Catalysts of PtSn/C Modified with Ru and Ta for Electrooxidation of Ethanol



Abstract
:1. Introduction
2. Results and Discussion
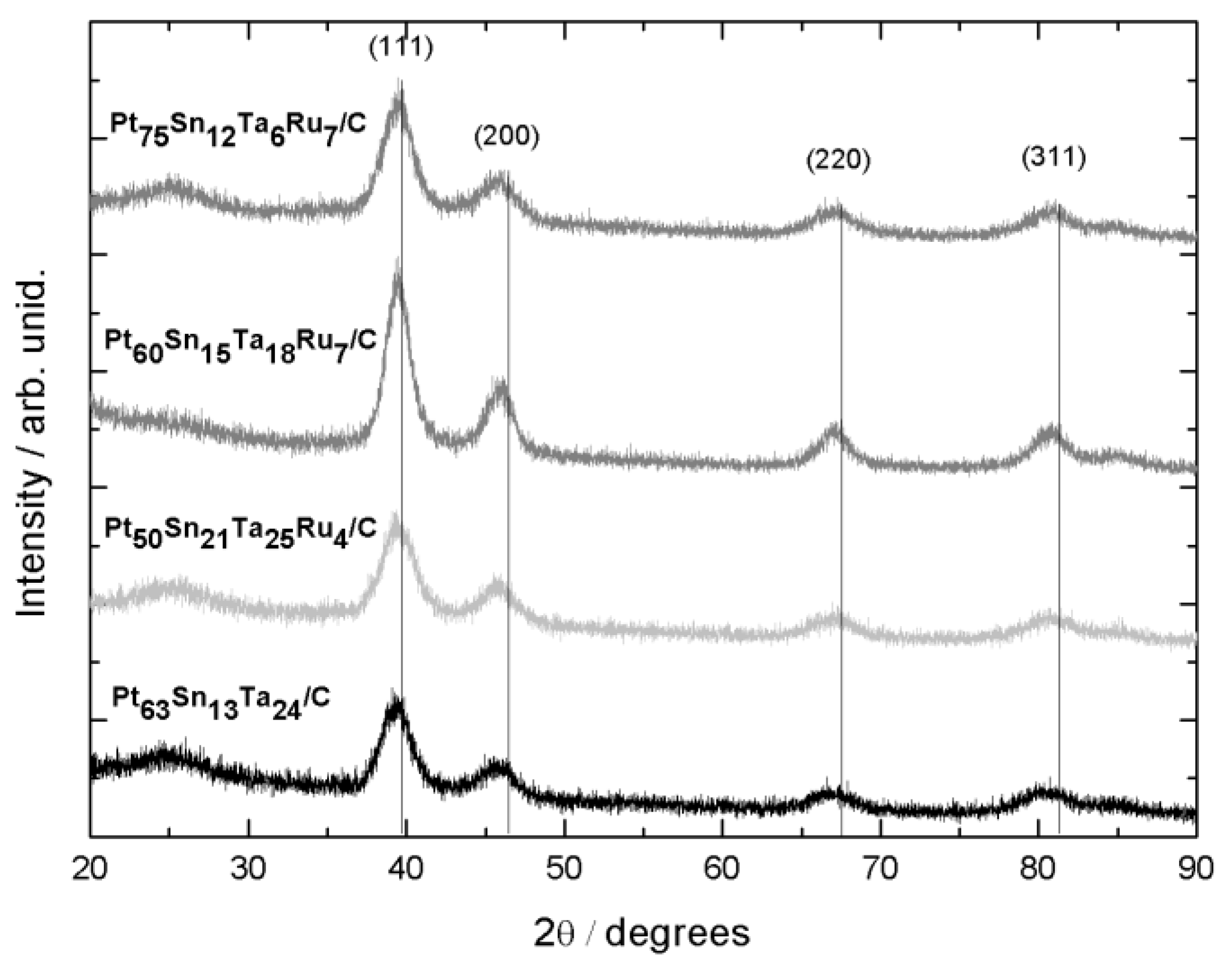
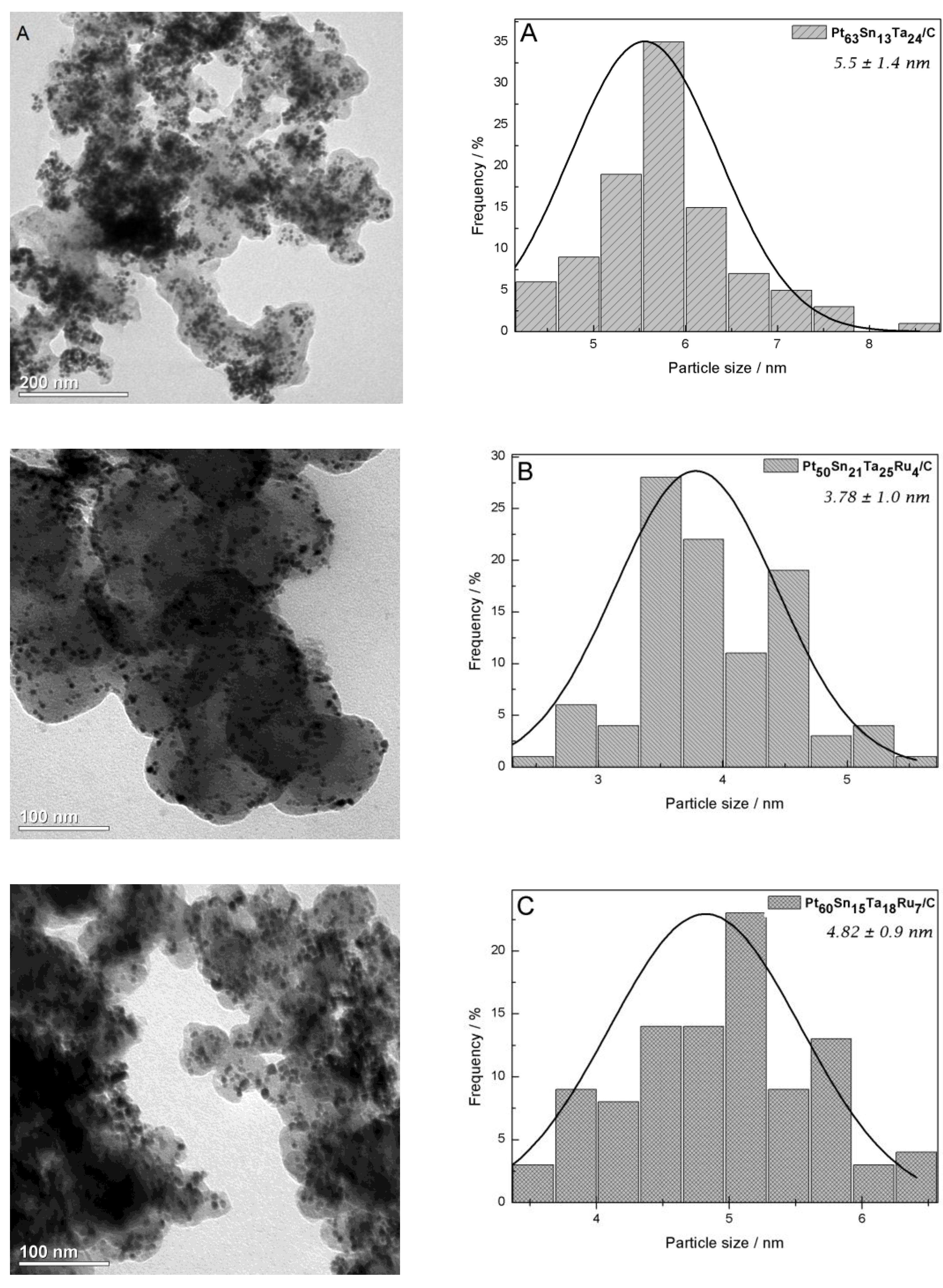
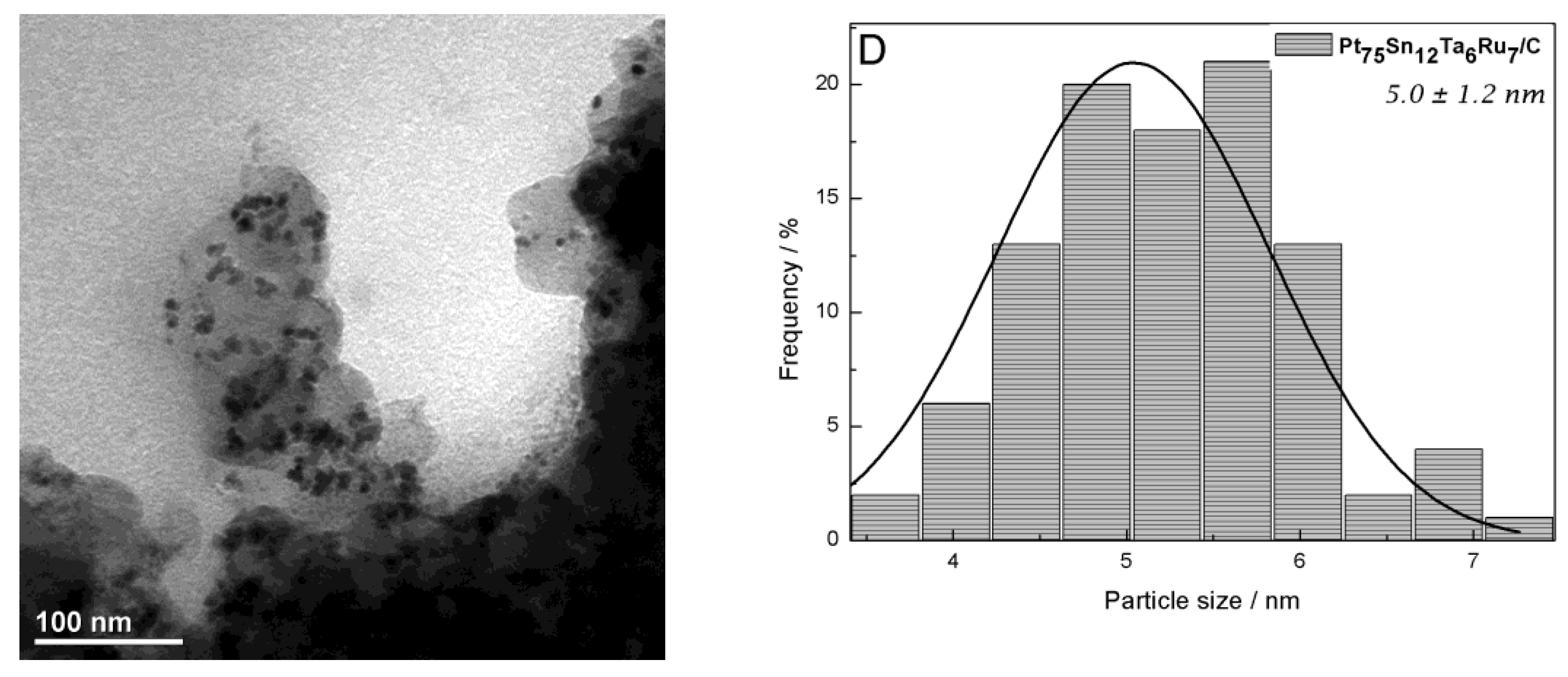
2.1. Physicochemical Characterizations
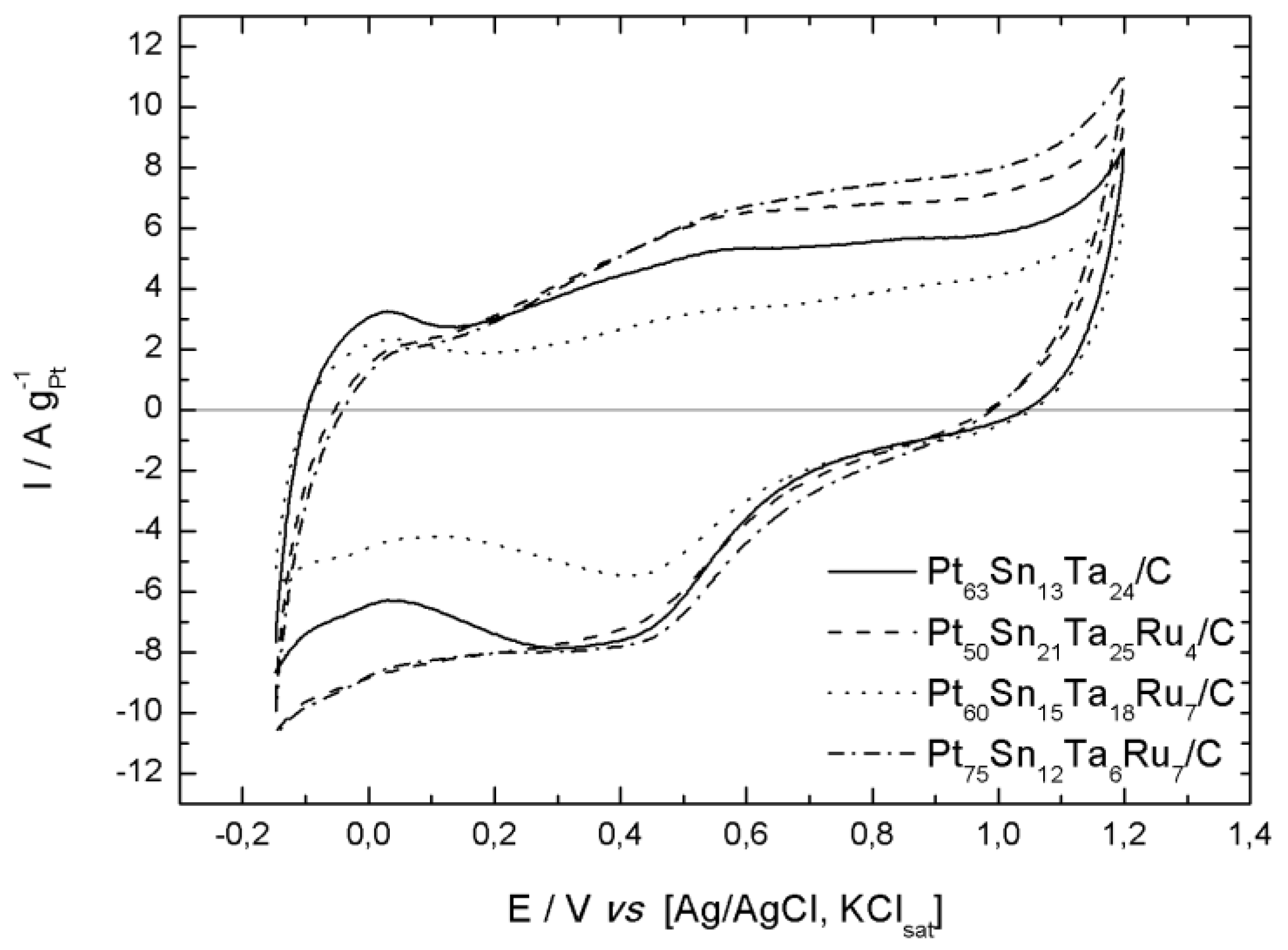
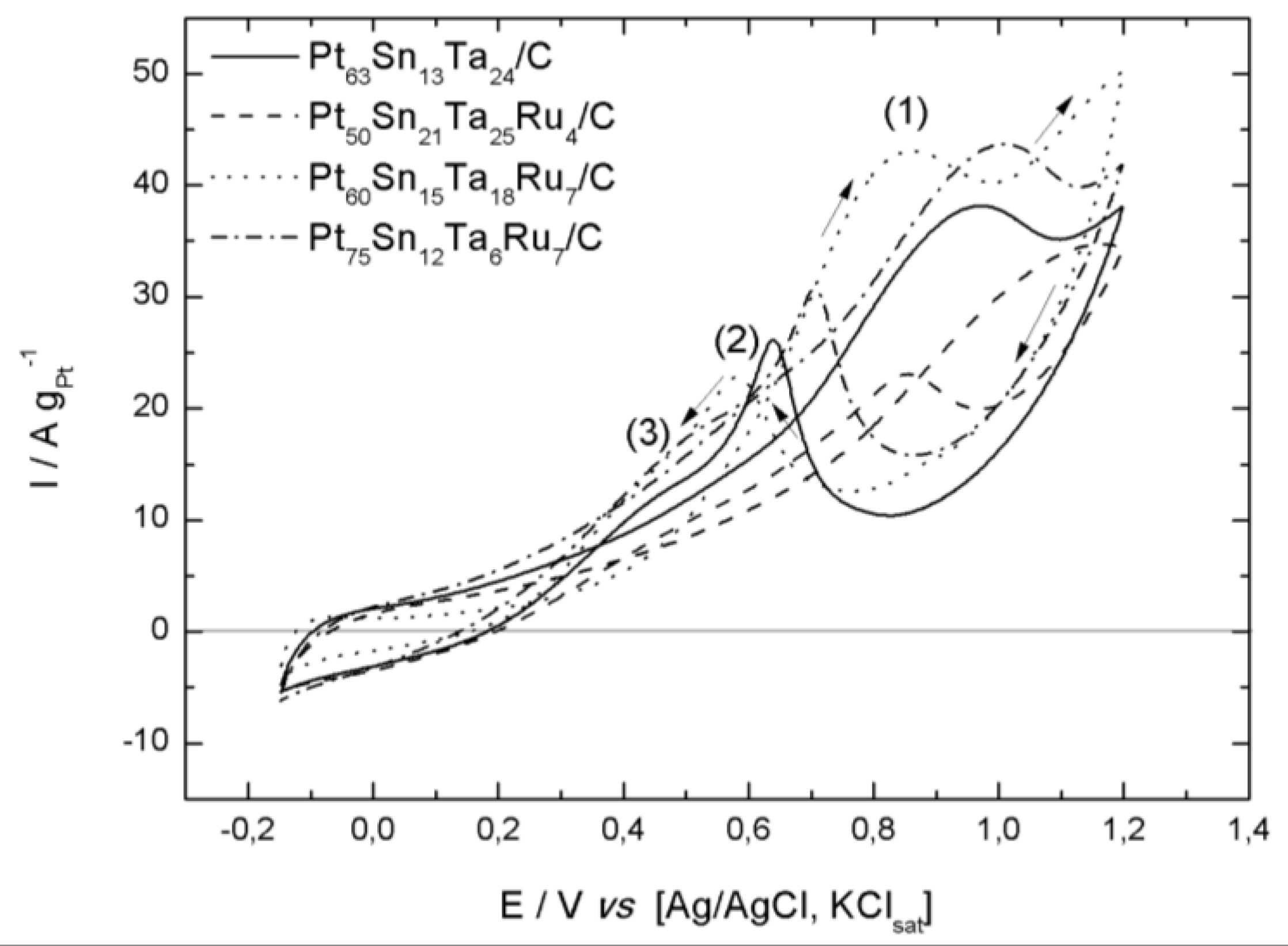
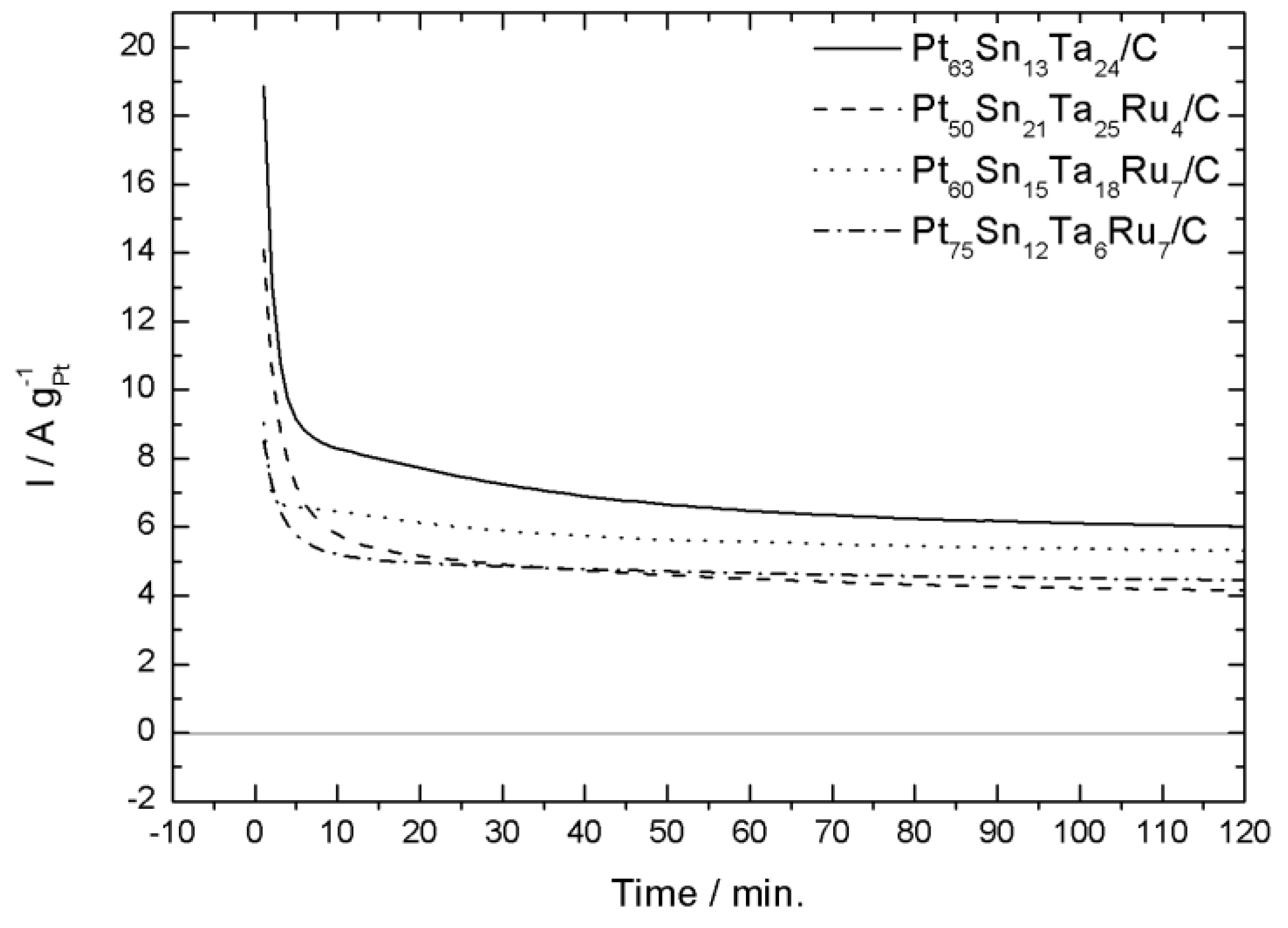
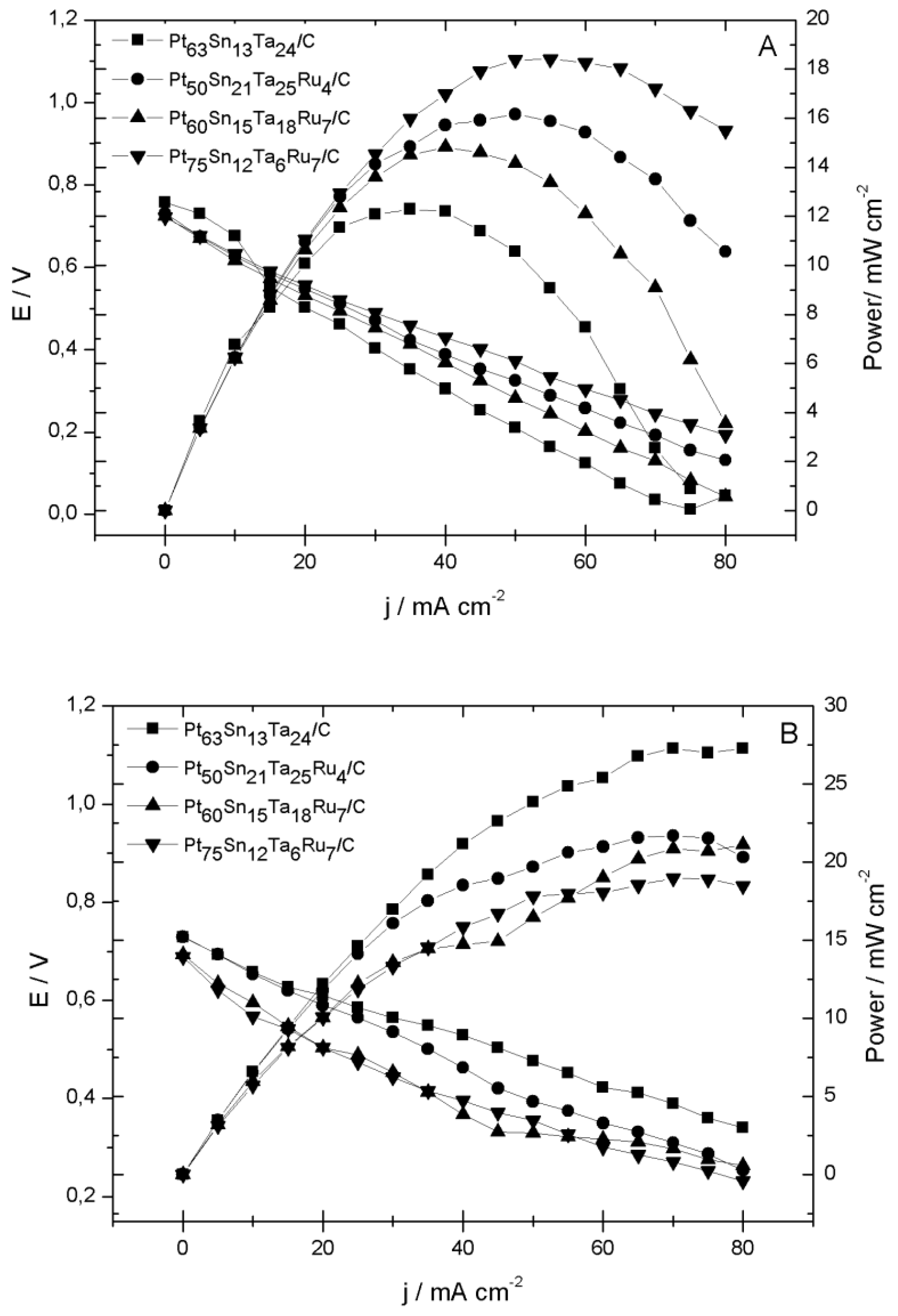
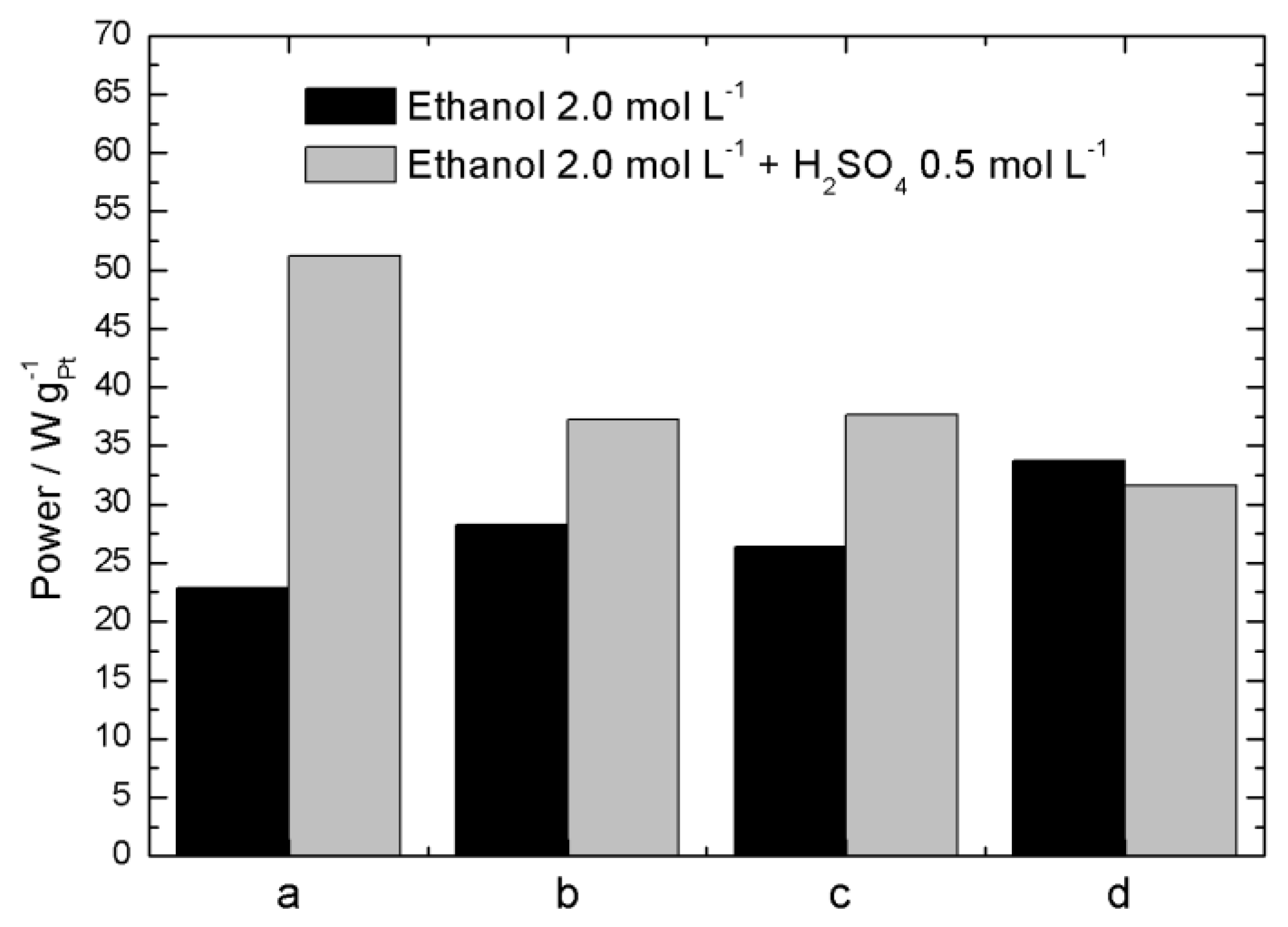
2.2. Electrochemical Characterization
3. Materials and Methods
3.1. Support Functionality
3.2. Preparation of PtSnTa/C and PtSnTaRu/C Catalysts
3.3. Physicochemical Characterization
3.4. Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| DAFC | Direct alcohol fuel cell |
| XRD | X-ray diffraction |
| D | Apparent size of the crystallite |
| K | Shape factor |
| λ | Wavelength |
| β | Diffraction full width at half-maximum intensity |
| FWHM | Full width at half-maximum intensity |
| θβ | Angle at maximum intensity and wavelength |
| θ | Angle of incidence of radiation |
| a | Lattice parameter |
| FCC | Face-centered cubic |
| TEM | Transmission electron microscopy |
| EDX | Energy dispersive X-ray measurements |
| ECSA | Electrochemical surface area |
| Ip | Peak current |
| Eonset-Ethanol | Ethanol oxidation initiation potential |
| QH | Hydrogen desorption charge |
| mPt | Mass of platinum deposited on the electrode |
| DPP | Decomposition of polymeric precursors |
| AC | Citric acid |
| EG | Ethylene glycol |
| MEA | Membrane electrode assemblies |
References
- Santos, M.C.; Parreira, L.S.; Souza, F.D.M.; Junior, J.C.; Gentil, T. Fuel Cells: Hydrogen and Ethanol Technologies. Sci. Mater. Eng. 2017, 2017, 1–22. [Google Scholar]
- Wu, H.-W. A review of recent development: Transport and performance modeling of PEM fuel cells. Appl. Energy 2016, 165, 81–106. [Google Scholar] [CrossRef]
- Ong, B.C.; Kamarudin, S.K.; Basri, S. Direct liquid fuel cells: A review. Int. J. Hydrog. Energy 2017, 42, 10142–10157. [Google Scholar] [CrossRef]
- Lin, R.H.; Xi, X.N.; Wang, P.N.; Wu, B.D.; Tian, S.M. Review on hydrogen fuel cell condition monitoring and prediction methods. Int. J. Hydrog. Energy 2018, 44, 5488–5498. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kaneda, Y.; Wagatsuma, M.; Tsujiguchi, T. Product distribution and the reaction kinetics at the anode of direct ethanol fuel cell with Pt/C, PtRu/C and PtRuRh/C. J. Power Sources 2012, 199, 103–109. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, R.; Han, Z.; Li, C.; Wang, Y.; Chi, B.; Li, J.; Wang, X. Electrooxidation of methanol and ethanol in acidic medium using a platinum electrode modified with lanthanum-doped tantalum oxide film. Electrochim. Acta 2015, 151, 544–551. [Google Scholar] [CrossRef]
- Badwal, S.P.S.; Giddey, S.; Kulkarni, A.; Goel, J.; Basu, S. Direct ethanol fuel cells for transport and stationary applications–A comprehensive review. Appl. Energy 2015, 145, 80–103. [Google Scholar] [CrossRef]
- Antolini, E. Catalyst for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Alcalá, R.; Mavrikakis, M.; Dumesic, J.A. DFT studies for cleavage of C-C and C-O bonds in surface species derived from ethanol on Pt(111). J. Catal. 2003, 218, 178–190. [Google Scholar] [CrossRef]
- Soloveichik, G.L. Liquid fuel cells. Beilstein J. Nanotechnol. 2014, 5, 1399–1418. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xiong, L.; Liu, Q.; Kui, H.; Wen, L.; Li, Y.; Kun, T.; Niu, L.; Yang, S.; Jian, X. Enhanced electrocatalytic activity for the oxidation of liquid fuels on PtSn nanoparticles. Electrochim. Acta 2011, 56, 9860–9867. [Google Scholar] [CrossRef]
- Lamy, C.; Coutanceau, C.; Leger, J.M. The Direct Ethanol Fuel Cell: A Challenge to Convert Bioethanol Cleanly into Electric Energy–Catalysis for Sustainable Energy Production. In Catalysis for Sustainable Energy Production, 1st ed.; Barbaro, P., Bianchini, C., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 13–27. [Google Scholar]
- Huang, S.; Shan, A.; Wang, R. Review Low Pt Alloyed Nanostructures for Fuel Cells Catalysts. Catalysts 2018, 8, 538. [Google Scholar] [CrossRef]
- González-Hernández, M.; Antolini, E.; Perez, J. Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts. Catalysts 2019, 9, 61. [Google Scholar] [CrossRef]
- Antolini, E. Pt-Ni and Pt-M-Ni (M = Ru, Sn) Anode Catalysts for Low-Temperature Acidic Direct Alcohol Fuel Cells: A Review. Energies 2017, 10, 42. [Google Scholar]
- Antoniassi, R.M.; Silva, J.C.M.; Oliveira, N.A.; Spinacé, E.V. Synthesis of Pt+SnO2/C electrocatalysts containing Pt nanoparticles with preferential (100) orientation for direct ethanol fuel cell. Appl. Catal. B 2017, 218, 91–100. [Google Scholar]
- Da Silva, E.L.; Cuna, A.; Vega, M.R.O.; Radtke, C.; Machado, G.; Tancredi, N.; de Malfatti, C.F. Influence of the support on PtSn electrocatalysts behavior: Ethanol electro-oxidation performance and in-situ ATR-FTIR studies. Appl. Catal. B 2016, 193, 170–179. [Google Scholar] [CrossRef]
- Jiang, L. Preparation and characterization of PtSn/C anode electrocatalysts for direct ethanol fuel cell. Catal. Today 2004, 93–95, 665–670. [Google Scholar] [CrossRef]
- Li, H.; Sun, G.; Cao, L.; Jiang, L.; Xin, Q. Comparison of different promotion effect of PtRu/C and PtSn/C electrocatalysts for ethanol electro-oxidation. Electrochim. Acta 2007, 52, 6622–6629. [Google Scholar]
- Neto, A.O.; Dias, R.R.; Tusi, M.M.; Linardi, M.; Spinacé, E.V. Electro-oxidation of methanol and ethanol using PtRu/C, PtSn/C and PtSnRu/C electrocatalysts prepared by an alcohol-reduction process. J. Power Sources 2007, 166, 87–91. [Google Scholar] [CrossRef]
- Cunha, E.M.; Ribeiro, J.; Kokoh, K.B.; de Andrade, A.R. Preparation, characterization and application of Pt-Ru-Sn/C trimetallic electrocatalysts for ethanol oxidation in direct fuel cell. Int. J. Hydrog. Energy 2011, 36, 11034–11042. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, X.; Xin, Q.; Sun, G. Polyol-synthesized Pt2.6Sn1Ru0.4/C as a high-performance anode catalyst for direct ethanol fuel cells. Chin. J. Catal. 2014, 35, 1394–1401. [Google Scholar] [CrossRef]
- Wendt, H.; Spinacé, E.V.; Neto, A.O.; Linardi, M. Electrocatalysis 881 and electrocatalysts for low temperature fuel cells: Fundamentaks, state of the art, research and development. Q. Nova 2005, 28, 1066–1075. [Google Scholar]
- Papageorgopoulos, D.C.; Keijzer, M.; de Bruijn, F.A. The inclusion of Mo, Nb and Ta in Pt and PtRu carbon supported electrocatalysts in the quest for improved CO tolerant PEMFC anodes. Electrochim. Acta 2002, 48, 197–204. [Google Scholar] [CrossRef]
- Anwar, M.T.; Yan, X.; Shen, S.; Husnain, N.; Zhu, F.; Luo, L.; Zhang, J. Enhanced durability of Pt electrocatalyst with tantalum doped titania as catalyst support. Int. J. Hydrog. Energy 2017, 42, 30750–30759. [Google Scholar] [CrossRef]
- Masud, J.; Alam, M.T.; Awaludin, Z.; El-Deab, M.S.; Okajima, T.; Ohsaka, T. Electrocatalytic oxidation of methanol at tantalum oxide-modified Pt electrodes. J. Power Sources 2012, 220, 399–404. [Google Scholar]
- Wang, D.; Lu, S.; Jiang, S.P. Tetrahydrofuran-functionalized multi-walled carbon nanotubes as effective support for Pt and PtSn electrocatalysts of fuel cells. Electrochim. Acta 2010, 55, 2964–2971. [Google Scholar]
- PDF-2; ICDD: Newtown Square, PA, 2011; PDF 00-004-0802 (Accessed 20 August 2018).
- Cullity, B.D. Elements of x-Ray Diffraction, 3rd ed.; Addison-Wesley: San Francisco, CA, USA, 2001; pp. 352–358. [Google Scholar]
- Reid, O.; Saleh, F.S.; Easton, E.B. Determining electrochemically active surface area in PEM fuel cell electrodes with electrochemical impedance spectroscopy and its application to catalyst durability. Electrochim. Acta 2013, 114, 278–284. [Google Scholar]
- Colmati, F.; Antolini, E.; Gonzalez, E.R. Ethanol oxidation on a carbon-supported Pt75Sn25 electrocatalyst prepared by reduction with formic acid: Effect of thermal treatment. Appl. Catal. B 2007, 73, 106–115. [Google Scholar] [CrossRef]
- Parreira, L.S.; Da Silva, J.C.M.; D’Villa–Silva, M.; Simões, F.C.; Garcia, S.; Gaubeur, I.; Cordeiro, M.A.L.; Leite, E.R.; dos Santos, M.C. PtSnNi/C nanoparticle electrocatalysts for the ethanol oxidation reaction: Ni stability study. Electrochim. Acta 2013, 96, 243–252. [Google Scholar] [CrossRef]
- Caliman, C.C.; Palma, L.M.; Ribeiro, J. Evaluation of Ni and Ti Addition in PtSn/C Catalysts for Ethanol and Glycerol Electrooxidation. J. Electrochem. Soc. 2013, 160, F853–F858. [Google Scholar]
- Crisafulli, R.; Antoniassi, R.M.; Oliveira, N.A.; Spinacé, E.V. Acid-treated PtSn/C and PtSnCu/C electrocatalysts for ethanol electro-oxidation. Int. J. Hydrog. Energy 2014, 39, 5671–5677. [Google Scholar] [CrossRef]
- Oliveira, N.A.; Dias, R.R.; Ribeiro, V.A.; Spinacé, E.V.; Linardi, M. Eletro-oxidação de etanol sobre eletrocatalisadores PtRh/C, PtSn/C e PtSnRh/C preparados pelo método da redução por álcool. Eclética Química 2006, 31, 81–88. [Google Scholar]
- Martínez-Huerta, M.V.; Tsiouvaras, N.; García, G.; Peña, M.A.; Pastor, E.; Rodriguez, J.L.; Fierro, J.L.G. Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells. Catalysts 2013, 3, 811–838. [Google Scholar] [CrossRef]
- Maillard, F. Infrared Spectroscopic Study of CO Adsorption and Electro-oxidation on Carbon-Supported Pt Nanoparticles: Interparticle versus Intraparticle Heterogeneity. J. Phys. Chem. 2004, 108, 17893–17904. [Google Scholar] [CrossRef]
- Almeida, T.S.; Kokoh, K.B.; de Andrade, A.R. Effect of Ni on Pt/C and PtSn/C prepared by the Pechini method. Int. J. Hydrog. Energy 2011, 36, 3803–38810. [Google Scholar] [CrossRef]
- Santos, D.M.; Paganoto, G.T.; Queiroz, M.A.R.; Guimarães, M.C.C.; Ribeiro, J. Influence of Support material of PtSnNiGa/C Electrocatalysts for Ethanol Oxidation. Orbital Electron. J. Chem. 2017, 9, 140–150. [Google Scholar] [CrossRef]
- Meier, J.C.; Galeano, C.; Katsounaros, I. Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 2014, 5, 44–67. [Google Scholar] [CrossRef]
- Rose, A.; Bilsborrow, R.; King, C.R. In situ Ru K-edge EXAFS of CO adsorption on a Ru modified Pt/C fuel cell catalyst. Electrochim. Acta 2009, 54, 5262–5266. [Google Scholar] [CrossRef]
- Almeida, T.S.; Wassen, A.R.V.; Vandover, R.B.; de Andrade, A.R. Combinatorial PtSnM (M ¼ Fe, Ni, Ru and Pd ) nanoparticle catalyst library toward ethanol electrooxidation. J. Power Sources 2015, 284, 623–630. [Google Scholar]
- Arenz, M.; Stamenkovic, V.; Blizanac, B.B.; Mayrhofer, K.J.; Markovic, N.M.; Ross, P.N. Carbon-supported Pt–Sn electrocatalysts for the anodic oxidation of H2, CO, and H2/CO mixtures. Part II: The structure–activity relationship. J. Catal. 2005, 232, 402–410. [Google Scholar]
- Binninger, T.; Fabbri, E.; Kotz, R.; Schmidt, T.J. Determination of the Electrochemically Active Surface Area of Metal-Oxide Supported Platinum Catalyst. J. Electrochim. Soc. 2013, 161, H121–H128. [Google Scholar] [CrossRef]
- De Souza, R.F.B.; Silva, J.C.M.; Assumpção, M.H.M.T.; Neto, A.O.; Santos, M.C. Ethanol oxidation reaction using PtSn/C+Ce/C Electrocatalysts: Aspects of ceria contribution. Electrochim. Acta 2014, 117, 292–298. [Google Scholar] [CrossRef]
- Teran, F.E.; Santos, D.M.; Ribeiro, J.; Kokoh, K.B. Activity of PtSnRh/C nanoparticles for the electrooxidation of C1 and C2 alcohols. Thin Solid Film 2012, 520, 5846–5850. [Google Scholar] [CrossRef]
- De Souza, M.M.; Gomes, R.S.; de Bortoli, A.L. A model for direct ethanol fuel cells considering variations in the concentration of the species. Int. J. Hydrog. Energy 2018, 43, 13475–13488. [Google Scholar] [CrossRef]
- Mai, P.T.; Haze, A.; Chiku, M.; Higuchi, E.; Inoue, H. Ethanol Oxidation Reaction on Tandem Pt/Rh/SnOx Catalyst. Catalysts 2017, 246, 1–10. [Google Scholar]
- Song, S.Q.; Zhou, W.J.; Zhou, Z.H. Direct ethanol PEM fuel cells: The case of platinum based anodes. Int. J. Hydrog. Energy 2005, 30, 995–1001. [Google Scholar] [CrossRef]
- Sadhasivam, T.; Palanisamy, G.; Roh, S.H.; Kurkuri, M.D.; Kim, S.C.; Jung, H.Y. Electro-analytical performance of bifunctional electrocatalyst materials in unitized regenerative fuel cell system. Int. J. Hydrog. Energy 2018, 43, 18169–18184. [Google Scholar] [CrossRef]
- Azam, A.M.I.N.; Lee, S.H.; Masdar, M.S.; Zainoodin, A.M.; Kamarudin, S.K. ScienceDirect Parametric study on direct ethanol fuel cell (DEFC) performance and fuel crossover. Int. J. Hydrogen. Energ. 2019, 44, 8566–8574. [Google Scholar] [CrossRef]
- Purgato, F.L.S.; Proniera, S.; Olivi, P.; de Andrade, A.R.; Légera, J.M.; Tremiliosi-Filho, G.; Kokoh, K.B. Direct ethanol fuel cell: Electrochemical performance at 90 °C on Pt and PtSn/C electrocatalysts. J. Power Sources 2012, 198, 95–99. [Google Scholar] [CrossRef]
- Fatih, K.; Neburchilov, V.; Alzate, V.; Neagu, R.; Wang, H. Synthesis and characterization of quaternary PtRuIrSn/C electrocatalysts for direct ethanol fuel cells. J. Power Sources 2010, 195, 7168–7175. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi- and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Ribeiro, J.; dos Anjos, D.M.; Kokoh, K.B.; Coutanceau, C.; Leger, J.-M.; Olivi, P.; de Andrade, A.R.; Tremiliosi-Filho, G. Carbon-supported ternary PtSnIr catalysts for direct ethanol fuel cell. Electrochim. Acta 2007, 52, 6997–7006. [Google Scholar] [CrossRef]
- Artem, L.M.; Santos, D.M.; De Andrade, A.R.; Kokoh, K.B.; Ribeiro, J. Development of ternary and quaternary catalysts for the electrooxidation of glycerol. Sci. World J. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Yadav, A.P. Estimation of Active Surface Area of Platinum by Electrochemical Impedance Spectroscopy. Int. J. Chem. Stud. 2013, 1, 1–4. [Google Scholar]
- Stevens, D.A.; Dahn, J.R. Electrochemical Characterization of the Active Surface in Carbon-Supported Platinum Electrocatalysts for PEM Fuel Cells. J. Electrochim. Soc. 2003, 150, A770–A775. [Google Scholar] [CrossRef]
- Caliman, C.C. Estudo da Eletro-Oxidação de Alcoóis em Catalisadores do Tipo PtSnNiTi para Aplicação em Células a Combustível; Universidade Federal do Espírito Santo: Vitória-ES, Brazil, 2013. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Composition (%mol) | Experimental Composition (%mol) 1 | a/Å | V/Å 3 | D 2/nm | Average Particle Size 3/nm | ECSA-H2 4/m2 gPt−1 | |||
|---|---|---|---|---|---|---|---|---|---|
| (111) | (200) | (220) | (311) | ||||||
| Pt70Sn10Ta20/C | Pt63Sn13Ta24/C | 3.941 ± 0.002 | 61.20 ± 0.08 | 4.3 | 4.6 | 3.4 | 2.9 | 5.5 | 14.6 |
| Pt70Sn10Ta15Ru5/C | Pt50Sn21Ta25Ru4/C | 3.944 ± 0.003 | 61.35 ± 0.15 | 3.9 | 3.7 | 2.9 | 3.6 | 3.8 | 8.7 |
| Pt70Sn10Ta10Ru10/C | Pt60Sn15Ta18Ru7/C | 3.942 ± 0.002 | 61.24 ± 0.11 | 5.3 | 5.2 | 4.8 | 4.7 | 4.8 | 10.7 |
| Pt70Sn10Ta5Ru15/C | Pt75Sn12Ta6Ru7/C | 3.944 ± 0.005 | 61.36 ± 0.28 | 4.2 | 3.7 | 3.3 | 3.4 | 5.0 | 7.5 |
| Oxidation Peaks | |||||||
|---|---|---|---|---|---|---|---|
| Catalysts | Eonset-Ethanol | (1) | (2) | (3) | |||
| E/V | E/V | E/V | E/V | ||||
| Pt63Sn13Ta24/C | 0.26 | 0.97 | 38.11 | 0.63 | 25.80 | 0.45 | 12.95 |
| Pt50Sn21Ta25Ru4/C | 0.33 | 1.16 | 34.64 | 0.86 | 23.08 | 0.47 | 9.23 |
| Pt60Sn15Ta18Ru7/C | 0.36 | 0.86 | 42.80 | 0.58 | 22.83 | 0.46 | 15.91 |
| Pt75Sn12Ta6Ru7/C | 0.20 | 1.00 | 43.55 | 0.70 | 30.75 | 0.46 | 15.91 |
| Catalyst | Loading Pt | Open Circuit Potential | Power Density | Ref. |
|---|---|---|---|---|
| Pt/C | 100% | 0.55 V | 10 mW cm−2 | [16] |
| 0.48 V | 7 mW cm−2 | [53] | ||
| PtSn/C | 80 wt% | 0.75 V | 15 W gPt−1 | [52] |
| 75 wt% | 0.78 V | 27 mW cm−2 | [42] | |
| 70 wt% | 0.70 V | 25 mW cm−2 | [53] | |
| 45 wt% | 0.83 V | 32 W gPt−1 | [19] | |
| PtRu/C | 70 wt% | 0.60 V | 8 W gPt−1 | [52] |
| 50 wt% | 0.70 V | 30 mW cm−2 | [54] | |
| 45 wt% | 0.75 V | 30 W gPt−1 | [19] | |
| PtSnRu/C | 30 wt% | 0.6 V | 17 W gPt−1 | [52] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queiroz, M.A.R.; Ribeiro, J. Catalysts of PtSn/C Modified with Ru and Ta for Electrooxidation of Ethanol. Catalysts 2019, 9, 277. https://doi.org/10.3390/catal9030277
Queiroz MAR, Ribeiro J. Catalysts of PtSn/C Modified with Ru and Ta for Electrooxidation of Ethanol. Catalysts. 2019; 9(3):277. https://doi.org/10.3390/catal9030277
Chicago/Turabian StyleQueiroz, Maria Aparecida Ribeiro, and Josimar Ribeiro. 2019. "Catalysts of PtSn/C Modified with Ru and Ta for Electrooxidation of Ethanol" Catalysts 9, no. 3: 277. https://doi.org/10.3390/catal9030277
APA StyleQueiroz, M. A. R., & Ribeiro, J. (2019). Catalysts of PtSn/C Modified with Ru and Ta for Electrooxidation of Ethanol. Catalysts, 9(3), 277. https://doi.org/10.3390/catal9030277