Atomic Layer Deposition for Preparation of Highly Efficient Catalysts for Dry Reforming of Methane



Abstract
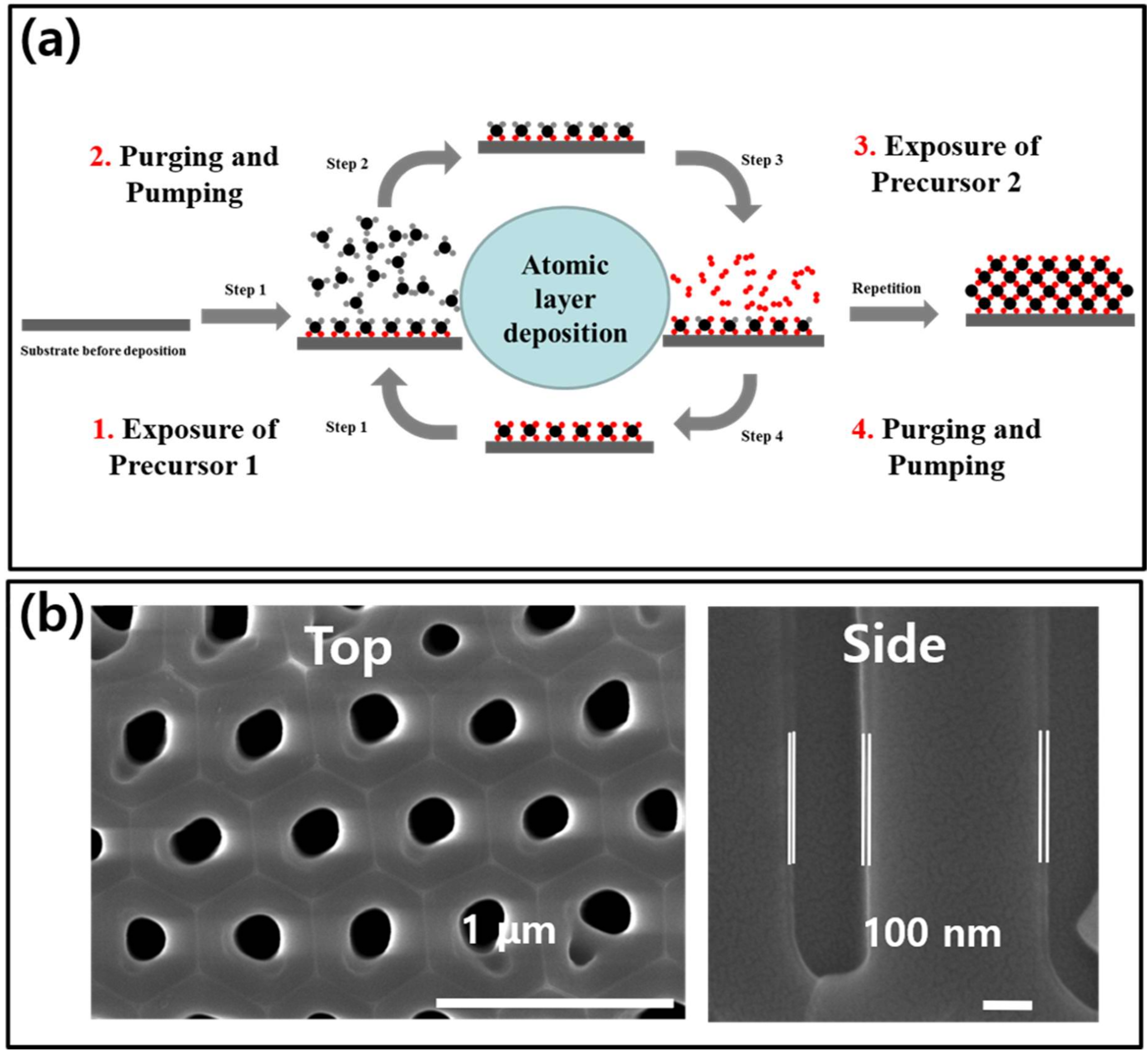
1. Introduction
2. Shell-Core-Type DRM Catalysts Prepared by ALD
3. DRM Catalysts Consisting of Nanoparticles Implemented in a Mesoporous Template Prepared by ALD
4. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Bowes, G. Facing the inevitable: Plants and increasing atmospheric CO2. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1993, 44, 309–332. [Google Scholar] [CrossRef]
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in carbon dioxide separation and capture: A review. J. Environ. Sci. 2008, 20, 14–27. [Google Scholar] [CrossRef]
- King, A.W.; Emanuel, W.R.; Post, W.M. Projecting future concentrations of atmospheric CO2 with global carbon cycle models: The importance of simulating historical changes. Environ. Manag. 1992, 16, 91–108. [Google Scholar] [CrossRef]
- Sozzani, P.; Bracco, S.; Comotti, A.; Ferretti, L.; Simonutti, R. Methane and carbon dioxide storage in a porous van der waals crystal. Angew. Chem. Int. Ed. 2005, 44, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Thallapally, P.K.; McGrail, B.P.; Brown, D.R.; Liu, J. Progress in adsorption-based CO-2 capture by metal–organic frameworks. Chem. Soc. Rev. 2012, 41, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Z.U.; Zhou, H.-C. Recent advances in carbon dioxide capture with metal-organic frameworks. Greenh. Gases 2012, 2, 239–259. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon dioxide capture in metal–organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-R.; Ma, Y.; McCarthy, M.C.; Sculley, J.; Yu, J.; Jeong, H.-K.; Balbuena, P.B.; Zhou, H.-C. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011, 255, 1791–1823. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, S.; Ma, X.; Gong, J. Recent advances in capture of carbon dioxide using alkali-metal-based oxides. Energy Environ. Sci. 2011, 4, 3805–3819. [Google Scholar] [CrossRef]
- Bhagiyalakshmi, M.; Lee, J.Y.; Jang, H.T. Synthesis of mesoporous magnesium oxide: Its application to CO2 chemisorption. Int. J. Greenh. Gas Control 2010, 4, 51–56. [Google Scholar] [CrossRef]
- Millward, A.R.; Yaghi, O.M. Metal−organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-G.; Kim, S.Y.; Kim, D.H.; Han, S.W.; Kim, I.H.; Lee, M.; Hwang, Y.K.; Kim, Y.D. High-performing and durable MgO/Ni catalysts via atomic layer deposition for CO2 reforming of methane (CRM). Appl. Catal. A 2016, 515, 45–50. [Google Scholar] [CrossRef]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Jitaru, M.; Lowy, D.A.; Toma, M.; Toma, B.C.; Oniciu, L. Electrochemical reduction of carbon dioxide on flat metallic cathodes. J. Appl. Electrochem. 1997, 27, 875–889. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward solar fuels: Photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Holtcamp, M.W. Catalysts for the reactions of epoxides and carbon dioxide. Coord. Chem. Rev. 1996, 153, 155–174. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Omae, I. Recent developments in carbon dioxide utilization for the production of organic chemicals. Coord. Chem. Rev. 2012, 256, 1384–1405. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Soltanieh, M.; Azar, K.M.; Saber, M. Development of a zero emission integrated system for co-production of electricity and methanol through renewable hydrogen and CO2 capture. Int. J. Greenh. Gas Control 2012, 7, 145–152. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S.; Mazaheri, H. A review on novel processes of biodiesel production from waste cooking oil. Appl. Energy 2013, 104, 683–710. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. The changing paradigm in CO2 utilization. J. CO2 Util. 2013, 3–4, 65–73. [Google Scholar] [CrossRef]
- Seo, O.H. Recent scientific progress on developing supported Ni catalysts for dry (CO2) reforming of methane. Catalysts 2018, 8, 110. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Rostrupnielsen, J.R.; Hansen, J.H.B. CO2-reforming of methane over transition metals. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, S.Y.; Han, S.W.; Cho, Y.K.; Jeong, M.-G.; Park, E.J.; Kim, Y.D. The catalytic stability of TiO2-shell/Ni-core catalysts for CO2 reforming of CH4. Appl. Catal. A 2015, 495, 184–191. [Google Scholar] [CrossRef]
- García, V.; Fernández, J.J.; Ruíz, W.; Mondragón, F.; Moreno, A. Effect of MgO addition on the basicity of Ni/ZrO2 and on its catalytic activity in carbon dioxide reforming of methane. Catal. Commun. 2009, 11, 240–246. [Google Scholar] [CrossRef]
- Gould, T.D.; Montemore, M.M.; Lubers, A.M.; Ellis, L.D.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Enhanced dry reforming of methane on Ni and Ni-Pt catalysts synthesized by atomic layer deposition. Appl. Catal. A 2015, 492, 107–116. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. Catalytic reforming of methane with carbon dioxide over nickel catalysts I. Catalyst characterization and activity. Appl. Catal. A 1996, 142, 73–96. [Google Scholar] [CrossRef]
- Kroll, V.C.H.; Swaan, H.M.; Mirodatos, C. Methane reforming reaction with carbon dioxide over Ni/SiO2 catalyst: I. deactivation studies. J. Catal. 1996, 161, 409–422. [Google Scholar] [CrossRef]
- Guczi, L.; Stefler, G.; Geszti, O.; Sajó, I.; Pászti, Z.; Tompos, A.; Schay, Z. Methane dry reforming with CO2: A study on surface carbon species. Appl. Catal. A 2010, 375, 236–246. [Google Scholar] [CrossRef]
- Liu, C.-J.; Ye, J.; Jiang, J.; Pan, Y. Progresses in the preparation of coke resistant Ni-based catalyst for steam and CO2 reforming of methane. ChemCatChem 2011, 3, 529–541. [Google Scholar] [CrossRef]
- Bouarab, R.; Akdim, O.; Auroux, A.; Cherifi, O.; Mirodatos, C. Effect of MgO additive on catalytic properties of Co/SiO2 in the dry reforming of methane. Appl. Catal. A 2004, 264, 161–168. [Google Scholar] [CrossRef]
- Xu, L.; Song, H.; Chou, L. One-pot synthesis of ordered mesoporous NiO–CaO–Al2O3 composite oxides for catalyzing CO2 reforming of CH4. ACS Catal. 2012, 2, 1331–1342. [Google Scholar] [CrossRef]
- Wang, S.-G.; Liao, X.-Y.; Cao, D.-B.; Huo, C.-F.; Li, Y.-W.; Wang, J.; Jiao, H. Factors controlling the Interaction of CO2 with transition metal surfaces. J. Phys. Chem. C 2007, 111, 16934–16940. [Google Scholar] [CrossRef]
- Quincoces, C.E.; Dicundo, S.; Alvarez, A.M.; González, M.G. Effect of addition of CaO on Ni/Al2O3 catalysts over CO2 reforming of methane. Mater. Lett. 2001, 50, 21–27. [Google Scholar] [CrossRef]
- Baudouin, D.; Rodemerck, U.; Krumeich, F.; Mallmann, A.D.; Szeto, K.C.; Ménard, H.; Veyre, L.; Candy, J.-P.; Webb, P.B.; Thieuleux, C.; et al. Particle size effect in the low temperature reforming of methane by carbon dioxide on silica-supported Ni nanoparticles. J. Catal. 2013, 297, 27–34. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Battocchio, C.; Lo Mastro, S.; Sodo, A. Ni/CeO2–Al2O3 catalysts for the dry reforming of methane: The effect of CeAlO3 content and nickel crystallite size on catalytic activity and coke resistance. Appl. Catal. A 2015, 500, 12–22. [Google Scholar] [CrossRef]
- Kim, J.-H.; Suh, D.J.; Park, T.-J.; Kim, K.-L. Effect of metal particle size on coking during CO2 reforming of CH4 over Ni–alumina aerogel catalysts. Appl. Catal. A 2000, 197, 191–200. [Google Scholar] [CrossRef]
- Slagtern, s.; Olsbye, U.; Blom, R.; Dahl, I.M. The influence of rare earth oxides on Ni/Al2O3 catalysts during CO2 reforming of CH4. Stud. Surf. Sci. Catal. 1997, 107, 497–502. [Google Scholar]
- Wang, S.; Lu, G.Q. Reforming of methane with carbon dioxide over Ni/Al2O3 catalysts: Effect of nickel precursor. Appl. Catal. A 1998, 169, 271–280. [Google Scholar] [CrossRef]
- Lemonidou, A.A.; Vasalos, I.A. Carbon dioxide reforming of methane over 5 wt.% Ni/CaO-Al2O3 catalyst. Appl. Catal. A 2002, 228, 227–235. [Google Scholar] [CrossRef]
- Sneh, O.; Clark-Phelps, R.B.; Londergan, A.R.; Winkler, J.; Seidel, T.E. Thin film atomic layer deposition equipment for semiconductor processing. Thin Solid Films 2002, 402, 248–261. [Google Scholar] [CrossRef]
- Kim, H. Atomic layer deposition of metal and nitride thin films: Current research efforts and applications for semiconductor device processing. J. Vac. Sci. Technol. B 2003, 21, 2231–2261. [Google Scholar] [CrossRef]
- Sheng, J.; Han, K.-L.; Hong, T.; Choi, W.-H.; Park, J.-S. Review of recent progresses on flexible oxide semiconductor thin film transistors based on atomic layer deposition processes. J. Semicond. 2018, 39, 011008. [Google Scholar] [CrossRef]
- Yan, B.; Li, X.; Bai, Z.; Song, X.; Xiong, D.; Zhao, M.; Li, D.; Lu, S. A review of atomic layer deposition providing high performance lithium sulfur batteries. J. Power Sources 2017, 338, 34–48. [Google Scholar] [CrossRef]
- Palmstrom, A.F.; Santra, P.K.; Bent, S.F. Atomic layer deposition in nanostructured photovoltaics: Tuning optical, electronic and surface properties. Nanoscale 2015, 7, 12266–12283. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.P.; Meng, X.; Elam, J.W.; Martinson, A.B.F. Atomic layer deposition of metal sulfide materials. Acc. Chem. Res. 2015, 48, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.R.; Hoogland, S.; Adachi, M.M.; Kanjanaboos, P.; Wong, C.T.O.; McDowell, J.J.; Xu, J.; Voznyy, O.; Ning, Z.; Houtepen, A.J.; et al. Perovskite thin films via atomic layer deposition. Adv. Mater. 2015, 27, 53–58. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.J.; Jackson, D.H.K.; Lee, J.; Canlas, C.; Stair, P.C.; Marshall, C.L.; Elam, J.W.; Kuech, T.F.; Dumesic, J.A.; Huber, G.W. Catalyst design with atomic layer deposition. ACS Catal. 2015, 5, 1804–1825. [Google Scholar] [CrossRef]
- Marichy, C.; Bechelany, M.; Pinna, N. Atomic layer deposition of nanostructured materials for energy and environmental applications. Adv. Mater. 2012, 24, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, X.; King, D.M.; Jiang, Y.-B.; Weimer, A.W. Highly dispersed Pt nanoparticle catalyst prepared by atomic layer deposition. Appl. Catal. B 2010, 97, 220–226. [Google Scholar] [CrossRef]
- Kim, D.H.; Sim, J.K.; Lee, J.; Seo, H.O.; Jeong, M.-G.; Kim, Y.D.; Kim, S.H. Carbon dioxide reforming of methane over mesoporous Ni/SiO2. Fuel 2013, 112, 111–116. [Google Scholar] [CrossRef]
- Kim, D.H.; Seo, H.O.; Jeong, M.-G.; Kim, Y.D. Reactivity and stability of Ni nanoparticles supported by mesoporous SiO2 and TiO2/SiO2 for CO2 Reforming of CH4. Catal. Lett. 2014, 144, 56–61. [Google Scholar] [CrossRef]
- Seo, H.O.; Sim, J.K.; Kim, K.-D.; Kim, Y.D.; Lim, D.C.; Kim, S.H. Carbon dioxide reforming of methane to synthesis gas over a TiO2–Ni inverse catalyst. Appl. Catal. A 2013, 451, 43–49. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Hultqvist, A.; Bent, S.F. A brief review of atomic layer deposition: From fundamentals to applications. Mater. Today 2014, 17, 236–246. [Google Scholar] [CrossRef]
- Ritala, M.; Leskelä, M.; Dekker, J.-P.; Mutsaers, C.; Soininen, P.J.; Skarp, J. Perfectly conformal TiN and Al2O3 films deposited by atomic layer deposition. Chem. Vap. Depos. 1999, 5, 7–9. [Google Scholar] [CrossRef]
- Singh, J.A.; Yang, N.; Bent, S.F. Nanoengineering heterogeneous catalysts by atomic layer deposition. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Qin, Y. Design and properties of confined nanocatalysts by atomic layer deposition. Acc. Chem. Res. 2017, 50, 2309–2316. [Google Scholar] [CrossRef] [PubMed]
- Leskelä, M.; Ritala, M. Atomic layer deposition (ALD): From precursors to thin film structures. Thin Solid Films 2002, 409, 138–146. [Google Scholar] [CrossRef]
- Puurunen, R.L.; Vandervorst, W. Island growth as a growth mode in atomic layer deposition: A phenomenological model. J. Appl. Phys. 2004, 96, 7686–7695. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, J.-Y.; Kang, S.-W. Film growth model of atomic layer deposition for multicomponent thin films. J. Appl. Phys. 2005, 97, 093505. [Google Scholar] [CrossRef]
- Aaltonen, T.; Alén, P.; Ritala, M.; Leskelä, M. Ruthenium thin films grown by atomic layer deposition. Chem. Vap. Depos. 2003, 9, 45–49. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, H.O.; Kim, D.W.; Kim, K.-D.; Luo, Y.; Lim, D.C.; Ju, H.; Kim, J.W.; Lee, J.; Kim, Y.D. A high-performing nanostructured TiO2 filter for volatile organic compounds using atomic layer deposition. Chem. Commun. 2011, 47, 5605–5607. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Mane, A.U.; Elam, J.W.; Croy, J.R. Amorphous metal fluoride passivation coatings prepared by atomic layer deposition on LiCoO2 for Li-ion batteries. Chem. Mater. 2015, 27, 1917–1920. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Lee, K.-T.; Huang, W.-K.; Siao, H.-Y.; Chang, Y.-C. High-performance, air-stable, low-temperature processed semitransparent perovskite solar cells enabled by atomic layer deposition. Chem. Mater. 2015, 27, 5122–5130. [Google Scholar] [CrossRef]
- Kozen, A.C.; Lin, C.-F.; Pearse, A.J.; Schroeder, M.A.; Han, X.; Hu, L.; Lee, S.-B.; Rubloff, G.W.; Noked, M. Next-generation lithium metal anode engineering via atomic layer deposition. ACS Nano 2015, 9, 5884–5892. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, Y.D. Oxidation behaviors of poly(3-hexylthiophene-2,5-diyl) on indium tin oxide surfaces without and with additional TiO2 thin films. Bull. Korean Chem. Soc. 2015, 36, 798–803. [Google Scholar]
- Kim, D.H.; Jeong, M.-G.; Seo, H.O.; Kim, Y.D. Oxidation behavior of P3HT layers on bare and TiO2-covered ZnO ripple structures evaluated by photoelectron spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-D.; Im, D.-C.; Jeong, M.-G.; Seo, H.O.; Seo, B.Y.; Lee, J.-Y.; Song, Y.-S.; Cho, S.; Lim, J.-H.; Kim, Y.D. Enhanced stability of organic photovoltaics by additional ZnO layers on rippled ZnO electron-collecting layer using atomic layer deposition. Bull. Korean Chem. Soc. 2014, 35, 353–356. [Google Scholar] [CrossRef]
- Kim, K.-D.; Lim, D.C.; Hu, J.; Kwon, J.-D.; Jeong, M.-G.; Seo, H.O.; Lee, J.Y.; Jang, K.-Y.; Lim, J.-H.; Lee, K.H.; et al. Surface modification of a ZnO electron-collecting layer using atomic layer deposition to fabricate high-Performing inverted organic photovoltaics. ACS Appl. Mater. Interfaces 2013, 5, 8718–8723. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-D.; Lim, D.C.; Seo, H.O.; Lee, J.Y.; Seo, B.Y.; Lee, D.J.; Song, Y.; Cho, S.; Lim, J.-H.; Kim, Y.D. Enhanced performance of organic photovoltaics by TiO2-interlayer with precisely controlled thickness between ZnO electron collecting and active layers. Appl. Surf. Sci. 2013, 279, 380–383. [Google Scholar] [CrossRef]
- Lim, D.C.; Kim, K.-D.; Park, S.-Y.; Hong, E.M.; Seo, H.O.; Lim, J.H.; Lee, K.H.; Jeong, Y.; Song, C.; Lee, E.; et al. Towards fabrication of high-performing organic photovoltaics: New donor-polymer, atomic layer deposited thin buffer layer and plasmonic effects. Energy Environ. Sci. 2012, 5, 9803–9807. [Google Scholar] [CrossRef]
- Park, S.-Y.; Seo, H.O.; Kim, K.-D.; Shim, W.H.; Heo, J.; Cho, S.; Kim, Y.D.; Lee, K.H.; Lim, D.C. Organic solar cells fabricated by one-step deposition of a bulk heterojunction mixture and TiO2/NiO hole-collecting agents. J. Phys. Chem. C 2012, 116, 15348–15352. [Google Scholar] [CrossRef]
- Park, S.-Y.; Seo, H.O.; Kim, K.-D.; Lee, J.E.; Kwon, J.-D.; Kim, Y.D.; Lim, D.C. Organic photovoltaics with high stability sustained for 100 days without encapsulation fabricated using atomic layer deposition. Phys. Status Solidi Rapid Res. Lett. 2012, 6, 196–198. [Google Scholar] [CrossRef]
- Seo, H.O.; Sim, C.W.; Kim, K.-D.; Kim, Y.D.; Park, J.H.; Lee, B.C.; Lee, K.H.; Lim, D.C. Influence of high-energy electron-beam on photocatalytic activity of TiO2 films on carbon-fiber deposited by atomic layer deposition. Radiat. Phys. Chem. 2012, 81, 290–294. [Google Scholar] [CrossRef]
- Kim, K.-D.; Dey, N.K.; Seo, H.O.; Kim, Y.D.; Lim, D.C.; Lee, M. Photocatalytic decomposition of toluene by nanodiamond-supported TiO2 prepared using atomic layer deposition. Appl. Catal. A 2011, 408, 148–155. [Google Scholar] [CrossRef]
- Schwab, G.-M. Electronics of Supported Catalysts. Adv. Catal. 1979, 27, 1–22. [Google Scholar]
- Rodríguez, J.A.; Hrbek, J. Inverse oxide/metal catalysts: A versatile approach for activity tests and mechanistic studies. Surf. Sci. 2010, 604, 241–244. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Graciani, J.; Senanayake, S.D.; Grinter, D.C.; Stacchiola, D.; Hrbek, J.; Fernández-Sanz, J. Inverse oxide/metal catalysts in fundamental studies and practical applications: A perspective of recent developments. J. Phys. Chem. Lett. 2016, 7, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Medlin, J.W. Catalyst design using an inverse strategy: From mechanistic studies on inverted model catalysts to applications of oxide-coated metal nanoparticles. Surf. Sci. Rep. 2018, 73, 117–152. [Google Scholar] [CrossRef]
- Mahapatra, M.; Gutiérrez, R.A.; Kang, J.; Rui, N.; Hamlyn, R.; Liu, Z.; Orozco, I.; Ramírez, P.J.; Senanayake, S.D.; Rodriguez, J.A. The behavior of inverse oxide/metal catalysts: CO oxidation and water-gas shift reactions over ZnO/Cu(111) surfaces. Surf. Sci. 2019, 681, 116–121. [Google Scholar] [CrossRef]
- Dey, N.K.; Kim, M.J.; Kim, K.-D.; Seo, H.O.; Kim, D.; Kim, Y.D.; Lim, D.C.; Lee, K.H. Adsorption and photocatalytic degradation of methylene blue over TiO2 films on carbon fiber prepared by atomic layer deposition. J. Mol. Catal. A Chem. 2011, 337, 33–38. [Google Scholar] [CrossRef]
- Barroso-Quiroga, M.M.; Castro-Luna, A.E. Catalytic activity and effect of modifiers on Ni-based catalysts for the dry reforming of methane. Int. J. Hydrogen Energy 2010, 35, 6052–6056. [Google Scholar] [CrossRef]
- Ginsburg, J.M.; Piña, J.; El Solh, T.; de Lasa, H.I. Coke formation over a nickel catalyst under methane dry reforming conditions: Thermodynamic and kinetic models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Chen, D.; Lødeng, R.; Anundskås, A.; Olsvik, O.; Holmen, A. Deactivation during carbon dioxide reforming of methane over Ni catalyst: Microkinetic analysis. Chem. Eng. Sci. 2001, 56, 1371–1379. [Google Scholar] [CrossRef]
- Pechimuthu, N.A.; Pant, K.K.; Dhingra, S.C. Deactivation studies over Ni−K/CeO2−Al2O3 catalyst for dry reforming of methane. Ind. Eng. Chem. Res. 2007, 46, 1731–1736. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Li, J.; Zhang, Y.; Hu, J. Carbon dioxide reforming of methane over nickel catalyst supported on MgO(111) nanosheets. Top. Catal. 2014, 57, 619–626. [Google Scholar] [CrossRef]
- Koo, K.Y.; Roh, H.-S.; Seo, Y.T.; Seo, D.J.; Yoon, W.L.; Park, S.B. Coke study on MgO-promoted Ni/Al2O3 catalyst in combined H2O and CO2 reforming of methane for gas to liquid (GTL) process. Appl. Catal. A 2008, 340, 183–190. [Google Scholar] [CrossRef]
- Xu, L.; Song, H.; Chou, L. Carbon dioxide reforming of methane over ordered mesoporous NiO–MgO–Al2O3 composite oxides. Appl. Catal. B 2011, 108–109, 177–190. [Google Scholar] [CrossRef]
- Wang, H.Y.; Ruckenstein, E. Carbon dioxide reforming of methane to synthesis gas over supported rhodium catalysts: The effect of support. Appl. Catal. A 2000, 204, 143–152. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Rodríguez-Ramos, I.; Anderson, J.A.; Guerrero-Ruiz, A. Mechanistic aspects of the dry reforming of methane over ruthenium catalysts. Appl. Catal. A 2000, 202, 183–196. [Google Scholar] [CrossRef]
- Amenomiya, Y.; Morikawa, Y.; Pleizier, G. Infrared spectroscopy of C18O2 on alumina. J. Catal. 1977, 46, 431–433. [Google Scholar] [CrossRef]
- Daza, C.E.; Gallego, J.; Moreno, J.A.; Mondragón, F.; Moreno, S.; Molina, R. CO2 reforming of methane over Ni/Mg/Al/Ce mixed oxides. Catal. Today 2008, 133–135, 357–366. [Google Scholar] [CrossRef]
- King, J.S.; Wittstock, A.; Biener, J.; Kucheyev, S.O.; Wang, Y.M.; Baumann, T.F.; Giri, S.K.; Hamza, A.V.; Baeumer, M.; Bent, S.F. Ultralow loading Pt nanocatalysts prepared by atomic layer deposition on carbon aerogels. Nano Lett. 2008, 8, 2405–2409. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-G.; Kim, D.H.; Lee, S.-K.; Lee, J.H.; Han, S.W.; Park, E.J.; Cychosz, K.A.; Thommes, M.; Hwang, Y.K.; Chang, J.-S.; et al. Decoration of the internal structure of mesoporous chromium terephthalate MIL-101 with NiO using atomic layer deposition. Microporous Mesoporous Mater. 2016, 221, 101–107. [Google Scholar] [CrossRef]
- Ferguson, J.D.; Weimer, A.W.; George, S.M. Atomic layer deposition of ultrathin and conformal Al2O3 films on BN particles. Thin Solid Films 2000, 371, 95–104. [Google Scholar] [CrossRef]
- Kaiser, N. Review of the fundamentals of thin-film growth. Appl. Opt. Appl. Opt. 2002, 41, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, T.; Ritala, M.; Sajavaara, T.; Keinonen, J.; Leskelä, M. Atomic layer deposition of platinum thin films. Chem. Mater. 2003, 15, 1924–1928. [Google Scholar] [CrossRef]
- Jeong, M.-G.; Kim, I.H.; Han, S.W.; Kim, D.H.; Kim, Y.D. Room temperature CO oxidation catalyzed by NiO particles on mesoporous SiO2 prepared via atomic layer deposition: Influence of pre-annealing temperature on catalytic activity. J. Mol. Catal. A Chem. 2016, 414, 87–93. [Google Scholar] [CrossRef]
- Kaydouh, M.N.; El Hassan, N.; Davidson, A.; Casale, S.; El Zakhem, H.; Massiani, P. Highly active and stable Ni/SBA-15 catalysts prepared by a “two solvents” method for dry reforming of methane. Microporous Mesoporous Mater. 2016, 220, 99–109. [Google Scholar] [CrossRef]
- Kang, D.; Lim, H.S.; Lee, J.W. Enhanced catalytic activity of methane dry reforming by the confinement of Ni nanoparticles into mesoporous silica. Int. J. Hydrogen Energy 2017, 42, 11270–11282. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Li, L.; Liu, G.; Liang, X. Highly active and stable alumina supported nickel nanoparticle catalysts for dry reforming of methane. Appl. Catal. B 2017, 201, 302–309. [Google Scholar] [CrossRef]
- Han, S.W.; Kim, I.H.; Kim, D.H.; Park, K.J.; Park, E.J.; Jeong, M.-G.; Kim, Y.D. Temperature regulated-chemical vapor deposition for incorporating NiO nanoparticles into mesoporous media. Appl. Surf. Sci. 2016, 385, 597–604. [Google Scholar] [CrossRef]
- Kim, I.H.; Seo, H.O.; Park, E.J.; Han, S.W.; Kim, Y.D. Low temperature CO oxidation over Iron oxide nanoparticles decorating internal structures of a mesoporous alumina. Sci. Rep. 2017, 7, 40497. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Park, E.J.; Park, C.H.; Han, S.W.; Seo, H.O.; Kim, Y.D. Activity of catalysts consisting of Fe2O3 nanoparticles decorating entire internal structure of mesoporous Al2O3 bead for toluene total oxidation. Catal. Today 2017, 295, 56–64. [Google Scholar] [CrossRef]
- Kim, I.H.; Park, C.H.; Woo, T.G.; Jeong, J.H.; Jeon, C.S.; Kim, Y.D. Comparative Studies of Mesoporous Fe2O3/Al2O3 and Fe2O3/SiO2 fabricated by temperature-regulated chemical vapour deposition as catalysts for acetaldehyde oxidation. Catal. Lett. 2018, 148, 454–464. [Google Scholar] [CrossRef]
- Seo, M.; Kim, S.Y.; Kim, Y.D.; Park, E.D.; Uhm, S. Highly stable barium zirconate supported nickel oxide catalyst for dry reforming of methane: From powders toward shaped catalysts. Int. J. Hydrogen Energy 2018, 43, 11355–11362. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Number of Cycles | BET Surface Area (m2/g) | BJH Pore Volume (cm3/g) | Mean Thickness of Metal Oxide | Reference |
|---|---|---|---|---|---|
| Bare Ni | - | 3.5 | 5.5 × 10−3 | - | [13,30] |
| MgO/Ni | 50 | 3.5 | 4.3 × 10−3 | - | [13] |
| 200 | 1.9 | 7.5 × 10−3 | ~20 | ||
| TiO2/Ni | 100 | 4.9 | 5 × 10−3 | - | [30] |
| 500 | 25 | 1.3 × 10−3 | ~40 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.Y.; Cha, B.J.; Saqlain, S.; Seo, H.O.; Kim, Y.D. Atomic Layer Deposition for Preparation of Highly Efficient Catalysts for Dry Reforming of Methane. Catalysts 2019, 9, 266. https://doi.org/10.3390/catal9030266
Kim SY, Cha BJ, Saqlain S, Seo HO, Kim YD. Atomic Layer Deposition for Preparation of Highly Efficient Catalysts for Dry Reforming of Methane. Catalysts. 2019; 9(3):266. https://doi.org/10.3390/catal9030266
Chicago/Turabian StyleKim, Soong Yeon, Byeong Jun Cha, Shahid Saqlain, Hyun Ook Seo, and Young Dok Kim. 2019. "Atomic Layer Deposition for Preparation of Highly Efficient Catalysts for Dry Reforming of Methane" Catalysts 9, no. 3: 266. https://doi.org/10.3390/catal9030266
APA StyleKim, S. Y., Cha, B. J., Saqlain, S., Seo, H. O., & Kim, Y. D. (2019). Atomic Layer Deposition for Preparation of Highly Efficient Catalysts for Dry Reforming of Methane. Catalysts, 9(3), 266. https://doi.org/10.3390/catal9030266