Approximating Catalyst Effectiveness Factors with Reaction Rate Profiles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model Development
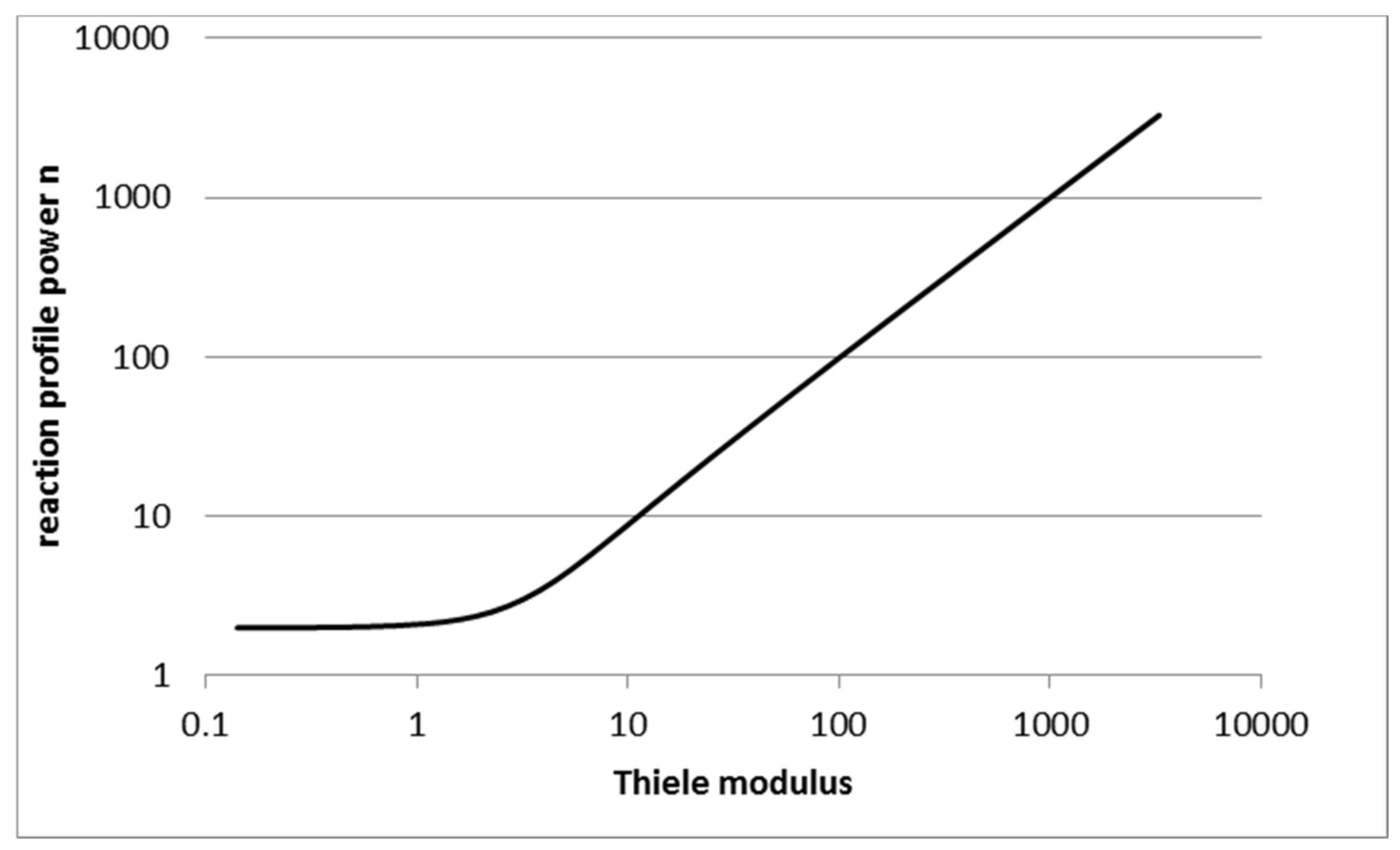
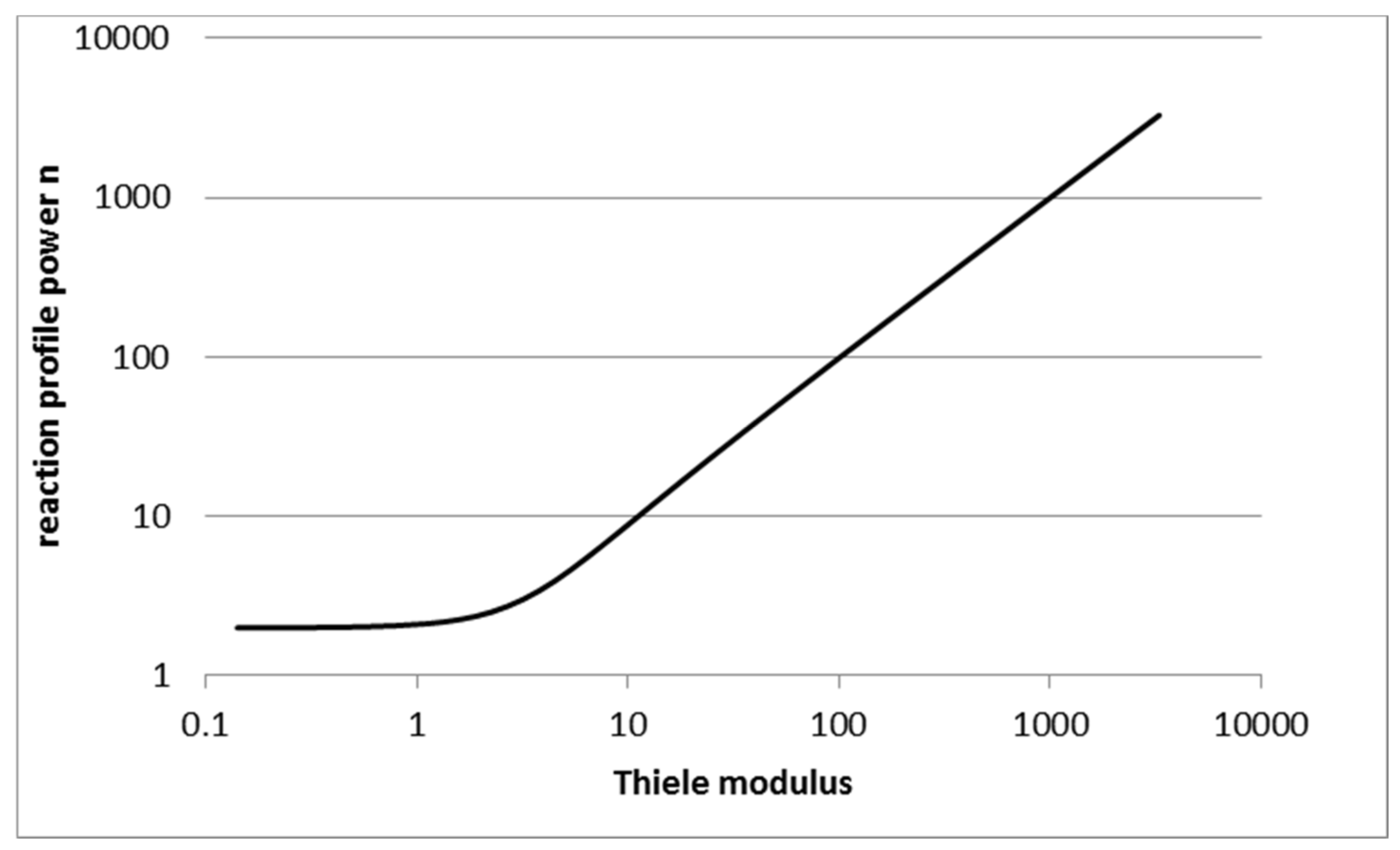
2.1. Reaction Rate Profile Approximation
2.2. Choosing the Reaction Rate Profile
2.3. Finite Volume Solution
3. Results and Discussion
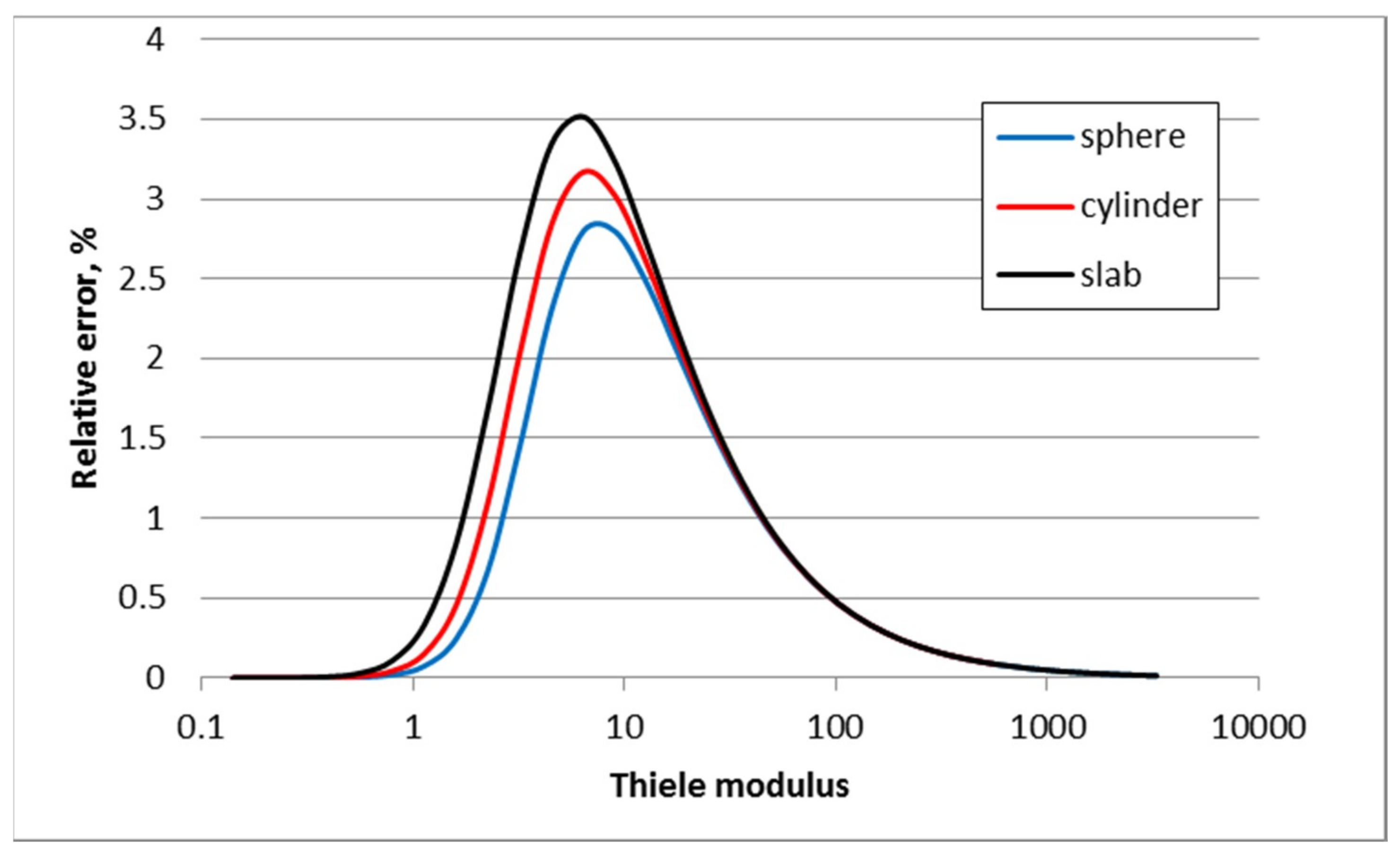
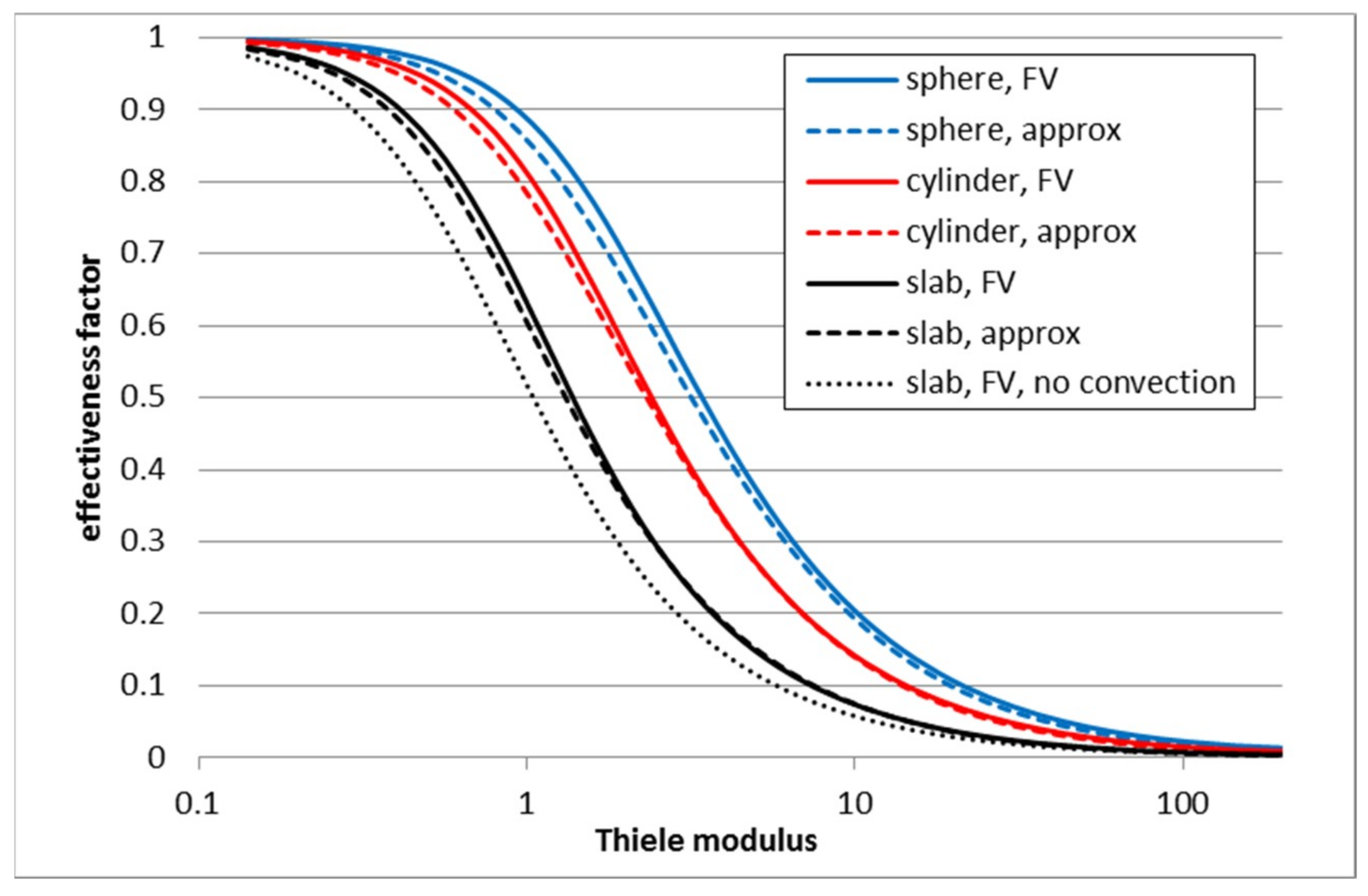
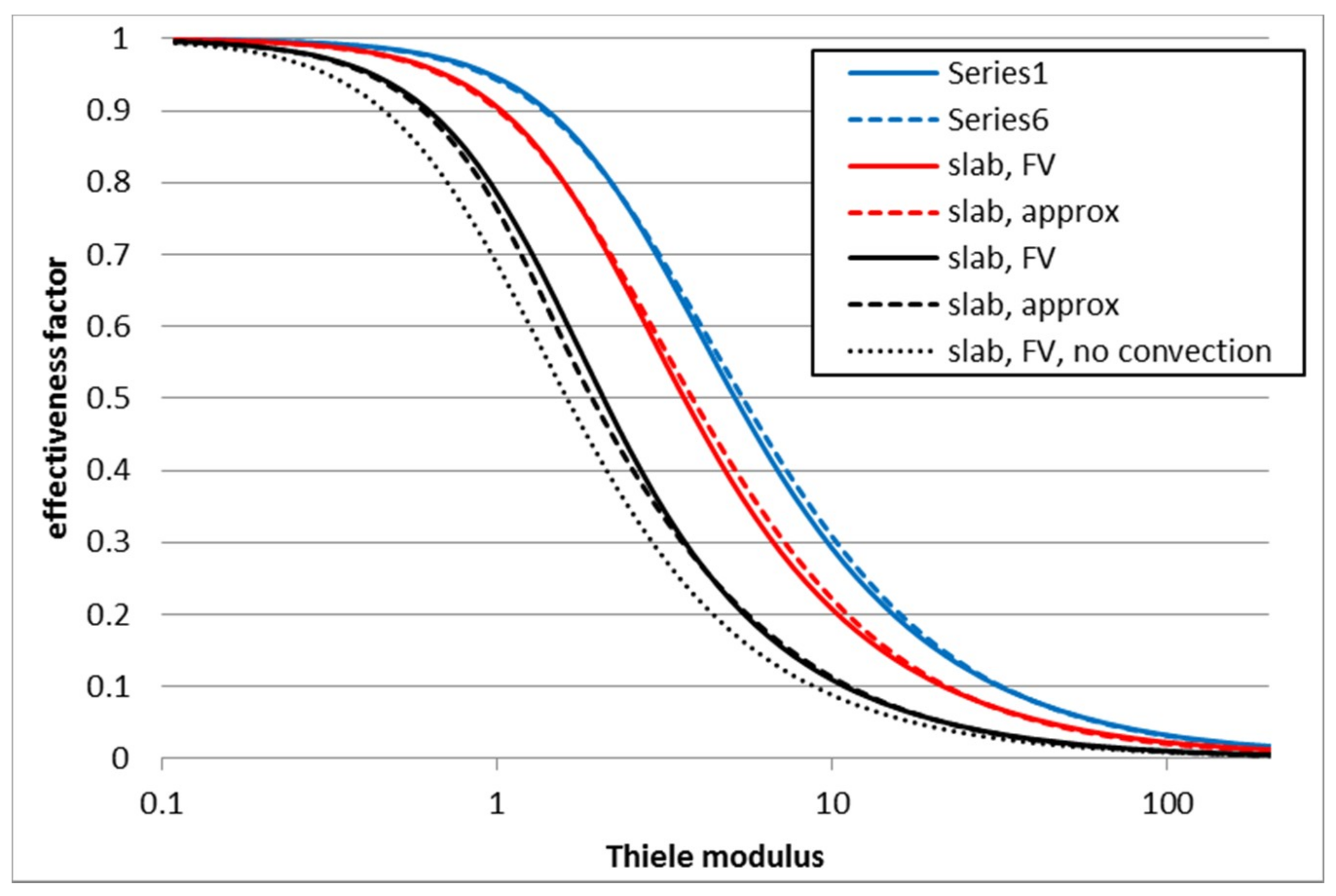
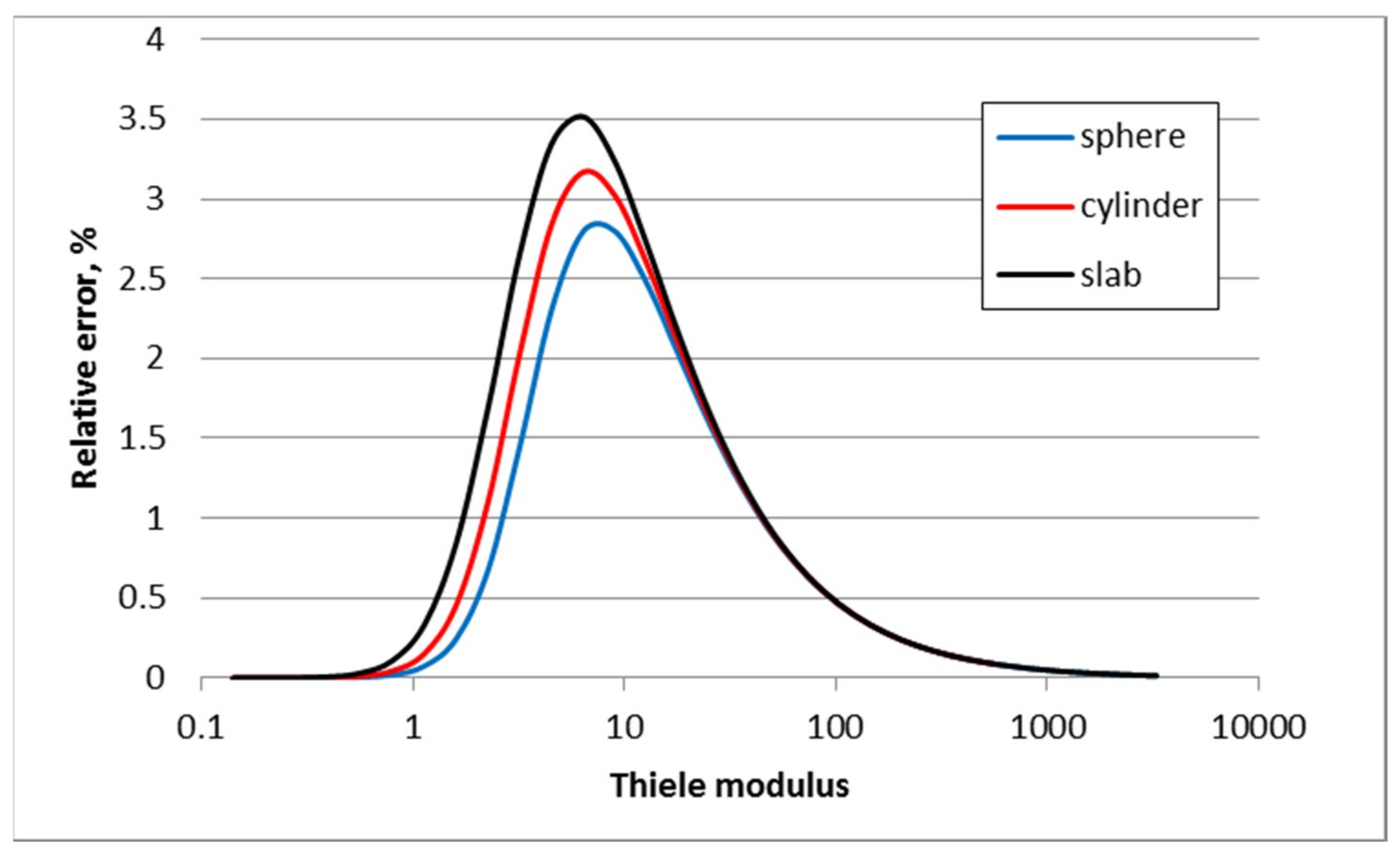
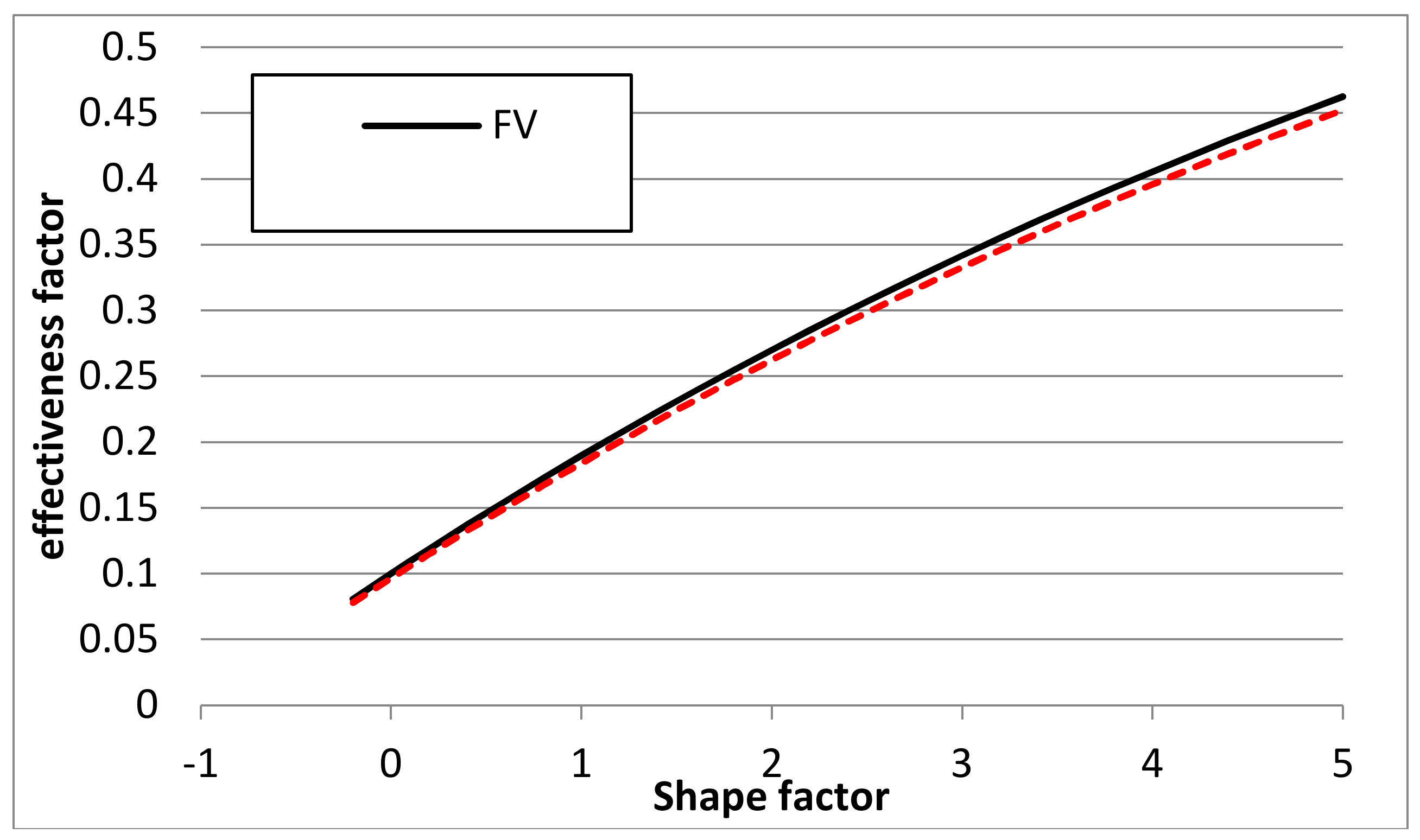
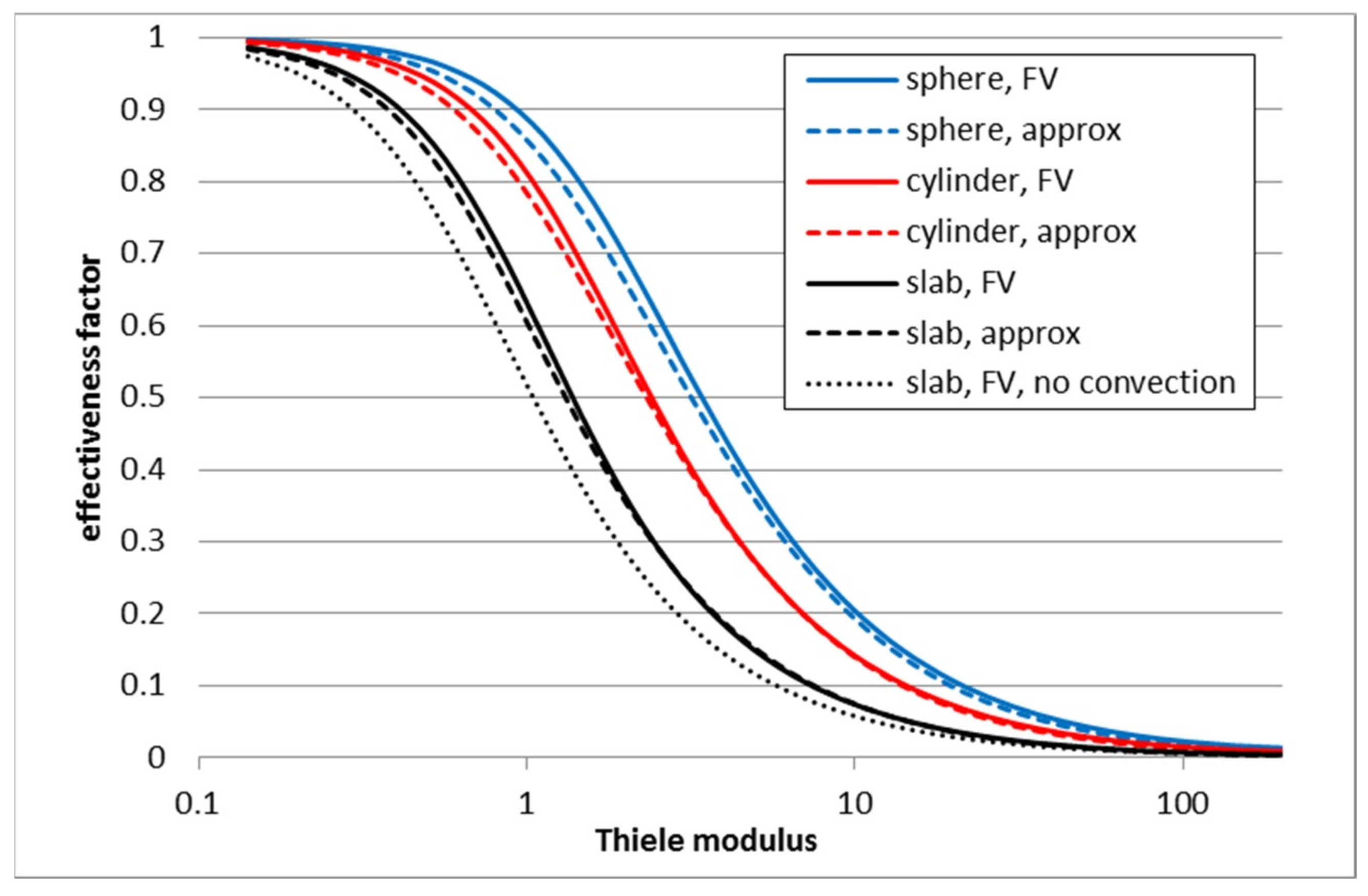
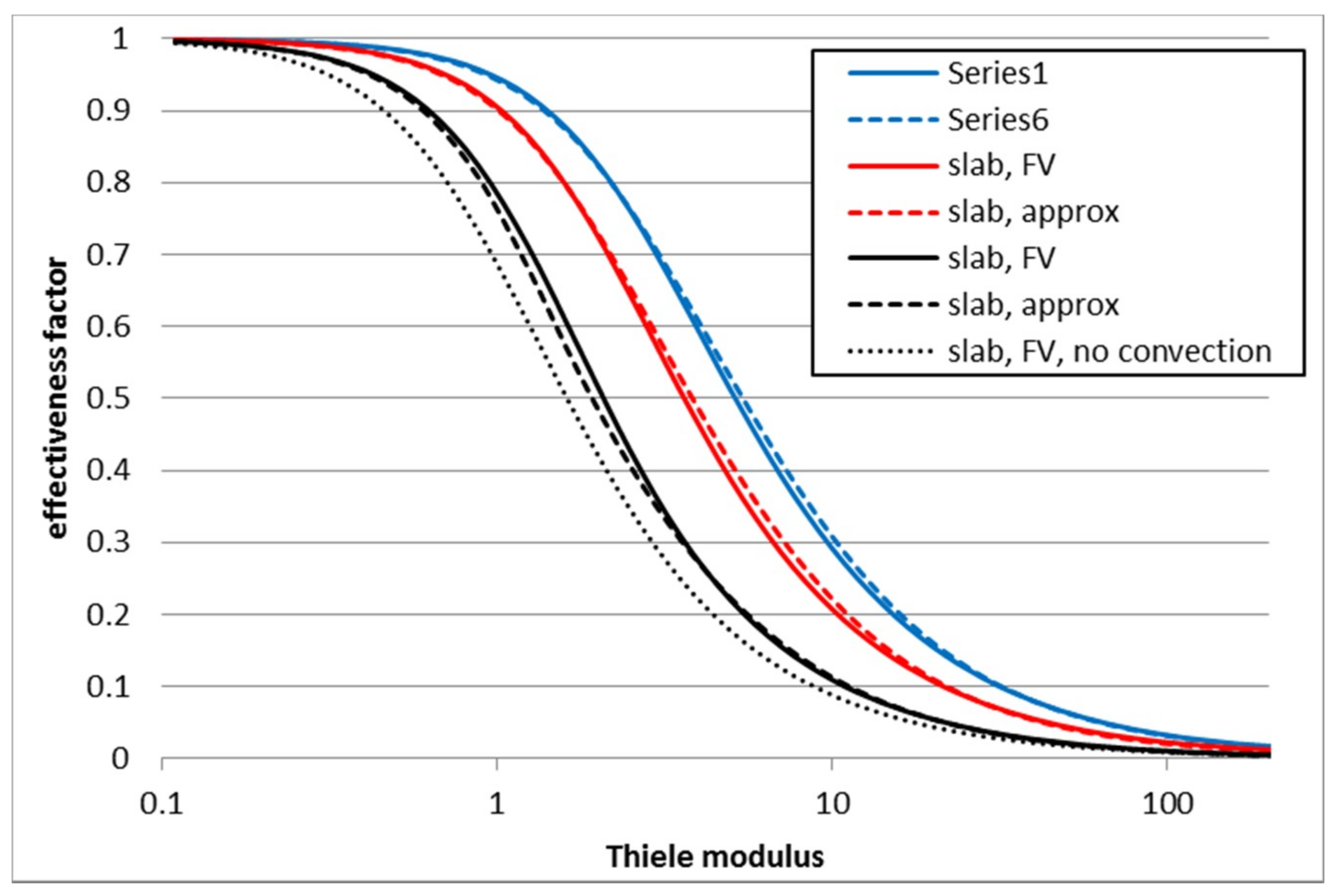
3.1. First Order Elementary Reaction A → B
3.2. Second Order Reaction 2A → B
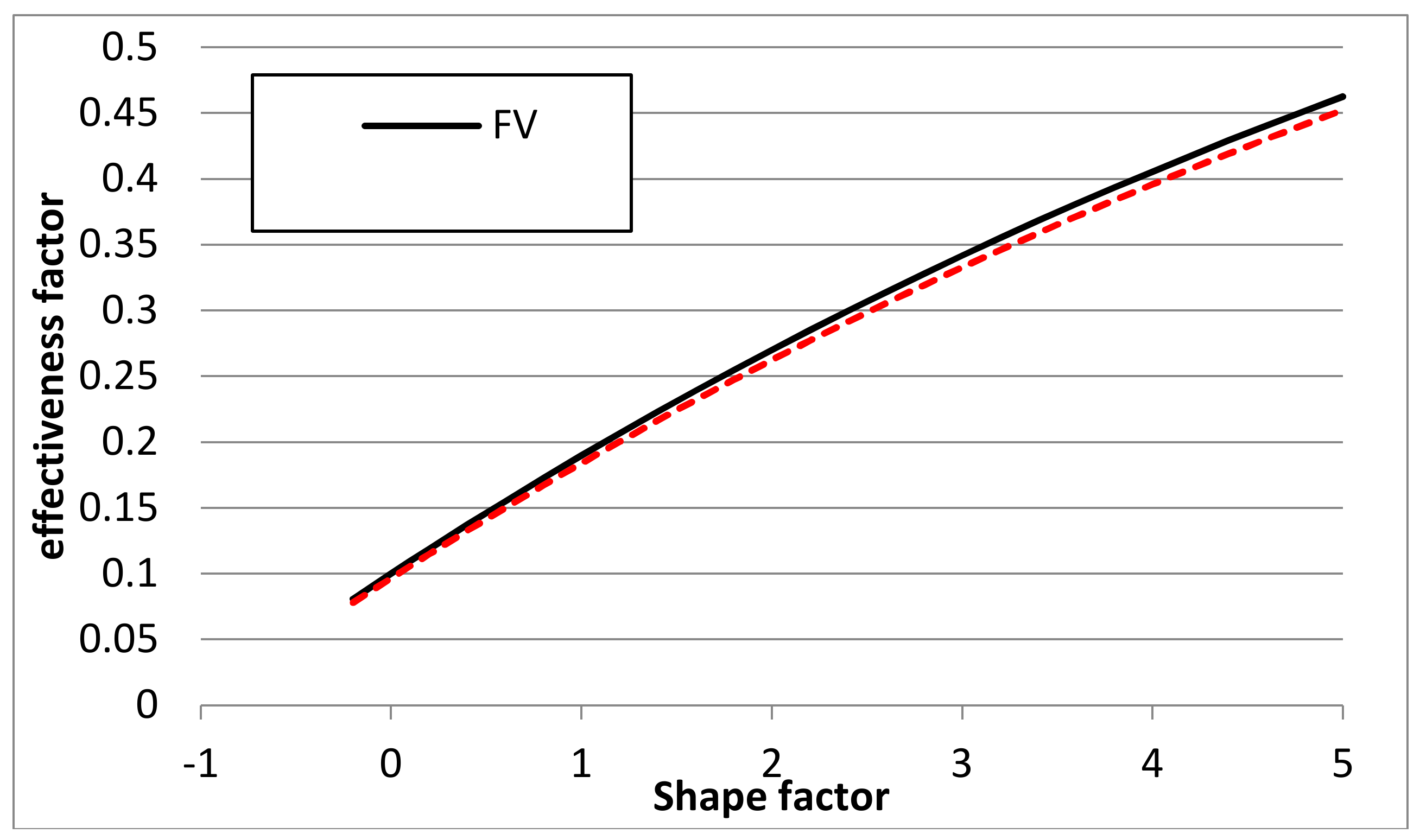
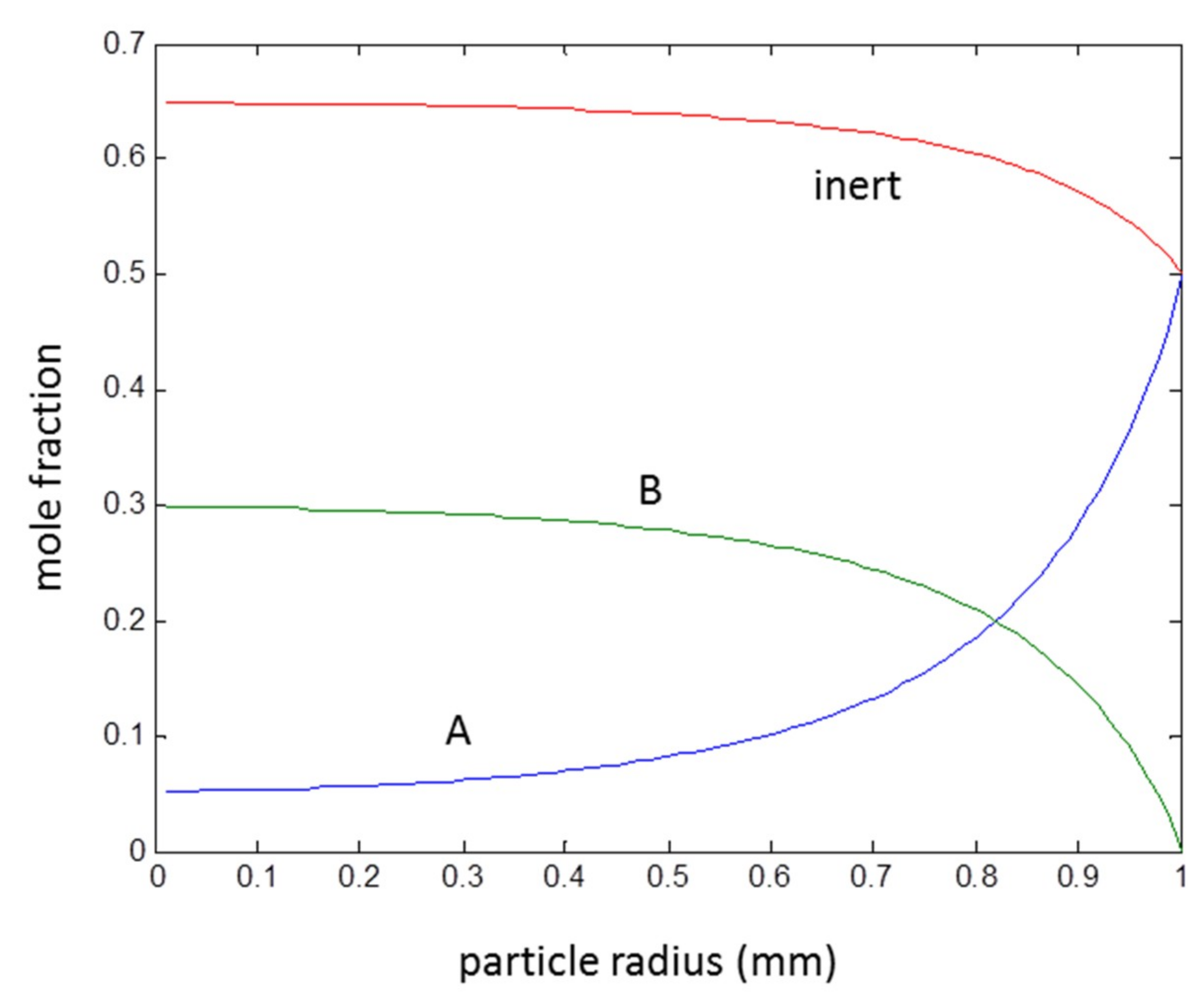
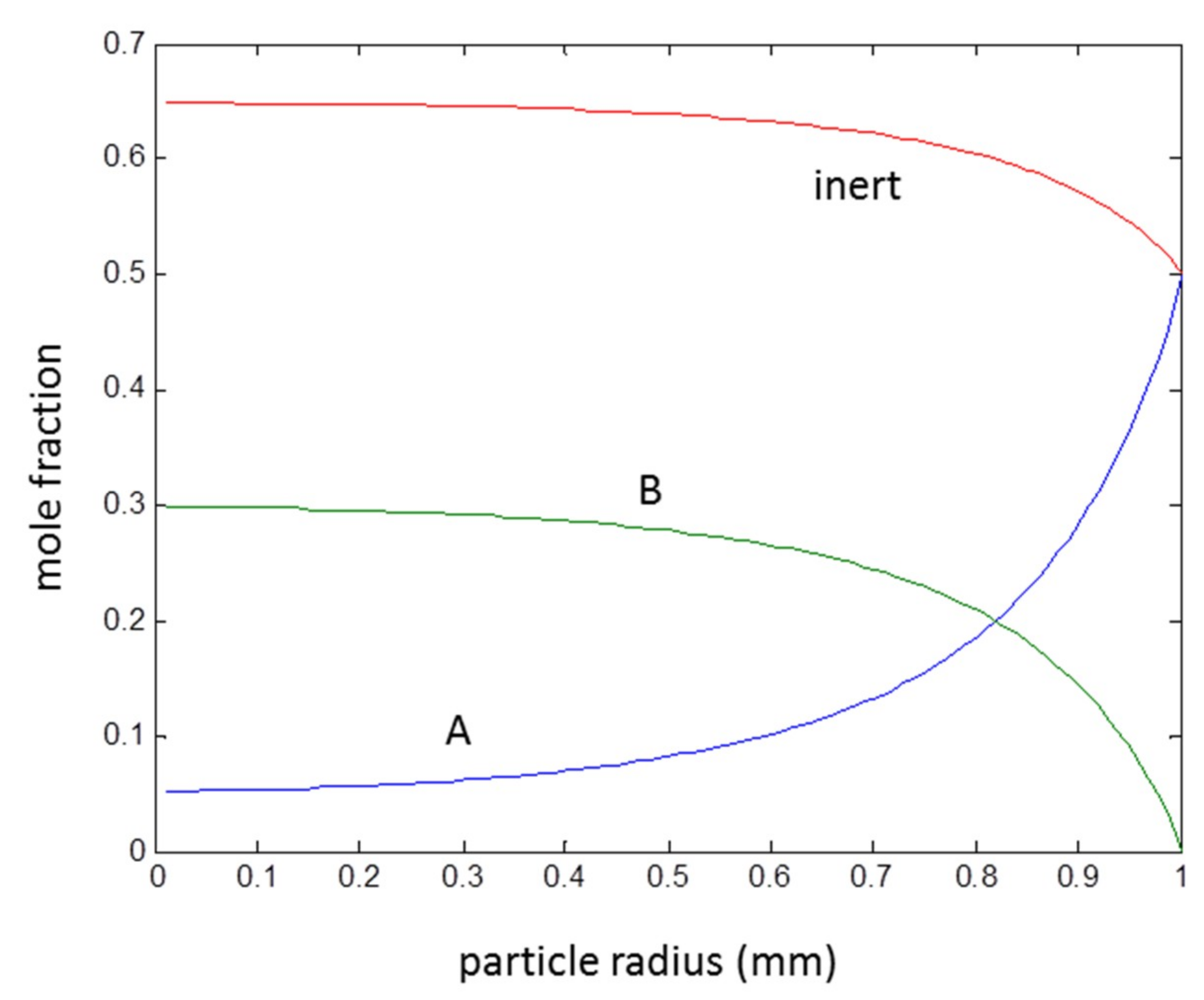
3.3. Second Order Reaction A + B → C
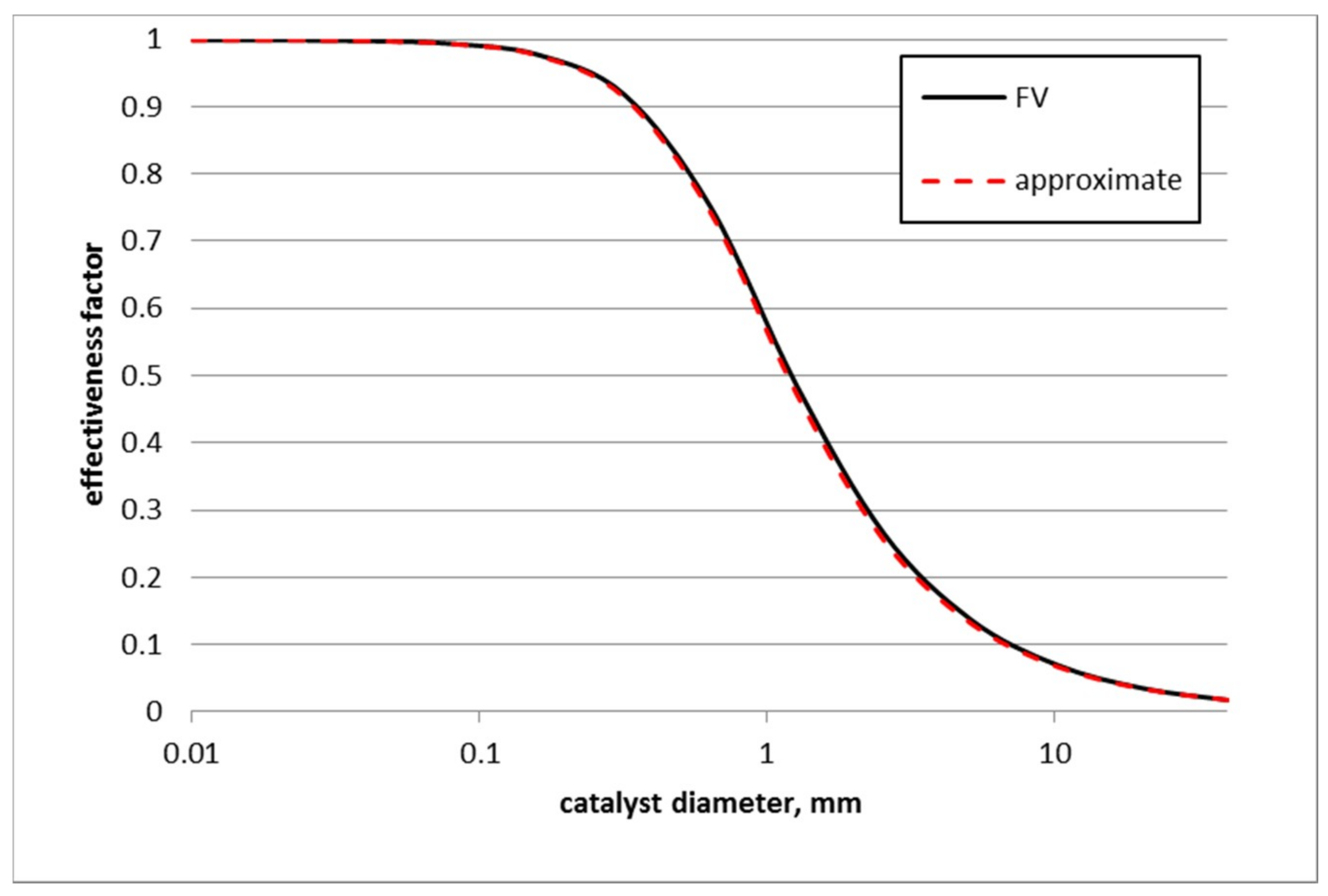
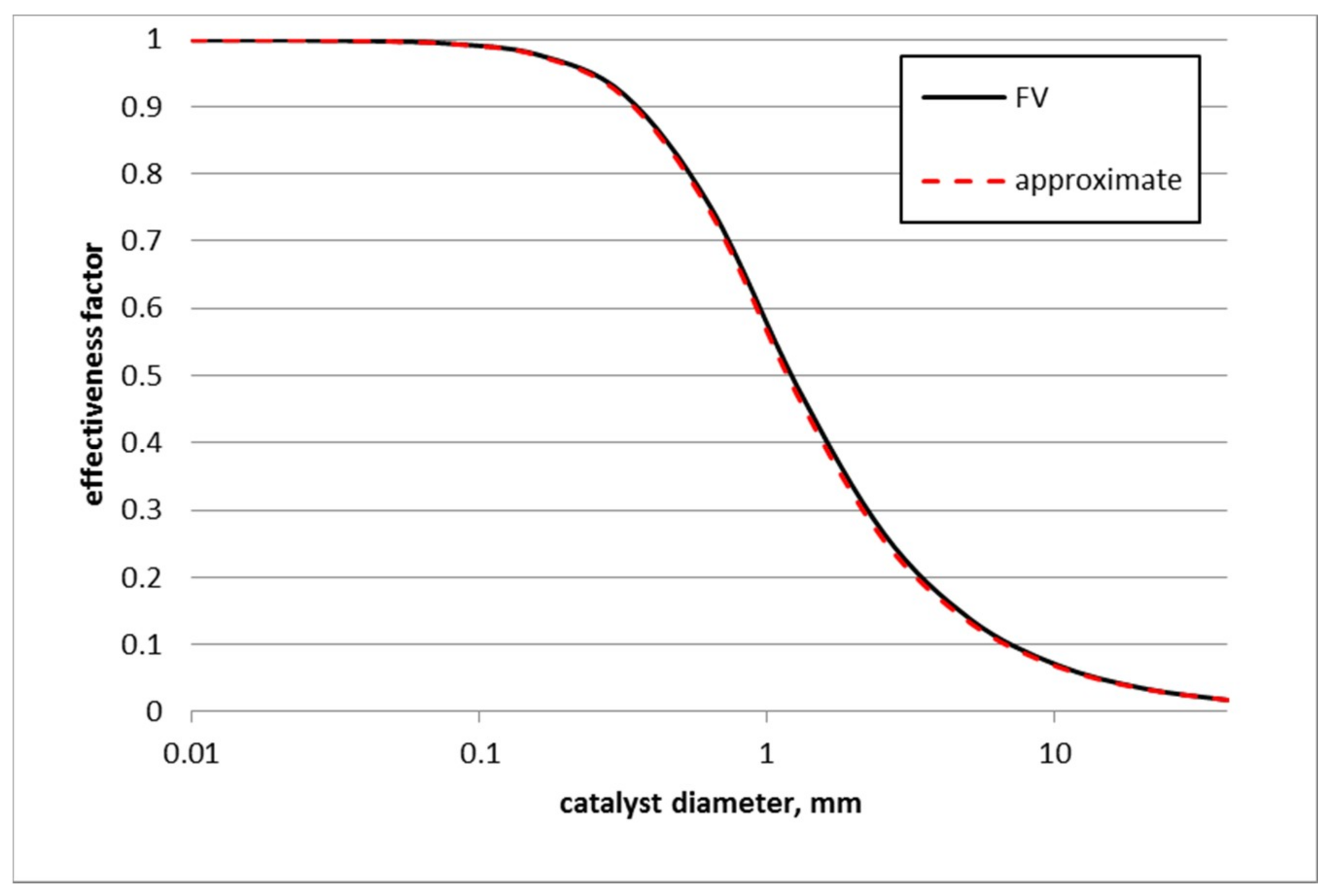
3.4. Effect of Catalyst Size on Toluene Hydrogenation
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| a | parameter in reaction profile | (mol/m3s) |
| A | area | (m2) |
| B | support variable, B = D−1/ct | (ms/mol) |
| b | parameter in reaction profile | various |
| c | concentration | (mol/m3) |
| ct | total concentration | (mol/m3) |
| D | diffusion coefficient | (m2/s) |
| J | diffusion flux (flux relative to molar average velocity frame) | (mol/m2s) |
| k | convection variable | (mol/m3s) |
| KA, KH | reaction rate constants (adsorption coefficients) | (m3/mol) |
| kr | reaction rate constant | (mol/kgs) |
| L | diameter of the reactive region (radius or half thickness) | (m) |
| m | geometry factor | ( ) |
| N | mass transfer flux (flux relative to stationary coordinates) | (mol/m2s) |
| n | parameter in reaction profile | ( ) |
| nc | number of components | ( ) |
| Nt | total flux | (mol/m2s) |
| r | length dimension | (m) |
| R, R0, RL | reaction rate, reaction rate at the center, reaction rate at the surface | (mol/m3s) |
| t | time | (s) |
| V | volume | (m3) |
| Vm | molar volume | (m3/mol) |
| x, x0, xL | mole fraction, mole fraction at the center, mole fraction at the surface | ( ) |
| xc | convective mole fractions | ( ) |
| ϕ | Thiele modulus, as defined by Equation (27) | ( ) |
| η | effectiveness factor | ( ) |
| [] | square matrix | various |
| ( ) | column matrix | various |
References
- Aris, R. The Mathematical Theory of Diffusion and Reaction in Permeable Catalysts. Vol I. The Theory of the Steady State; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Bird, R.B.; Stewart, W.E.; Lightfoot, E.N. Transport Phenomena; Wiley: New York, NY, USA, 1960. [Google Scholar]
- Fogler, S. Elements of Chemical Reaction Engineering, 4th ed.; Prentice Hall: Englewood Cliffs, NJ, USA, 2006. [Google Scholar]
- Levenspiel, O. Chemical Reaction Engineering, 2nd ed.; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Finlayson, B.A. Nonlinear Analysis in Chemical Engineering; McGraw-Hill: New York, NY, USA, 1980. [Google Scholar]
- Rice, R.G.; Do, D.D. Applied Mathematics and Modeling for Chemical Engineers; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Villadsen, J.; Michelsen, M.L. Solution of Differential Equation Models by Polynomial Approximation; Prentice-Hall: Englewood Cliffs, NJ, USA, 1978. [Google Scholar]
- Russo, V.; Kilpiö, T.; Di Serio, M.; Tesser, R.; Santacesaria, E.; Murzin, D.Y.; Salmi, T. Dynamic non-isothermal trickle bed reactor with both internal diffusion and heat conduction: Sugar hydrogenation as a case study. Chem. Eng. Res. Des. 2015, 102, 171–185. [Google Scholar] [CrossRef]
- Thiele, E.W. Relation between Catalytic Activity and Size of Particle. Ind. Eng. Chem. Res. 1939, 31, 916–920. [Google Scholar] [CrossRef]
- Gottifredi, J.C.; Gonzo, E.E.; Quiroga, O.D. Isothermal Effectiveness Factor—I Analytical expression for single reaction with arbitrary kinetics. Slab geometry. Chem. Eng. Sci. 1981, 36, 705–711. [Google Scholar]
- Gottifredi, J.C.; Gonzo, E.E.; Quiroga, O.D. Isothermal Effectiveness Factor—II Analytical expression for single reaction with arbitrary kinetics, geometry and activity distribution. Chem. Eng. Sci. 1981, 36, 713–719. [Google Scholar] [CrossRef]
- Haynes, H.W. An explicit approximation for the effectiveness factor in porous heterogeneous catalysts. Chem. Eng. Sci. 1986, 41, 412–415. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, J. A simple formula for estimation of the effectiveness factor in porous catalysts. AIChE J. 2006, 52, 3631–3635. [Google Scholar] [CrossRef]
- Kubota, H.; Yamanaka, Y. Remarks on approximate estimation of catalyst effectiveness factor. J. Chem. Eng. Jpn. 1969, 2, 238–240. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.H. An approximation method for the effectiveness factor in porous catalysts. Chem. Eng. Sci. 2006, 61, 5127–5136. [Google Scholar] [CrossRef]
- Marroquin de la Rosa, J.O.; Garcia, T.V.; Ochoa Tapia, J.A. A linear approximation method to evaluate isothermal effectiveness factors. Chem. Eng. Commun. 1999, 174, 53–60. [Google Scholar] [CrossRef]
- Wedel, S.; Luss, D. A rational approximation of the effectiveness factor. Chem. Eng. Commun. 1980, 7, 245–254. [Google Scholar] [CrossRef]
- Yin, Q.; Li, S. Rational Approximation of the Overall Effectiveness Factor for the Gas-Liquid-Solid Phase Catalytic Reaction. Ind. Eng. Chem. Res. 1995, 34, 3771–3776. [Google Scholar] [CrossRef]
- Aris, R. On the shape factors for irregular particles—I The steady state problem. Diffusion and reaction. Chem. Eng. Sci. 1957, 6, 262–268. [Google Scholar] [CrossRef]
- Burghardt, A.; Kubaczka, A. Generalization of the effectiveness factor for any shape of a catalyst pellet. Chem. Eng. Process. 1996, 35, 65–74. [Google Scholar] [CrossRef]
- Mariani, N.J.; Mocciaro, C.; Keegan, S.D.; Martinez, O.M.; Barreto, G.F. Evaluating the effectiveness factor from a 1D approximation fitted at high Thiele modulus: Spanning commercial pellet shapes with linear kinetics. Chem. Eng. Sci. 2009, 64, 2762–2766. [Google Scholar] [CrossRef]
- Liu, Z.; Suntio, V.; Kuitunen, S.; Roininen, J.; Alopaeus, V. Modeling of mass transfer and reactions in anisotropic biomass particles with reduced computational load. Ind. Eng. Chem. Res. 2014, 53, 4096–4103. [Google Scholar] [CrossRef]
- Salmi, T. Computer Aided Chemical Reaction Engineering. Lecture Notes; Åbo Akademi: Turku, Finland, 1996. [Google Scholar]
- Gorshkova, E.; Manninen, M.; Alopaeus, V.; Laavi, H.; Koskinen, J. Three-Phase CFD-Model for Trickle Bed Reactors. Int. J. Nonlinear Sci. Numer. Simul. 2012, 13, 397–404. [Google Scholar] [CrossRef]
- Taylor, R.; Krishna, R. Multicomponent Mass Transfer; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Jackson, R. On the limit of bulk diffusion control and high permeability in porous catalyst pellets. Chem. Eng. Sci. 1974, 29, 1413–1419. [Google Scholar] [CrossRef]
- Keegan, S.D.; Mariani, N.J.; Bressa, S.P.; Mazza, G.D.; Barreto, G.F. Approximation of the effectiveness factor in catalyst pellets. Chem. Eng. J. 2003, 94, 107–112. [Google Scholar] [CrossRef]
- Toppinen, S.; Rantakylä, T.-K.; Salmi, T.; Aittamaa, J. Kinetics of the Liquid-Phase Hydrogenation of Benzene and Some Monosubstituted Alkylbenzenes over a Nickel Catalyst. Ind. Eng. Chem. Res. 1996, 35, 1824–1833. [Google Scholar] [CrossRef]
- Reid, R.C.; Prausnitz, J.M.; Poling, B.E. Properties of Gases & Liquids, 4th ed.; McGraw-Hill: New York, NY, USA, 1987. [Google Scholar]
- Alopaeus, V.; Aittamaa, J. Appropriate simplifications in calculation of mass transfer in a multicomponent rate-based distillation tray model. Ind. Eng. Chem. Res. 2000, 39, 4336–4345. [Google Scholar] [CrossRef]
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alopaeus, V. Approximating Catalyst Effectiveness Factors with Reaction Rate Profiles. Catalysts 2019, 9, 255. https://doi.org/10.3390/catal9030255
Alopaeus V. Approximating Catalyst Effectiveness Factors with Reaction Rate Profiles. Catalysts. 2019; 9(3):255. https://doi.org/10.3390/catal9030255
Chicago/Turabian StyleAlopaeus, Ville. 2019. "Approximating Catalyst Effectiveness Factors with Reaction Rate Profiles" Catalysts 9, no. 3: 255. https://doi.org/10.3390/catal9030255
APA StyleAlopaeus, V. (2019). Approximating Catalyst Effectiveness Factors with Reaction Rate Profiles. Catalysts, 9(3), 255. https://doi.org/10.3390/catal9030255