 | 1.3 | 99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaki, A.; Hirose, K.; Moriwaki, Y.; Takumi, M.; Takahashi, Y.; Mitamura, K.; Matsukawa, K.; Ishizuka, N.; Yoshida, J.-i. Suzuki–Miyaura Coupling Using Monolithic Pd Reactors and Scaling-Up by Series Connection of the Reactors. Catalysts 2019, 9, 300. https://doi.org/10.3390/catal9030300
Nagaki A, Hirose K, Moriwaki Y, Takumi M, Takahashi Y, Mitamura K, Matsukawa K, Ishizuka N, Yoshida J-i. Suzuki–Miyaura Coupling Using Monolithic Pd Reactors and Scaling-Up by Series Connection of the Reactors. Catalysts. 2019; 9(3):300. https://doi.org/10.3390/catal9030300
Chicago/Turabian StyleNagaki, Aiichiro, Katsuyuki Hirose, Yuya Moriwaki, Masahiro Takumi, Yusuke Takahashi, Koji Mitamura, Kimihiro Matsukawa, Norio Ishizuka, and Jun-ichi Yoshida. 2019. "Suzuki–Miyaura Coupling Using Monolithic Pd Reactors and Scaling-Up by Series Connection of the Reactors" Catalysts 9, no. 3: 300. https://doi.org/10.3390/catal9030300
APA StyleNagaki, A., Hirose, K., Moriwaki, Y., Takumi, M., Takahashi, Y., Mitamura, K., Matsukawa, K., Ishizuka, N., & Yoshida, J.-i. (2019). Suzuki–Miyaura Coupling Using Monolithic Pd Reactors and Scaling-Up by Series Connection of the Reactors. Catalysts, 9(3), 300. https://doi.org/10.3390/catal9030300