Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs



Abstract
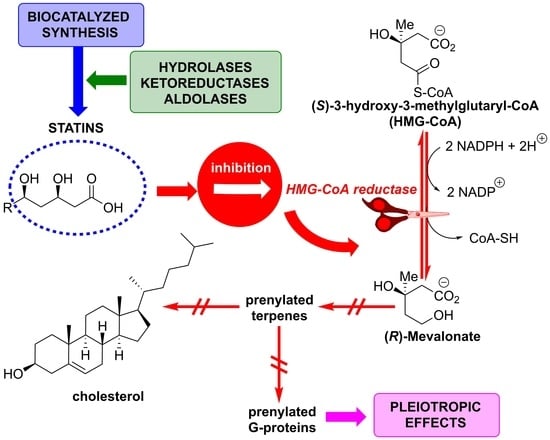
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. New Therapeutic Effects of Statins
2.1. Cardiovascular Effects
2.2. Immunomodulatory Effects
2.3. Neurological Disorders
2.3.1. Stroke
2.3.2. Alzheimer’s Disease (AD)
2.3.3. Parkinson’s Desease (PD)
2.3.4. Depression
2.3.5. Epilepsy
2.4. Cancer
3. Biocatalyzed Synthesis of Statins
3.1. Simvastatin
- Catalyst is produced efficiently from renewable feedstock.
- Reduced use of toxic and hazardous substances like tert-butyl dimethyl silane chloride, methyl iodide, and n-butyl lithium.
- Improved energy efficiency as the reaction is run at ambient temperature and at near atmospheric pressure.
- Reduction in solvent use because of the aqueous nature of the reaction conditions.
- The only by-product (methyl 3-mercaptopropionic acid) is recycled.
- The major waste streams generated are biodegraded in bio treatment facilities.
- Codexis’ process can produce simvastatin with yields of 97%, significant when compared with <70% with other manufacturing routes.
3.2. Biocatalyzed Synthesis of the Lateral Chain of Superstatins
3.2.1. Hydrolases as Catalysts for the Preparation of the Lateral Chain of Atorvastatin and Other Superstatins
3.2.2. Ketoreductases as Catalysts for the Preparation of the Lateral Chain of Atorvastatin and Other Superstatins
3.2.3. Aldolases for the Preparation of the Lateral Chain of Atorvastatin and Other Superstatins
4. Prognosis and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Raised Cholesterol. Situation and Trends. Available online: https://www.who.int/gho/ncd/risk_factors/cholesterol_text/en/ (accessed on 15 February 2019).
- Stein, E.A. The power of statins: Aggressive lipid lowering. Clin. Cardiol. 2003, 26, 25–31. [Google Scholar] [CrossRef]
- Bifulco, M.; Endo, A. Statin: New life for an old drug. Pharmacol. Res. 2014, 88, 1–2. [Google Scholar] [CrossRef]
- Stossel, T.P. The discovery of statins. Cell 2008, 134, 903–905. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinum. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef]
- Brown, A.G.; Smale, T.C.; King, T.J.; Hasenkamp, R.; Thompson, R.H. Crystal and molecular-structure of compactin, a new antifungal metabolite from Penicillium brevicompactum. J. Chem. Soc. Perkin Trans. 1 1976, 1165–1173. [Google Scholar] [CrossRef]
- Moore, R.N.; Bigam, G.; Chan, J.K.; Hogg, A.M.; Nakashima, T.T.; Vederas, J.C. Biosynthesis of the hypocholesterolemic agent mevinolin by Aspergillus terreus. Determination of the origin of carbon, hydrogen, and oxygen-atoms by 13C NMR and Mass Spectrometry. J. Am. Chem. Soc. 1985, 107, 3694–3701. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Cimerman, A. Pleurotus fruiting bodies contain the inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase—Lovastatin. Exp. Mycol. 1995, 19, 1–6. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Shi, Y.; Grimsgaard, S.; Alraek, T.; Fonnebo, V. Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: A meta-analysis of randomized controlled trials. Chin. Med. 2006, 1, 4. [Google Scholar] [CrossRef]
- Mol, M.; Erkelens, D.W.; Leuven, J.A.G.; Schouten, J.A.; Stalenhoef, A.F.H. Simvastatin (MK-733)—A potent cholesterol-synthesis inhibitor in heterozygous familial hypercholesterolemia. Atherosclerosis 1988, 69, 131–137. [Google Scholar] [CrossRef]
- Yoshino, G.; Kazumi, T.; Kasama, T.; Iwatani, I.; Iwai, M.; Inui, A.; Otsuki, M.; Baba, S. Effect of CS-514, an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, on lipoprotein and apolipoprotein in plasma of hypercholesterolemic diabetics. Diabetes Res. Clin. Pract. 1986, 2, 179–181. [Google Scholar] [CrossRef]
- Li, J.J. Triumph of the Heart: The Story of Statins; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Casar, Z. Historic Overview and Recent Advances in the Synthesis of Super-statins. Curr. Org. Chem. 2010, 14, 816–845. [Google Scholar] [CrossRef]
- Lindsley, C.W. The top prescription drugs of 2010 in the United States: Antipsychotics show strong growth. ACS Chem. Neurosci. 2011, 2, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. The top prescription drugs of 2011 in the United States: Antipsychotics and antidepressants once again lead CNS therapeutics. ACS Chem. Neurosci. 2012, 3, 630–631. [Google Scholar] [CrossRef]
- Lindsley, C.W. 2012 Trends and statistics for prescription medications in the United States: CNS therapeutics continue to hold leading positions. ACS Chem. Neurosci. 2013, 4, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. The top prescription drugs of 2012 globally: Biologics dominate, but small molecule CNS drugs hold on to top spots. ACS Chem. Neurosci. 2013, 4, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. 2013 Trends and statistics for prescription medications in the United States: CNS highest ranked and record number of prescriptions dispensed. ACS Chem. Neurosci. 2015, 6, 356–357. [Google Scholar] [CrossRef] [PubMed]
- Aitken, M.; Kleinrock, M.; Lyle, J.; Nass, D.; Caskey, L. Medicines Use and Spending Shifts: A Review of the Use of Medicines in the U.S. in 2014; IMS Institute for Healthcare Informatics: Parsippany, NJ, USA, 2015. [Google Scholar]
- Lindsley, C.W. 2014 Prescription medications in the United States: Tremendous growth, specialty/orphan drug expansion, and dispensed prescriptions continue to increase. ACS Chem. Neurosci. 2015, 6, 811–812. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. 2014 Global prescription medication statistics: Strong growth and CNS well represented. ACS Chem. Neurosci. 2015, 6, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.K.; Watanabe, K.; Wojcicki, W.A.; Wang, C.C.C.; Tang, Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem. Biol. 2006, 13, 1161–1169. [Google Scholar] [CrossRef]
- FiercePharma. The Cardiovascular Scene to Shift by 2024 as Next-Generation Drugs like Eliquis and Xarelto Eclipse Stalwarts. Available online: https://www.fiercepharma.com/pharma/cardiovascular-landscape-from-2017-to-2024-featuring-eliquis-xarelto-and-more (accessed on 14 February 2019).
- Watanabe, M.; Koike, H.; Ishiba, T.; Okada, T.; Seo, S.; Hirai, K. Synthesis and biological activity of methanesulfonamide pyrimidine- and N-methanesulfonyl pyrrole-substituted 3,5-dihydroxy-6-heptenoates, a novel series of HMG-CoA reductase inhibitors. Bioorg. Med. Chem. 1997, 5, 437–444. [Google Scholar] [CrossRef]
- Demasi, M. Statin wars: Have we been misled about the evidence? A narrative review. Br. J. Sports Med. 2018, 52, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A. Clinical evidence of statin therapy in non-dyslipidemic disorders. Pharmacol. Res. 2014, 88, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liu, P.Y.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Cordle, A.; Koenigsknecht-Talboo, J.; Wilkinson, B.; Limpert, A.; Landreth, G. Mechanisms of statin-mediated inhibition of small G-protein function. J. Biol. Chem. 2005, 280, 34202–34209. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Marasco, G.; Proto, M.C.; Laezza, C.; Gazzerro, P.; Bifulco, M. Statins in neurological disorders: An overview and update. Pharmacol. Res. 2014, 88, 74–83. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Delles, C.; Jacobi, J.; Schlaich, M.P.; Schneider, M.; Schmitz, G.; Schmieder, R.E. Rapid improvement of nitric oxide bioavailability after lipid-lowering therapy with cerivastatin within two weeks. J. Am. Coll. Cardiol. 2001, 37, 1351–1358. [Google Scholar] [CrossRef]
- Cheng, W.H.; Ho, W.Y.; Chang, C.F.; Lu, P.J.; Cheng, P.W.; Yeh, T.C.; Hong, L.Z.; Sun, G.C.; Hsiao, M.; Tseng, C.J. Simvastatin induces a central hypotensive effect via Ras-mediated signalling to cause eNOS up-regulation. Br. J. Pharmacol. 2013, 170, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Walter, M.F.; Jacob, R.F. Effects of HMG-CoA reductase inhibitors on endothelial function—Role of microdomains and oxidative stress. Circulation 2004, 109, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mihos, C.G.; Salas, M.J.; Santana, O. The pleiotropic effects of the hydroxy-methyl-glutaryl-CoA reductase inhibitors in cardiovascular disease a comprehensive review. Cardiol. Rev. 2010, 18, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Yanuck, D.; Mihos, C.G.; Santana, O. Mechanisms and clinical evidence of the pleiotropic effects of the Hydroxy-Methyl-Glutaryl-CoA Reductase inhibitors in Central Nervous System disorders: A comprehensive review. Int. J. Neurosci. 2012, 122, 619–629. [Google Scholar] [CrossRef]
- Mihos, C.G.; Artola, R.T.; Santana, O. The pleiotropic effects of the hydroxy-methyl-glutaryl-CoA reductase inhibitors in rheumatologic disorders: A comprehensive review. Rheumatol. Int. 2012, 32, 287–294. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Butterfield, D.A. Statins more than cholesterol lowering agents in Alzheimer disease: Their pleiotropic functions as potential therapeutic targets. Biochem. Pharmacol. 2014, 88, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Mihos, C.G.; Pineda, A.M.; Santana, O. Cardiovascular effects of statins, beyond lipid-lowering properties. Pharmacol. Res. 2014, 88, 12–19. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.M.; Downey, J.M. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct-sparing strategies. Cardiovasc. Res. 2006, 70, 231–239. [Google Scholar] [CrossRef]
- Mills, E.J.; Wu, P.; Chong, G.; Ghement, I.; Singh, S.; Akl, E.A.; Eyawo, O.; Guyatt, G.; Berwanger, O.; Briel, M. Efficacy and safety of statin treatment for cardiovascular disease: A network meta-analysis of 170 255 patients from 76 randomized trials. QJM-Int. J. Med. 2011, 104, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Lin, L.; Zhao, Y.J.; Khoo, A.L.; Davis, B.R.; Yong, Q.W.; Yeo, T.C.; Lim, B.P. Statins for primary prevention of cardiovascular disease in elderly patients: Systematic review and meta-analysis. Drugs Aging 2015, 32, 649–661. [Google Scholar] [CrossRef]
- Tervonen, T.; Naci, H.; van Valkenhoef, G.; Ades, A.E.; Angelis, A.; Hillege, H.L.; Postmus, D. Applying multiple criteria decision analysis to comparative benefit-risk assessment: Choosing among statins in primary prevention. Med. Decis. Mak. 2015, 35, 859–871. [Google Scholar] [CrossRef]
- Ulivieri, C.; Baldari, C.T. Statins: From cholesterol-lowering drugs to novel immunomodulators for the treatment of Th17-mediated autoimmune diseases. Pharmacol. Res. 2014, 88, 41–52. [Google Scholar] [CrossRef]
- Scheele, J.S.; Marks, R.E.; Boss, G.R. Signaling by small GTPases in the immune system. Immunol. Rev. 2007, 218, 92–101. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef]
- Chow, S.C. Immunomodulation by statins: Mechanisms and potential impact on autoimmune diseases. Arch. Immunol. Ther. Exp. (Warsz.) 2009, 57, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Khattri, S.; Zandman-Goddard, G. Statins and autoimmunity. Immunol. Res. 2013, 56, 348–357. [Google Scholar] [CrossRef]
- Ciurleo, R.; Bramanti, P.; Marino, S. Role of statins in the treatment of multiple sclerosis. Pharmacol. Res. 2014, 87, 133–143. [Google Scholar] [CrossRef]
- Pihl-Jensen, G.; Tsakiri, A.; Frederiksen, J.L. Statin treatment in multiple sclerosis: A systematic review and meta-analysis. CNS Drugs 2015, 29, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, M.; Mathieu, S.; Hermet, M.; Makarawiez, C.; Bruckert, E. Do all lupus patients need statins? Jt. Bone Spine 2013, 80, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-H.; Chen, P.-C.; Yang, Y.-H.; Wang, L.-C.; Lee, J.-H.; Lin, Y.-T.; Chiang, B.-L. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis 2015, 243, 11–18. [Google Scholar] [CrossRef]
- Ruiz-Limon, P.; Barbarroja, N.; Perez-Sanchez, C.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Almaden, Y.; Segui, P.; Khraiwesh, H.; et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: Effects of in vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, B.; Wang, W.; Zhang, C.; Zhang, M.; Zhang, Y.; Xia, Y.; Dong, Z.; Guo, Y.; An, F. Effects of HMG-CoA Reductase inhibitor on experimental autoimmune myocarditis. Cardiovasc. Drugs Ther. 2012, 26, 121–130. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. Statins as a new therapeutic perspective in myocarditis and postmyocarditis dilated cardiomyopathy. Cardiovasc. Drugs Ther. 2013, 27, 365–369. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimojo, N.; Sakai, S.; Machino-Ohtsuka, T.; Imanaka-Yoshida, K.; Hiroe, M.; Tsujimura, Y.; Kimura, T.; Sato, A.; Yasutomi, Y.; et al. Pitavastatin regulates helper T-cell differentiation and ameliorates autoimmune myocarditis in mice. Cardiovasc. Drugs Ther. 2013, 27, 413–424. [Google Scholar] [CrossRef]
- Lv, S.; Liu, Y.; Zou, Z.; Li, F.; Zhao, S.; Shi, R.; Bian, R.; Tian, H. The impact of statins therapy on disease activity and inflammatory factor in patients with rheumatoid arthritis: A meta-analysis. Clin. Exp. Rheumatol. 2015, 33, 69–76. [Google Scholar]
- Tascilar, K.; Dell’Aniello, S.; Hudson, M.; Suissa, S. Statins and risk of rheumatoid arthritis: A nested case-control study. Arthritis Rheumatol. 2016, 68, 2603–2611. [Google Scholar] [CrossRef]
- de Jong, H.J.I.; Tervaert, J.W.C.; Lalmohamed, A.; de Vries, F.; Vandebriel, R.J.; van Loveren, H.; Klungel, O.H.; van Staa, T.P. Pattern of risks of rheumatoid arthritis among patients using statins: A cohort study with the clinical practice research datalink. PLoS ONE 2018, 13, 1–17. [Google Scholar] [CrossRef]
- McFarland, A.J.; Anoopkumar-Dukie, S.; Arora, D.S.; Grant, G.D.; McDermott, C.M.; Perkins, A.V.; Davey, A.K. Molecular mechanisms underlying the effects of statins in the central nervous system. Int. J. Mol. Sci. 2014, 15, 20607–20637. [Google Scholar] [CrossRef]
- Qizilbash, N.; Lewington, S.; Duffy, S.; Peto, R.; Smith, T.; Spiegelhalter, D.; Iso, H.; Shimamoto, T.; Komachi, Y.; Iida, M.; et al. Cholesterol, diastolic blood pressure, and stroke: 13000 strokes in 450000 people in 45 prospective cohorts. Lancet 1995, 346, 1647–1653. [Google Scholar]
- O’Brien, E.C.; Greiner, M.A.; Xian, Y.; Fonarow, G.C.; Olson, D.M.; Schwamm, L.H.; Bhatt, D.L.; Smith, E.E.; Maisch, L.; Hannah, D.; et al. Clinical effectiveness of statin therapy after ischemic stroke: Primary results from the statin therapeutic area of the Patient-centered Research into Outcomes Stroke Patients prefer and Effectiveness Research (PROSPER) study. Circulation 2015, 132, 1404–1413. [Google Scholar] [CrossRef]
- Naci, H.; Brugts, J.J.; Fleurence, R.; Ades, A.E. Comparative effects of statins on major cerebrovascular events: A multiple-treatments meta-analysis of placebo-controlled and active-comparator trials. QJM-Int. J. Med. 2013, 106, 299–306. [Google Scholar] [CrossRef]
- Markel, A. Statins and peripheral arterial disease. Int. Angiol. 2015, 34, 416–427. [Google Scholar]
- Colivicchi, F.; Bassi, A.; Santini, M.; Caltagirone, C. Discontinuation of statin therapy and clinical outcome after ischemic stroke. Stroke 2007, 38, 2652–2657. [Google Scholar] [CrossRef]
- Laloux, P. Risk and benefit of statins in stroke secondary prevention. Curr. Vasc. Pharmacol. 2013, 11, 812–816. [Google Scholar] [CrossRef]
- Song, B.; Wang, Y.L.; Zhao, X.Q.; Liu, L.P.; Wang, C.X.; Wang, A.X.; Du, W.L.; Wang, Y.J. Association between statin use and short-term outcome based on severity of ischemic stroke: A cohort study. PLoS ONE 2014, 9, 7. [Google Scholar] [CrossRef]
- Gutierrez-Vargas, J.; Cespedes-Rubio, A.; Cardona-Gomez, G. Perspective of synaptic protection after post-infarction treatment with statins. J. Transl. Med. 2015, 13, 118. [Google Scholar] [CrossRef]
- Bustamante, A.; Montaner, J. Statin therapy should not be discontinued in patients with intracerebral hemorrhage. Stroke 2013, 44, 2060–2061. [Google Scholar] [CrossRef]
- Molina, C.A.; Selim, M.H. Continued statin treatment after acute intracranial hemorrhage fighting fire with fire. Stroke 2013, 44, 2062–2063. [Google Scholar] [CrossRef]
- Pan, Y.S.; Jing, J.; Wang, Y.L.; Zhao, X.Q.; Song, B.; Wang, W.J.; Wang, D.; Liu, G.F.; Liu, L.P.; Wang, C.X.; et al. Use of statin during hospitalization improves the outcome after intracerebral hemorrhage. CNS Neurosci. Ther. 2014, 20, 548–555. [Google Scholar] [CrossRef]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef]
- Silva, T.; Teixeira, J.; Remiao, F.; Borges, F. Alzheimer’s disease, cholesterol, and statins: The junctions of important metabolic pathways. Angew. Chem. Int. Ed. 2013, 52, 1110–1121. [Google Scholar] [CrossRef]
- Hottman, D.A.; Li, L. Protein prenylation and synaptic plasticity: Implications for Alzheimer’s disease. Mol. Neurobiol. 2014, 50, 177–185. [Google Scholar] [CrossRef]
- Mendoza-Oliva, A.; Zepeda, A.; Arias, C. The complex actions of statins in brain and their relevance for Alzheimer’s disease treatment: An analytical review. Curr. Alzheimer Res. 2014, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Wanamaker, B.L.; Swiger, K.J.; Blumenthal, R.S.; Martin, S.S. Cholesterol, statins, and dementia: What the cardiologist should know. Clin. Cardiol. 2015, 38, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Cullen, E.I.; Friedhoff, L.T. Pharmacological concentrations of the HMG-CoA reductase inhibitor lovastatin decrease the formation of the Alzheimer beta-amyloid peptide in vitro and in patients. Front. Biosci. 2002, 7, A50–A59. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, W.; Cheng, S.W.; Cao, D.F.; Parent, M. Isoprenoids and related pharmacological interventions: Potential application in Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Cibickova, L.; Palicka, V.; Cibicek, N.; Cermakova, E.; Micuda, S.; Bartosova, L.; Jun, D. Differential effects of statins and alendronate on cholinesterases in serum and brain of rats. Physiol. Res. 2007, 56, 765–770. [Google Scholar]
- Mozayan, M.; Lee, T.J.F. Statins prevent cholinesterase inhibitor blockade of sympathetic alpha 7-nAChR-mediated currents in rat superior cervical ganglion neurons. Am. J. Physiol.-Heart Circul. Physiol. 2007, 293, H1737–H1744. [Google Scholar] [CrossRef] [PubMed]
- Ghodke, R.M.; Tour, N.; Devi, K. Effects of statins and cholesterol on memory functions in mice. Metab. Brain Dis. 2012, 27, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Macan, M.; Vuksic, A.; Zunec, S.; Konjevoda, P.; Lovric, J.; Kelava, M.; Stambuk, N.; Vrkic, N.; Bradamante, V. Effects of simvastatin on malondialdehyde level and esterase activity in plasma and tissue of normolipidemic rats. Pharmacol. Rep. 2015, 67, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bosel, J.; Gandor, F.; Harms, C.; Synowitz, M.; Harms, U.; Djoufack, P.C.; Megow, D.; Dirnagl, U.; Hortnagl, H.; Fink, K.B.; et al. Neuroprotective effects of atorvastatin against glutamate-induced excitotoxicity in primary cortical neurones. J. Neurochem. 2005, 92, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Krisanova, N.; Sivko, R.; Kasatkina, L.; Borisova, T. Neuroprotection by lowering cholesterol: A decrease in membrane cholesterol content reduces transporter-mediated glutamate release from brain nerve terminals. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 1553–1561. [Google Scholar] [CrossRef]
- Sala, S.G.; Munoz, U.; Bartolome, F.; Bermejo, F.; Martin-Requero, A. HMG-CoA reductase inhibitor simvastatin inhibits cell cycle progression at the G(1)/S checkpoint in immortalized lymphocytes from Alzheimer’s disease patients independently of cholesterol-lowering effects. J. Pharmacol. Exp. Ther. 2008, 324, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Merla, R.; Ye, Y.; Lin, Y.; Manickavasagam, S.; Huang, M.-H.; Perez-Polo, R.J.; Uretsky, B.F.; Birnbaum, Y. The central role of adenosine in statin-induced ERK1/2, Akt, and eNOS phosphorylation. Am. J. Physiol.-Heart Circul. Physiol. 2007, 293, H1918–H1928. [Google Scholar] [CrossRef] [PubMed]
- Cordle, A.; Landreth, G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J. Neurosci. 2005, 25, 299–307. [Google Scholar] [CrossRef]
- Fonseca, A.C.R.G.; Resende, R.; Oliveira, C.R.; Pereira, C.M.F. Cholesterol and statins in Alzheimer’s disease: Current controversies. Exp. Neurol. 2010, 223, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Mans, R.A.; McMahon, L.L.; Li, L. Simvastatin-mediated enhancement of long-term potentiation is driven by farnesyl-pyrophosphate depletion and inhibition of farnesylation. Neuroscience 2012, 202, 1–9. [Google Scholar] [CrossRef]
- Mans, R.A.; Chowdhury, N.; Cao, D.; McMahon, L.L.; Li, L. Simvastatin enhances hippocampal long-term potentiation in C57BL/6 mice. Neuroscience 2010, 166, 435–444. [Google Scholar] [CrossRef]
- Parent, M.; Hottman, D.A.; Cheng, S.W.; Zhang, W.; McMahon, L.L.; Yuan, L.L.; Li, L. Simvastatin treatment enhances NMDAR-mediated synaptic transmission by upregulating the surface distribution of the GluN2B subunit. Cell. Mol. Neurobiol. 2014, 34, 693–705. [Google Scholar] [CrossRef]
- Wollmuth, L.P. Is cholesterol good or bad for your brain?—NMDARs have a say. J. Physiol.-Lond. 2015, 593, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, M.; Kundu, M.; Corbett, G.T.; Rangaswamy, S.B.; Mishra, R.K.; Luan, C.H.; Gonzalez, F.J.; Pahan, K. HMG-CoA Reductase Inhibitors bind to PPAR alpha to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab. 2015, 22, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Yum, K.S.; Chang, J.Y.; Kim, M.; Ahn, J.Y.; Kim, S.; Lapchak, P.A.; Han, M.K. Dose-specific effect of simvastatin on hypoxia-induced HIF-1 alpha and BACE expression in Alzheimer’s disease cybrid cells. BMC Neurol. 2015, 15, 7. [Google Scholar] [CrossRef]
- Power, M.C.; Weuve, J.; Sharrett, A.R.; Blacker, D.; Gottesman, R.F. Statins, cognition, and dementia-systematic review and methodological commentary. Nat. Rev. Neurol. 2015, 11, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Strom, B.L.; Schinnar, R.; Karlawish, J.; Hennessy, S.; Teal, V.; Bilker, W.B. Statin therapy and risk of acute memory impairment. JAMA Intern. Med. 2015, 175, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Oliva, A.; Ferrera, P.; Fragoso-Medina, J.; Arias, C. Lovastatin differentially affects neuronal cholesterol and amyloid- production invivo and invitro. CNS Neurosci. Ther. 2015, 21, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.S.; Lin, C.L.; Lin, M.C.; Sung, F.C.; Kao, C.H. Decreased prevalence of dementia associated with statins: A national population-based study. Eur. J. Neurol. 2015, 22, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. S1), S207–S209. [Google Scholar] [CrossRef]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lin, C.-H.; Wu, R.-M.; Lin, M.-S.; Lin, J.-W.; Chang, C.-H.; Lai, M.-S. Discontinuation of statin therapy associates with Parkinson disease. A population-based study. Neurology 2013, 81, 410–416. [Google Scholar] [CrossRef]
- Tison, F.; Negre-Pages, L.; Meissner, W.G.; Dupouy, S.; Li, Q.; Thiolat, M.-L.; Thiollier, T.; Galitzky, M.; Ory-Magne, F.; Milhet, A.; et al. Simvastatin decreases levodopa-induced dyskinesia in monkeys, but not in a randomized, placebo-controlled, multiple cross-over (“n-of-1”) exploratory trial of simvastatin against levodopa-induced dyskinesia in Parkinson’s disease patients. Parkinsonism Relat. Disord. 2013, 19, 416–421. [Google Scholar] [CrossRef]
- Selley, M.L. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Long, L.; Yan, J.Q.; Wei, L.; Pan, M.Q.; Gao, H.M.; Zhou, P.; Liu, M.; Zhu, C.S.; Tang, B.S.; et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci. Ther. 2013, 19, 170–177. [Google Scholar] [CrossRef]
- Yan, J.; Xu, Y.; Zhu, C.; Zhang, L.; Wu, A.; Yang, Y.; Xiong, Z.; Deng, C.; Huang, X.-F.; Yenari, M.A.; et al. Simvastatin prevents dopaminergic neurodegeneration in experimental parkinsonian models: The association with anti-inflammatory responses. PLoS ONE 2011, 6, e20945. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cao, X.B.; Zhang, T.; Shi, Q.Q.; Chen, Z.B.; Tang, B.S. Effect of simvastatin on L-DOPA-induced abnormal involuntary movements of hemiparkinsonian rats. Neurol. Sci. 2015, 36, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21(Ras) and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Jick, S.S.; Jick, H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch. Intern. Med. 2003, 163, 1926–1932. [Google Scholar] [CrossRef]
- Feng, L.; Tan, C.-H.; Merchant, R.A.; Ng, T.-P. Association between depressive symptoms and use of HMG-CoA reductase inhibitors (statins), corticosteroids and histamine H-2 receptor antagonists in community-dwelling older persons—Cross-sectional analysis of a population-based cohort. Drugs Aging 2008, 25, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Stafford, L.; Berk, M. The use of statins after a cardiac intervention is associated with reduced risk of subsequent depression: Proof of concept for the inflammatory and oxidative hypotheses of depression? J. Clin. Psychiatry 2011, 72, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Zhao, S.; Whooley, M.A. Statin use and risk of depression in patients with coronary heart disease: Longitudinal data from the heart and soul study. J. Clin. Psychiatry 2012, 73, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Stewart, R.; Kang, H.-J.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, J.-T.; Park, M.-S.; Cho, K.-H.; Yoon, J.-S. A prospective study of statin use and poststroke depression. J. Clin. Psychopharmacol. 2014, 34, 72–79. [Google Scholar] [CrossRef]
- Chuang, C.-S.; Yang, T.-Y.; Muo, C.-H.; Su, H.-L.; Sung, F.-C.; Kao, C.-H. Hyperlipidemia, statin use and the risk of developing depression: A nationwide retrospective cohort study. Gen. Hosp. Psych. 2014, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-Y.; Chang, A.Y.W.; Lin, T.-K. Simvastatin treatment exerts antidepressant-like effect in rats exposed to chronic mild stress. Pharmacol. Biochem. Behav. 2014, 124, 174–179. [Google Scholar] [CrossRef]
- ElBatsh, M.M. Antidepressant-like effect of simvastatin in diabetic rats. Can. J. Physiol. Pharmacol. 2015, 93, 649–656. [Google Scholar] [CrossRef]
- Abbasi, S.H.; Mohammadinejad, P.; Shahmansouri, N.; Salehiomran, A.; Beglar, A.A.; Zeinoddini, A.; Forghani, S.; Akhondzadeh, S. Simvastatin versus atorvastatin for improving mild to moderate depression in post-coronary artery bypass graft patients: A double-blind, placebo-controlled, randomized trial. J. Affect. Disord. 2015, 183, 149–155. [Google Scholar] [CrossRef]
- Gougol, A.; Zareh-Mohammadi, N.; Raheb, S.; Farokhnia, M.; Salimi, S.; Iranpour, N.; Yekehtaz, H.; Akhondzadeh, S. Simvastatin as an adjuvant therapy to fluoxetine in patients with moderate to severe major depression: A double-blind placebo-controlled trial. J. Psychopharmacol. 2015, 29, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Agostini, J.V.; Tinetti, M.E.; Han, L.; McAvay, G.; Foody, J.M.; Concato, J. Effects of statin use on muscle strength, cognition, and depressive symptoms in older adults. J. Am. Geriatr. Soc. 2007, 55, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yap, K.B.; Kua, E.H.; Ng, T.P. Statin use and depressive symptoms in a prospective study of community-living older persons. Pharmacoepidemiol. Drug Saf. 2010, 19, 942–948. [Google Scholar] [CrossRef]
- Al Badarin, F.J.; Spertus, J.A.; Gosch, K.L.; Buchanan, D.M.; Chan, P.S. Initiation of statin therapy after acute myocardial infarction is not associated with worsening depressive symptoms: Insights from the Prospective Registry Evaluating Outcomes After Myocardial Infarctions: Events and Recovery (PREMIER) and Translational Research Investigating Underlying Disparities in Acute Myocardial Infarction Patients’ Health Status (TRIUMPH) registries. Am. Heart J. 2013, 166, 879–886. [Google Scholar] [PubMed]
- Ludka, F.K.; Zomkowski, A.D.E.; Cunha, M.P.; Dal-Cim, T.; Zeni, A.L.B.; Rodrigues, A.L.S.; Tasca, C.I. Acute atorvastatin treatment exerts antidepressant-like effect in mice via the L-arginine-nitric oxide-cyclic guanosine monophosphate pathway and increases BDNF levels. Eur. Neuropsychopharmacol. 2013, 23, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Shahsavarian, A.; Javadi, S.; Jahanabadi, S.; Khoshnoodi, M.; Shamsaee, J.; Shafaroodi, H.; Mehr, S.E.; Dehpour, A. Antidepressant-like effect of atorvastatin in the forced swimming test in mice: The role of PPAR-gamma receptor and nitric oxide pathway. Eur. J. Pharmacol. 2014, 745, 52–58. [Google Scholar] [CrossRef]
- Citraro, R.; Chimirri, S.; Aiello, R.; Gallelli, L.; Trimboli, F.; Britti, D.; De Sarro, G.; Russo, E. Protective effects of some statins on epileptogenesis and depressive-like behavior in WAG/Rij rats, a genetic animal model of absence epilepsy. Epilepsia 2014, 55, 1284–1291. [Google Scholar] [CrossRef]
- Etminan, M.; Samii, A.; Brophy, J.M. Statin use and risk of epilepsy A nested case-control study. Neurology 2010, 75, 1496–1500. [Google Scholar] [CrossRef]
- Piermartiri, T.C.B.; Vandresen-Filho, S.; Herculano, B.d.A.; Martins, W.C.; Dal’Agnolo, D.; Stroeh, E.; Carqueja, C.L.; Boeck, C.R.; Tasca, C.I. Atorvastatin prevents hippocampal cell death due to quinolinic acid-induced seizures in mice by increasing Akt phosphorylation and glutamate uptake. Neurotox. Res. 2009, 16, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhao, Y.; Kwak, Y.-D.; Yang, Z.; Thompson, R.; Luo, Z.; Xu, H.; Liao, F.-F. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via Rho-ROCK signaling. J. Neurosci. 2009, 29, 11226–11236. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Won, J.-S.; Singh, A.K.; Singh, I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci. Lett. 2008, 440, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Siniscalchi, A.; Mintzer, S. Statins for poststroke seizures The first antiepileptogenic agent? Neurology 2015, 85, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.; de la Ossa, N.P.; Hurtado, O.; Millan, M.; Arenillas, J.F.; Davalos, A.; Gasull, T. Simvastatin reduces the association of NMDA receptors to lipid rafts: A cholesterol-mediated effect in neuroprotection. Stroke 2008, 39, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Seker, F.B.; Kilic, U.; Caglayan, B.; Ethemoglu, M.S.; Caglayan, A.B.; Ekimci, N.; Demirci, S.; Dogan, A.; Oztezcan, S.; Sahin, F.; et al. HMG-CoA reductase inhibitor rosuvastatin improves abnormal brain electrical activity via mechanisms involving eNOs. Neuroscience 2015, 284, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, F.; Constanti, A.; Citraro, R.; De Sarro, G.; Russo, E. Statins and epilepsy: Preclinical studies, clinical trials and statin-anticonvulsant drug interactions. Curr. Drug Targets 2015, 16, 747–756. [Google Scholar] [CrossRef]
- Sierra-Marcos, A.; Alvarez, V.; Faouzi, M.; Burnand, B.; Rossetti, A.O. Statins are associated with decreased mortality risk after status epilepticus. Eur. J. Neurol. 2015, 22, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.M.; Coleman, C.I.; Henyan, N.N.; Kluger, J.; White, C.M. Statins and cancer risk—A meta-analysis. JAMA-J. Am. Med. Assoc. 2006, 295, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, N.E. Statins and Cancer-Related Mortality—Let’s Work Together. N. Engl. J. Med. 2012, 367, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Osmak, M. Statins and cancer: Current and future prospects. Cancer Lett. 2012, 324, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Picardi, P.; Ciaglia, E.; D’Alessandro, A.; Bifulco, M. Novel prospects of statins as therapeutic agents in cancer. Pharmacol. Res. 2014, 88, 84–98. [Google Scholar] [CrossRef]
- Kubatka, P.; Kruzliak, P.; Rotrekl, V.; Jelinkova, S.; Mladosievicova, B. Statins in oncological research: From experimental studies to clinical practice. Crit. Rev. Oncol. Hematol. 2014, 92, 296–311. [Google Scholar] [CrossRef]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells-review. Tumor Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; Simes, J.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.C.; Lin, C.L.; Hsu, W.Y.; Chang, C.S.; Yeh, H.Z.; Tung, C.F.; Wu, Y.L.; Sung, F.C.; Kao, C.H. Statins are associated with a reduced risk of cholangiocarcinoma: A population-based case-control study. Br. J. Clin. Pharmacol. 2015, 80, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Archibugi, L.; Maisonneuve, P.; Piciucchi, M.; Valente, R.; Delle Fave, G.; Capurso, G. Statins but not aspirin nor their combination have a chemopreventive effect on pancreatic cancer occurrence. Pancreas 2015, 44, 1359. [Google Scholar]
- Sivaprasad, U.; Abbas, T.; Dutta, A. Differential efficacy of 3-hydroxy-3-methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol. Cancer Ther. 2006, 5, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, B.-C.; Lu, X.-Y.; Yang, L.-L.; Zhai, Y.-J.; Eaton, A.F.; Thai, T.L.; Eaton, D.C.; Ma, H.-P.; Shen, B.-Z. Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.-S.; Kang, X.-L.; Zhou, J.-G.; Lv, X.-F.; Tang, Y.-B.; Guan, Y.-Y. Involvement of Chk1-Cdc25A-cyclin A/CDk2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells. Eur. J. Pharmacol. 2011, 670, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Luo, Y.; Zhou, Y.; Zhang, Q.; Wang, J.; Wei, N.; Mi, M.; Zhu, J.; Wang, B.; Chang, H.; et al. BRCA1 overexpression sensitizes cancer cells to lovastatin via regulation of cyclin D1-CDK4-p21(WAF1/CIP1) pathway: Analyses using a breast cancer cell line and tumoral xenograft model. Int. J. Oncol. 2008, 33, 555–563. [Google Scholar] [PubMed]
- Rao, S.; Porter, D.C.; Chen, X.M.; Herliczek, T.; Lowe, M.; Keyomarsi, K. Lovastatin-mediated G(1) arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7797–7802. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Asano, T.; Hayakawa, M. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, fluvastatin, as a novel agent for prophylaxis of renal cancer metastasis. Clin. Cancer Res. 2004, 10, 8648–8655. [Google Scholar] [CrossRef]
- Denoyelle, C.; Vasse, M.; Korner, M.; Mishal, Z.; Ganne, F.; Vannier, J.P.; Soria, J.; Soria, C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: An in vitro study. Carcinogenesis 2001, 22, 1139–1148. [Google Scholar] [CrossRef]
- Spampanato, C.; De Maria, S.; Sarnataro, M.; Giordano, E.; Zanfardino, M.; Baiano, S.; Carteni, M.; Morelli, F. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int. J. Oncol. 2012, 40, 935–941. [Google Scholar] [CrossRef]
- Herrero-Martin, G.; Lopez-Rivas, A. Statins activate a mitochondria-operated pathway of apoptosis in breast tumor cells by a mechanism regulated by ErbB2 and dependent on the prenylation of proteins. FEBS Lett. 2008, 582, 2589–2594. [Google Scholar] [CrossRef]
- Zhu, Y.; Casey, P.J.; Kumar, A.P.; Pervaiz, S. Deciphering the signaling networks underlying simvastatin-induced apoptosis in human cancer cells: Evidence for non-canonical activation of RhoA and Rac1 GTPases. Cell Death Dis. 2013, 4, e568. [Google Scholar] [CrossRef]
- Miller, T.; Yang, F.; Wise, C.E.; Meng, F.; Priester, S.; Munshi, M.K.; Guerrier, M.; Dostal, D.E.; Glaser, S.S. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig. Liver Dis. 2011, 43, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Gniadecki, R. Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem. Biophys. Res. Commun. 2004, 320, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.Y.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Boscher, C.; Nabi, I.R. CAVEOLIN-1: Role in Cell Signaling. In Caveolins and Caveolae: Roles in Signaling and Disease Mechanisms; Jasmin, J.F., Frank, P.G., Lisanti, M.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 729, pp. 29–50. [Google Scholar]
- Altwairgi, A.K. Statins are potential anticancerous agents (Review). Oncol. Rep. 2015, 33, 1019–1039. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Gonzalez, J.; Miranda, R.U. Biotechnological production and applications of statins. Appl. Microbiol. Biotechnol. 2010, 85, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.F.; Alberts, A.W.; Anderson, P.S.; Chen, J.S.; Smith, R.L.; Willard, A.K. 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors. 4. Side-chain ester derivatives of mevinolin. J. Med. Chem. 1986, 29, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Askin, D.; Verhoeven, T.R.; Liu, T.M.H.; Shinkai, I. Synthesis of synvinolin—Extremely high conversion alkylation of an ester enolate. J. Org. Chem. 1991, 56, 4929–4932. [Google Scholar] [CrossRef]
- Xie, X.K.; Tang, Y. Efficient synthesis of simvastatin by use of whole-cell biocatalysis. Appl. Environ. Microbiol. 2007, 73, 2054–2060. [Google Scholar] [CrossRef]
- Schimmel, T.G.; Borneman, W.S.; Conder, M.J. Purification and characterization of a lovastatin esterase from Clonostachys compactiuscula. Appl. Environ. Microbiol. 1997, 63, 1307–1311. [Google Scholar] [PubMed]
- Chen, L.C.; Lai, Y.K.; Wu, S.C.; Lin, C.C.; Guo, J.H. Production by Clonostachys compactiuscula of a lovastatin esterase that converts lovastatin to monacolin J. Enzyme Microb. Technol. 2006, 39, 1051–1059. [Google Scholar] [CrossRef]
- Gao, X.; Xie, X.K.; Pashkov, I.; Sawaya, M.R.; Laidman, J.; Zhang, W.J.; Cacho, R.; Yeates, T.O.; Tang, Y. Directed Evolution and Structural Characterization of a Simvastatin Synthase. Chem. Biol. 2009, 16, 1064–1074. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Presidential Green Chemistry Challenge: 2012 Greener Synthetic Pathways Award. Available online: http://www2.epa.gov/green-chemistry/2012-greener-synthetic-pathways-award (accessed on 14 February 2019).
- Xu, W.; Chooi, Y.H.; Choi, J.W.; Li, S.; Vederas, J.C.; Da Silva, N.A.; Tang, Y. LovG: The thioesterase required for dihydromonacolin L release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew. Chem. Int. Ed. 2013, 52, 6472–6475. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.E.; Le, T.V.; Millar, A.; Nanninga, T.N. Process for the synthesis of (5R)-1,1-dimethylethyl-6-cyano-5-hydroxy-3-oxo-hexanoate. US Patent 5155251, 13 October 1992. [Google Scholar]
- Turner, N.J.; O’Reilly, E. Biocatalytic retrosynthesis. Nat. Chem. Biol. 2013, 9, 285–288. [Google Scholar] [CrossRef]
- Brady, D. Biocatalytic Hydrolysis of Nitriles. In Handbook of Green Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2010; Volume 3, (Biocatalysis). [Google Scholar]
- Faber, K. Biotransformations of non-natural compounds: State of the art and future development. Pure Appl. Chem. 1997, 69, 1613–1632. [Google Scholar] [CrossRef]
- DeSantis, G.; Zhu, Z.L.; Greenberg, W.A.; Wong, K.V.; Chaplin, J.; Hanson, S.R.; Farwell, B.; Nicholson, L.W.; Rand, C.L.; Weiner, D.P.; et al. An enzyme library approach to biocatalysis: Development of nitrilases for enantioselective production of carboxylic acid derivatives. J. Am. Chem. Soc. 2002, 124, 9024–9025. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, G.; Wong, K.; Farwell, B.; Chatman, K.; Zhu, Z.L.; Tomlinson, G.; Huang, H.J.; Tan, X.Q.; Bibbs, L.; Chen, P.; et al. Creation of a productive, highly enantioselective nitrilase through gene site saturation mutagenesis (GSSM). J. Am. Chem. Soc. 2003, 125, 11476–11477. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, S.; Chaplin, D.A.; Edwards, J.H.; Ellis, B.S.W.; Hill, C.L.; Holt-Tiffin, K.; Knight, J.R.; Mahoney, T.; Osborne, A.P.; Ruecroft, G. Nitrilase-catalysed desymmetrisation of 3-hydroxyglutaronitrile: Preparation of a statin side-chain intermediate. Org. Process Res. Dev. 2006, 10, 661–665. [Google Scholar] [CrossRef]
- Yao, P.; Li, J.; Yuan, J.; Han, C.; Liu, X.; Feng, J.; Wu, Q.; Zhu, D. Enzymatic synthesis of a key intermediate for rosuvastatin by nitrilase-catalyzed hydrolysis of ethyl (R)-4-cyano-3-hydroxybutyate at high substrate concentration. ChemCatChem 2015, 7, 271–275. [Google Scholar] [CrossRef]
- Ma, S.K.; Gruber, J.; Davis, C.; Newman, L.; Gray, D.; Wang, A.; Grate, J.; Huisman, G.W.; Sheldon, R.A. A green-by-design biocatalytic process for atorvastatin intermediate. Green Chem. 2010, 12, 81–86. [Google Scholar] [CrossRef]
- Chung, S.; Hwang, Y. Stereoselective hydrolysis of racemic ethyl 4-chloro-3-hydroxybutyrate by a lipase. Biocatal. Biotransform. 2008, 26, 327–330. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, O.J.; Uh, H.S. A chemoenzymatic approach to the synthesis of enantiomerically pure (S)-3-hydroxy-gamma-butyrolactone. Appl. Microbiol. Biotechnol. 2008, 79, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.M. Biocatalytic synthesis of atorvastatin intermediates. J. Mol. Catal. B-Enzym. 2009, 61, 123–128. [Google Scholar] [CrossRef]
- Martin, C.H.; Dhamankar, H.; Tseng, H.C.; Sheppard, M.J.; Reisch, C.R.; Prather, K.L.J. A platform pathway for production of 3-hydroxyacids provides a biosynthetic route to 3-hydroxy-gamma-butyrolactone. Nat. Commun. 2013, 4, 1414. [Google Scholar] [CrossRef] [PubMed]
- Narasaka, K.; Pai, F.C. Stereoselective reduction of beta-hydroxyketones to 1,3-diols highly selective 1,3-asymmetric induction via boron chelates. Tetrahedron 1984, 40, 2233–2238. [Google Scholar] [CrossRef]
- Chen, K.M.; Hardtmann, G.E.; Prasad, K.; Repic, O.; Shapiro, M.J. 1,3-syn diastereoselective reduction of beta-hydroxyketones utilizing alkoxydialkylboranes. Tetrahedron Lett. 1987, 28, 155–158. [Google Scholar] [CrossRef]
- Sterk, D.; Casar, Z.; Jukic, M.; Kosmrlj, J. Concise and highly efficient approach to three key pyrimidine precursors for rosuvastatin synthesis. Tetrahedron 2012, 68, 2155–2160. [Google Scholar] [CrossRef]
- Ohrlein, R.; Baisch, G. Chemo-enzymatic approach to statin side-chain building blocks. Adv. Synth. Catal. 2003, 345, 713–715. [Google Scholar] [CrossRef]
- Santaniello, E.; Chiari, M.; Ferraboschi, P.; Trave, S. Enhanced and reversed enantioselectivity of enzymatic-hydrolysis by simple substrate modifications—The case of 3-hydroxyglutarate diesters. J. Org. Chem. 1988, 53, 1567–1569. [Google Scholar] [CrossRef]
- Metzner, R.; Hummel, W.; Wetterich, F.; Konig, B.; Groger, H. Integrated biocatalysis in multistep drug synthesis without intermediate isolation: A de novo approach toward a rosuvastatin key building block. Org. Process Res. Dev. 2015, 19, 635–638. [Google Scholar] [CrossRef]
- König, B.; Wetterich, F.; Gröger, H.; Metzner, R. Process for enantioselective synthesis of 3-hydroxy-glutaric acid monoesters and use thereof. WO 2014/140006, 18 September 2014. [Google Scholar]
- You, Z.Y.; Liu, Z.Q.; Zheng, Y.G. Chemical and enzymatic approaches to the synthesis of optically pure ethyl (R)-4-cyano-3-hydroxybutanoate. Appl. Microbiol. Biotechnol. 2014, 98, 11–21. [Google Scholar] [CrossRef]
- Asako, H.; Shimizu, M.; Itoh, N. Biocatalytic production of (S)-4-bromo-3-hydroxybutyrate and structurally related chemicals and their applications. Appl. Microbiol. Biotechnol. 2009, 84, 397–405. [Google Scholar] [CrossRef]
- Asako, H.; Shimizu, M.; Makino, Y.; Itoh, N. Biocatalytic reduction system for the production of chiral methyl (R)/(S)-4-bromo-3-hydroxybutyrate. Tetrahedron Lett. 2010, 51, 2664–2666. [Google Scholar] [CrossRef]
- Patel, R.N.; McNamee, C.G.; Banerjee, A.; Howell, J.M.; Robison, R.S.; Szarka, L.J. Stereoselective reduction of beta-keto-esters by Geotrichum candidum. Enzyme Microb. Technol. 1992, 14, 731–738. [Google Scholar] [CrossRef]
- Ye, Q.; Ouyang, P.K.; Ying, H.J. A review-biosynthesis of optically pure ethyl (S)-4-chloro-3-hydroxybutanoate ester: Recent advances and future perspectives. Appl. Microbiol. Biotechnol. 2011, 89, 513–522. [Google Scholar] [CrossRef]
- Yasohara, Y.; Kizaki, N.; Hasegawa, J.; Takahashi, S.; Wada, M.; Kataoka, M.; Shimizu, S. Synthesis of optically active ethyl 4-chloro-3-hydroxybutanoate by microbial reduction. Appl. Microbiol. Biotechnol. 1999, 51, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Kataoka, M.; Shimizu, S. Diversity of 4-chloroacetoacetate ethyl ester-reducing enzymes in yeasts and their application to chiral alcohol synthesis. J. Biosci. Bioeng. 1999, 88, 591–598. [Google Scholar] [CrossRef]
- Wada, M.; Kataoka, M.; Kawabata, H.; Yasohara, Y.; Kizaki, N.; Hasegawa, J.; Shimizu, S. Purification and characterization of NADPH-dependent carbonyl reductase, involved in stereoselective reduction of ethyl 4-chloro-3-oxobutanoate, from Candida magnoliae. Biosci. Biotechnol. Biochem. 1998, 62, 280–285. [Google Scholar] [CrossRef]
- Kataoka, M.; Doi, Y.; Sim, T.S.; Shimizu, S.; Yamada, H. A novel NADPH-dependent carbonyl reductase of Candida macedoniensis. Purification and characterization. Arch. Biochem. Biophys. 1992, 294, 469–474. [Google Scholar] [CrossRef]
- He, J.Y.; Sun, Z.H.; Ruan, W.Q.; Xu, Y. Biocatalytic synthesis of ethyl (S)-4-chloro-3-hydroxy-butanoate in an aqueous-organic solvent biphasic system using Aureobasidium pullulans CGMCC 1244. Process Biochem. 2006, 41, 244–249. [Google Scholar] [CrossRef]
- Saratani, Y.; Uheda, E.; Yamamoto, H.; Nishimura, A.; Yoshizako, F. Stereoselective reduction of ethyl 4-chloro-3-oxobutanoate by fungi. Biosci. Biochem. Biophys. 2001, 65, 1676–1679. [Google Scholar] [CrossRef]
- Ye, Q.; Yan, M.; Xu, L.; Cao, H.; Li, Z.; Chen, Y.; Li, S.; Ying, H. A novel carbonyl reductase from Pichia stipitis for the production of ethyl (S)-4-chloro-3-hydroxybutanoate. Biotechnol. Lett. 2009, 31, 537–542. [Google Scholar] [CrossRef]
- Cai, P.; An, M.D.; Xu, L.; Xu, S.; Hao, N.; Li, Y.; Guo, K.; Yan, M. Development of a substrate-coupled biocatalytic process driven by an NADPH-dependent sorbose reductase from Candida albicans for the asymmetric reduction of ethyl 4-chloro-3-oxobutanoate. Biotechnol. Lett. 2012, 34, 2223–2227. [Google Scholar] [CrossRef]
- Pan, J.; Zheng, G.-W.; Ye, Q.; Xu, J.-H. Optimization and scale-up of a bioreduction process for preparation of ethyl (S)-4-chloro-3-hydroxybutanoate. Org. Process Res. Dev. 2014, 18, 739–743. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Ye, J.-J.; Shen, Z.-Y.; Hong, H.-B.; Yan, J.-B.; Lin, Y.; Chen, Z.-X.; Zheng, Y.-G.; Shen, Y.-C. Upscale production of ethyl (S)-4-chloro-3-hydroxybutanoate by using carbonyl reductase coupled with glucose dehydrogenase in aqueous-organic solvent system. Appl. Microbiol. Biotechnol. 2015, 99, 2119–2129. [Google Scholar] [CrossRef]
- Schallmey, M.; Floor, R.J.; Hauer, B.; Breuer, M.; Jekel, P.A.; Wijma, H.J.; Dijkstra, B.W.; Janssen, D.B. Biocatalytic and structural properties of a highly engineered halohydrin dehalogenase. ChemBioChem 2013, 14, 870–881. [Google Scholar] [CrossRef]
- Ritter, S.K. Going green keeps getting easier. Chem. Eng. News 2006, 84, 24–27. [Google Scholar] [CrossRef]
- Li, C.-J.; Anastas, P.T. Green Chemistry: Present and future. Chem. Soc. Rev. 2012, 41, 1413–1414. [Google Scholar] [CrossRef]
- Chen, X.F.; Xiong, F.J.; Chen, W.X.; He, Q.Q.; Chen, F.E. Asymmetric Synthesis of the HMG-CoA Reductase Inhibitor Atorvastatin Calcium: An Organocatalytic Anhydride Desymmetrization and Cyanide-Free Side Chain Elongation Approach. J. Org. Chem. 2014, 79, 2723–2728. [Google Scholar] [CrossRef]
- Everaere, K.; Franceschini, N.; Mortreux, A.; Carpentier, J.F. Diastereoselective synthesis of syn-3,5-dihydroxyesters via ruthenium-catalyzed asymmetric transfer hydrogenation. Tetrahedron Lett. 2002, 43, 2569–2571. [Google Scholar] [CrossRef]
- Wolberg, M.; Hummel, W.; Muller, M. Biocatalytic reduction of beta, delta-diketo esters: A highly stereoselective approach to all four stereoisomers of a chlorinated beta, delta-dihydroxy hexanoate. Chem. Eur. J. 2001, 7, 4562–4571. [Google Scholar] [CrossRef]
- Wolberg, M.; Hummel, W.; Wandrey, C.; Muller, M. Highly regio- and enantioselective reduction of 3,5-dioxocarboxylates. Angew. Chem. Int. Ed. 2000, 39, 4306–4308. [Google Scholar] [CrossRef]
- Wolberg, M.; Villela, M.; Bode, S.; Geilenkirchen, P.; Feldmann, R.; Liese, A.; Hummel, W.; Muller, M. Chemoenzymatic synthesis of the chiral side-chain of statins: Application of an alcohol dehydrogenase catalysed ketone reduction on a large scale. Bioproc. Biosyst. Eng. 2008, 31, 183–191. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Hu, Z.L.; Zhang, X.J.; Tang, X.L.; Cheng, F.; Xue, Y.P.; Wang, Y.J.; Wu, L.; Yao, D.K.; Zhou, Y.T.; et al. Large-scale synthesis of tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate by a stereoselective carbonyl reductase with high substrate concentration and product yield. Biotechnol. Progr. 2017, 33, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Wu, L.; Zhang, X.J.; Xue, Y.P.; Zheng, Y.G. Directed evolution of carbonyl reductase from Rhodosporidium toruloides and its application in stereoselective synthesis of tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate. J. Agric. Food Chem. 2017, 65, 3721–3729. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Yin, H.H.; Zhang, X.J.; Zhou, R.; Wang, Y.M.; Zheng, Y.G. Improvement of carbonyl reductase activity for the bioproduction of tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate. Bioorg. Chem. 2018, 80, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Wu, L.; Zheng, L.; Wang, W.Z.; Zhang, X.J.; Jin, L.Q.; Zheng, Y.G. Biosynthesis of tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate by carbonyl reductase from Rhodosporidium toruloides in mono and biphasic media. Bioresour. Technol. 2018, 249, 161–167. [Google Scholar] [CrossRef]
- Xu, T.T.; Wang, C.; Zhu, S.Z.; Zheng, G.J. Enzymatic preparation of optically pure t-butyl 6-chloro-(3R,5S)-dihydroxyhexanoate by a novel alcohol dehydrogenase discovered from Klebsiella oxytoca. Process Biochem. 2017, 57, 72–79. [Google Scholar] [CrossRef]
- Gong, X.M.; Zheng, G.W.; Liu, Y.Y.; Xu, J.H. Identification of a robust carbonyl reductase for diastereoselectively building syn-3,5-dihydroxy hexanoate: A bulky side chain of atorvastatin. Org. Process Res. Dev. 2017, 21, 1349–1354. [Google Scholar] [CrossRef]
- Gong, X.M.; Qin, Z.; Li, F.L.; Zeng, B.B.; Zheng, G.W.; Xu, J.H. Development of an engineered ketoreductase with simultaneously improved thermostability and activity for making a bulky atorvastatin precursor. ACS Catal. 2019, 9, 147–153. [Google Scholar] [CrossRef]
- Luo, X.; Wang, Y.-J.; Zheng, Y.-G. Improved stereoselective bioreduction of t-butyl 6-cyano-(5R)-hydroxy-3-oxohexanoate by Rhodotorula glutinis through heat treatment. Biotechnol. Appl. Biochem. 2016, 63, 795–804. [Google Scholar] [CrossRef]
- Wu, X.; Gou, X.; Chen, Y. Enzymatic preparation of t-butyl-6-cyano-(3R,5R)-dihydroxyhexanoate by a whole-cell biocatalyst co-expressing carbonyl reductase and glucose dehydrogenase. Process Biochem. 2015, 50, 104–110. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, X.P.; Shen, W.; Liu, Z.Q.; Zheng, Y.G. Chiral diol t-butyl 6-cyano-(3R,5R)-dihydroxylhexanoate synthesis catalyzed by immobilized cells of carbonyl reductase and glucose dehydrogenase co-expression E. coli. Biochem. Eng. J. 2017, 128, 54–62. [Google Scholar] [CrossRef]
- Luo, X.; Wang, Y.-J.; Zheng, Y.-G. Cloning and characterization of a NADH-dependent aldo-keto reductase from a newly isolated Kluyveromyces lactis XP1461. Enzyme Microb. Technol. 2015, 77, 68–77. [Google Scholar] [CrossRef]
- Luo, X.; Wang, Y.-J.; Shen, W.; Zheng, Y.-G. Activity improvement of a Kluyveromyces lactis aldo-keto reductase KlAKR via rational design. J. Biotechnol. 2016, 224, 20–26. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Liu, X.-Q.; Luo, X.; Liu, Z.-Q.; Zheng, Y.-G. Cloning, expression and enzymatic characterization of an aldo-keto reductase from Candida albicans XP1463. J. Mol. Catal. B-Enzym. 2015, 122, 44–50. [Google Scholar] [CrossRef]
- Pfruender, H.; Amidjojo, M.; Hang, F.; Weuster-Botz, D. Production of Lactobacillus kefir cells for asymmetric synthesis of a 3,5-dihydroxycarboxylate. Appl. Microbiol. Biotechnol. 2005, 67, 619–622. [Google Scholar] [CrossRef]
- Patel, R.N.; Banerjee, A.; McNamee, C.G.; Brzozowski, D.; Hanson, R.L.; Szarka, L.J. Enantioselective microbial reduction of 3,5-dioxo-6-(benzyloxy) hexanoic acid, ethyl-ester. Enzyme Microb. Technol. 1993, 15, 1014–1021. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Goswami, A.; Hanson, R.L.; Patel, R.N. Synthesis of ethyl and t-butyl (3R,5S)-dihydroxy-6-benzyloxy hexanoates via diastereo- and enantioselective microbial reduction. Tetrahedron Asymm. 2006, 17, 1589–1602. [Google Scholar] [CrossRef]
- Goldberg, S.; Guo, Z.; Chen, S.; Goswami, A.; Patel, R.N. Synthesis of ethyl-(3R,5S)-dihydroxy-6-benzyloxyhexanoates via diastereo- and enantioselective microbial reduction: Cloning and expression of ketoreductase III from Acinetobacter sp. SC 13874. Enzyme Microb. Technol. 2008, 43, 544–549. [Google Scholar] [CrossRef]
- Wu, X.; Liu, N.; He, Y.; Chen, Y. Cloning, expression, and characterization of a novel diketoreductase from Acinetobacter baylyi. Acta Biochim. Biophys. Sin. 2009, 41, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.; Wu, X. Dicarbonyl reduction by single enzyme for the preparation of chiral diols. Chem. Soc. Rev. 2012, 41, 1742–1753. [Google Scholar] [CrossRef]
- Lu, M.; Huang, Y.; White, M.A.; Wu, X.; Liu, N.; Cheng, X.; Chen, Y. Dual catalysis mode for the dicarbonyl reduction catalyzed by diketoreductase. Chem. Commun. 2012, 48, 11352–11354. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lu, Z.; Ma, M.; Liu, N.; Chen, Y. Functional roles of tryptophan residues in diketoreductase from Acinetobacter baylyi. BMB Rep. 2012, 45, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lu, Z.; Liu, N.; Chen, Y. Identification of important residues in diketoreductase from Acinetobacter baylyi by molecular modeling and site-directed mutagenesis. Biochimie 2012, 94, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; Wong, C.H. Sequential 3-substrate and 4-substrate aldol reactions catalyzed by aldolases. J. Am. Chem. Soc. 1995, 117, 7585–7591. [Google Scholar] [CrossRef]
- Gijsen, H.J.M.; Wong, C.H. Unprecedented asymmetric aldol reactions with 3 aldehyde substrates catalyzed by 2-deoxyribose-5-phosphate aldolase. J. Am. Chem. Soc. 1994, 116, 8422–8423. [Google Scholar] [CrossRef]
- Liu, J.J.; Hsu, C.C.; Wong, C.H. Sequential aldol condensation catalyzed by DERA mutant Ser238Asp and a formal total synthesis of atorvastatin. Tetrahedron Lett. 2004, 45, 2439–2441. [Google Scholar] [CrossRef]
- Greenberg, W.A.; Varvak, A.; Hanson, S.R.; Wong, K.; Huang, H.J.; Chen, P.; Burk, M.J. Development of an efficient, scalable, aldolase-catalyzed process for enantioselective synthesis of statin intermediates. Proc. Natl. Acad. Sci. USA 2004, 101, 5788–5793. [Google Scholar] [CrossRef]
- Jennewein, S.; Schurmann, M.; Wolberg, M.; Hilker, I.; Luiten, R.; Wubbolts, M.; Mink, D. Directed evolution of an industrial biocatalyst: 2-deoxy-d-ribose 5-phosphate aldolase. Biotechnol. J. 2006, 1, 537–548. [Google Scholar] [CrossRef]
- Oslaj, M.; Cluzeau, J.; Orkic, D.; Kopitar, G.; Mrak, P.; Casar, Z. A highly productive, whole-cell DERA chemoenzymatic process for production of key lactonized side-chain intermediates in statin synthesis. PLoS ONE 2013, 8, e62250. [Google Scholar] [CrossRef]
- Ručigaj, A.; Krajnc, M. Optimization of a crude deoxyribose-5-phosphate aldolase lyzate-catalyzed process in synthesis of statin intermediates. Org. Process Res. Dev. 2013, 17, 854–862. [Google Scholar] [CrossRef]
- You, Z.Y.; Liu, Z.Q.; Zheng, Y.G.; Shen, Y.C. Characterization and application of a newly synthesized 2-deoxyribose-5-phosphate aldolase. J. Ind. Microbiol. Biotechnol. 2013, 40, 29–39. [Google Scholar] [CrossRef]
- Casar, Z.; Steinbucher, M.; Kosmrlj, J. Lactone pathway to statins utilizing the Wittig reaction. The synthesis of rosuvastatin. J. Org. Chem. 2010, 75, 6681–6684. [Google Scholar] [CrossRef]
- Vajdic, T.; Oslaj, M.; Kopitar, G.; Mrak, P. Engineered, highly productive biosynthesis of artificial, lactonized statin side-chain building blocks: The hidden potential of Escherichia coli unleashed. Metab. Eng. 2014, 24, 160–172. [Google Scholar] [CrossRef]
- Sakuraba, H.; Yoneda, K.; Yoshihara, K.; Satoh, K.; Kawakami, R.; Uto, Y.; Tsuge, H.; Takahashi, K.; Hori, H.; Ohshima, T. Sequential aldol condensation catalyzed by hyperthermophilic 2-deoxy-d-ribose-5-phosphate aldolase. Appl. Environ. Microbiol. 2007, 73, 7427–7434. [Google Scholar] [CrossRef]
- Fei, H.; Zheng, C.C.; Liu, X.Y.; Li, Q. An industrially applied biocatalyst: 2-Deoxy-d-ribose-5-phosphate aldolase. Process Biochem. 2017, 63, 55–59. [Google Scholar] [CrossRef]
- Haridas, M.; Abdelraheem, E.M.M.; Hanefeld, U. 2-Deoxy-d-ribose-5-phosphate aldolase (DERA): Applications and modifications. Appl. Microbiol. Biotechnol. 2018, 102, 9959–9971. [Google Scholar] [CrossRef]
- Muller, M. Chemoenzymatic synthesis of building blocks for statin side chains. Angew. Chem. Int. Ed. 2005, 44, 362–365. [Google Scholar] [CrossRef]
- Garattini, L.; Padula, A. Cholesterol-lowering drugs: Science and marketing. J. R. Soc. Med. 2017, 110, 57–64. [Google Scholar] [CrossRef]
- Reiner, Z. Resistance and intolerance to statins. Nutr. Metab. Carbiovasc. Dis. 2014, 24, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. PCSK9 inhibitors: A new era of lipid lowering therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C. New directions in managing dyslipidemia. J. Nurse Pract. 2019, 15, 73–79. [Google Scholar] [CrossRef]
- Xu, S.T.; Luo, S.S.; Zhu, Z.Y.; Xu, J.Y. Small molecules as inhibitors of PCSK9: Current status and future challenges. Eur. J. Med. Chem. 2019, 162, 212–233. [Google Scholar] [CrossRef] [PubMed]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoyos, P.; Pace, V.; Alcántara, A.R. Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs. Catalysts 2019, 9, 260. https://doi.org/10.3390/catal9030260
Hoyos P, Pace V, Alcántara AR. Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs. Catalysts. 2019; 9(3):260. https://doi.org/10.3390/catal9030260
Chicago/Turabian StyleHoyos, Pilar, Vittorio Pace, and Andrés R. Alcántara. 2019. "Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs" Catalysts 9, no. 3: 260. https://doi.org/10.3390/catal9030260
APA StyleHoyos, P., Pace, V., & Alcántara, A. R. (2019). Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs. Catalysts, 9(3), 260. https://doi.org/10.3390/catal9030260