Change of the Product Specificity of a Cyclodextrin Glucanotransferase by Semi-Rational Mutagenesis to Synthesize Large-Ring Cyclodextrins


Abstract
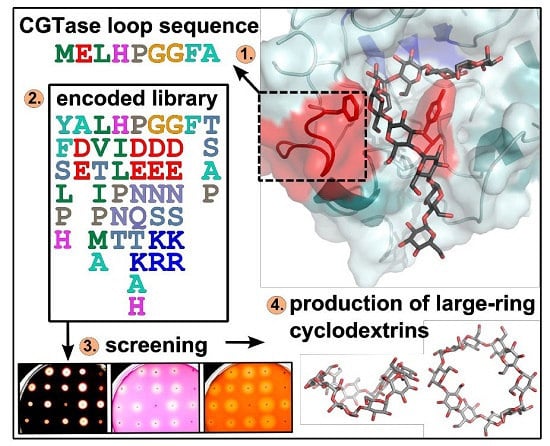
1. Introduction
2. Results
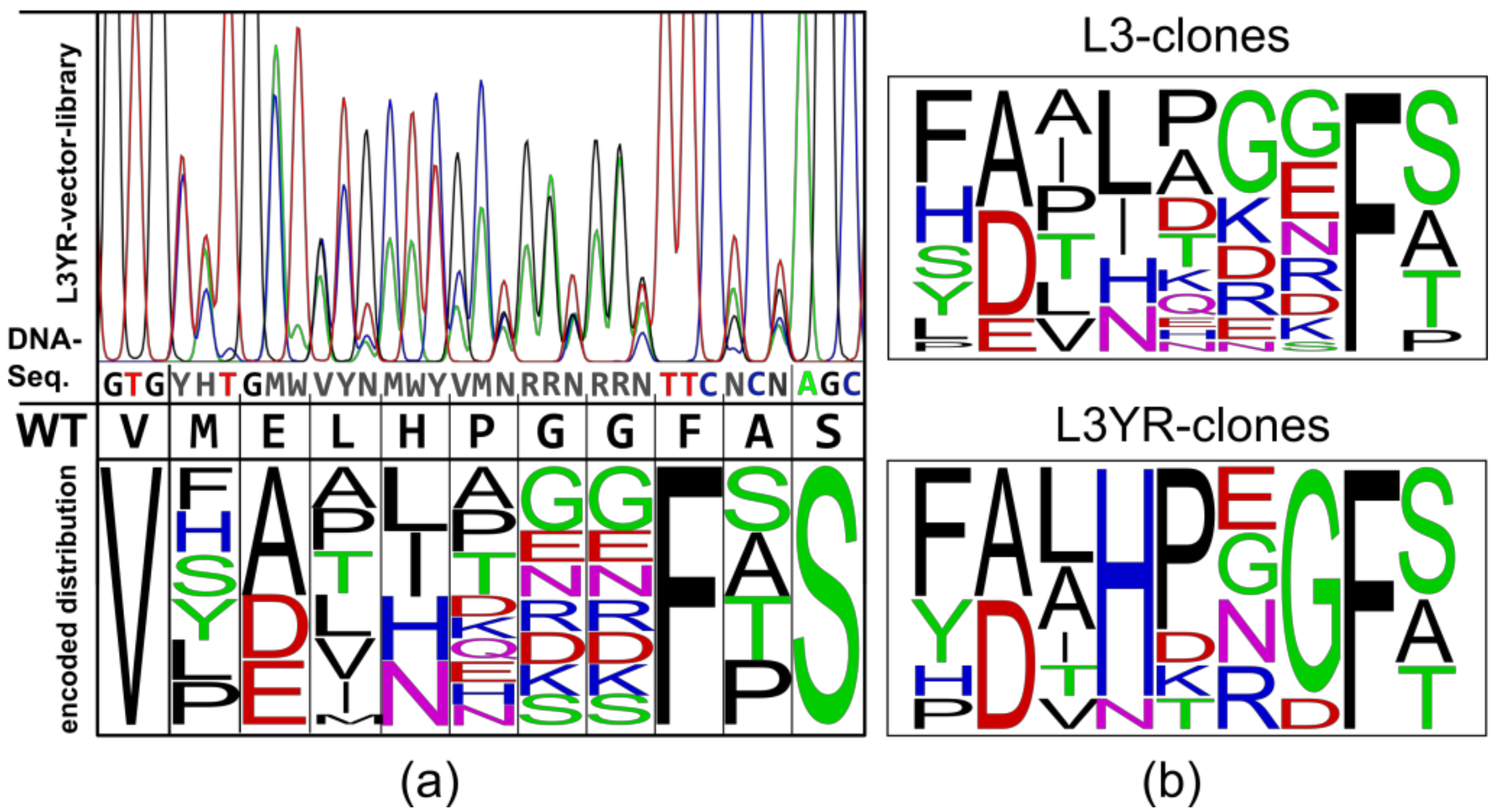
2.1. Construction of Vector Libraries
2.2. Agar Plate Sceening of the CGTase Variants
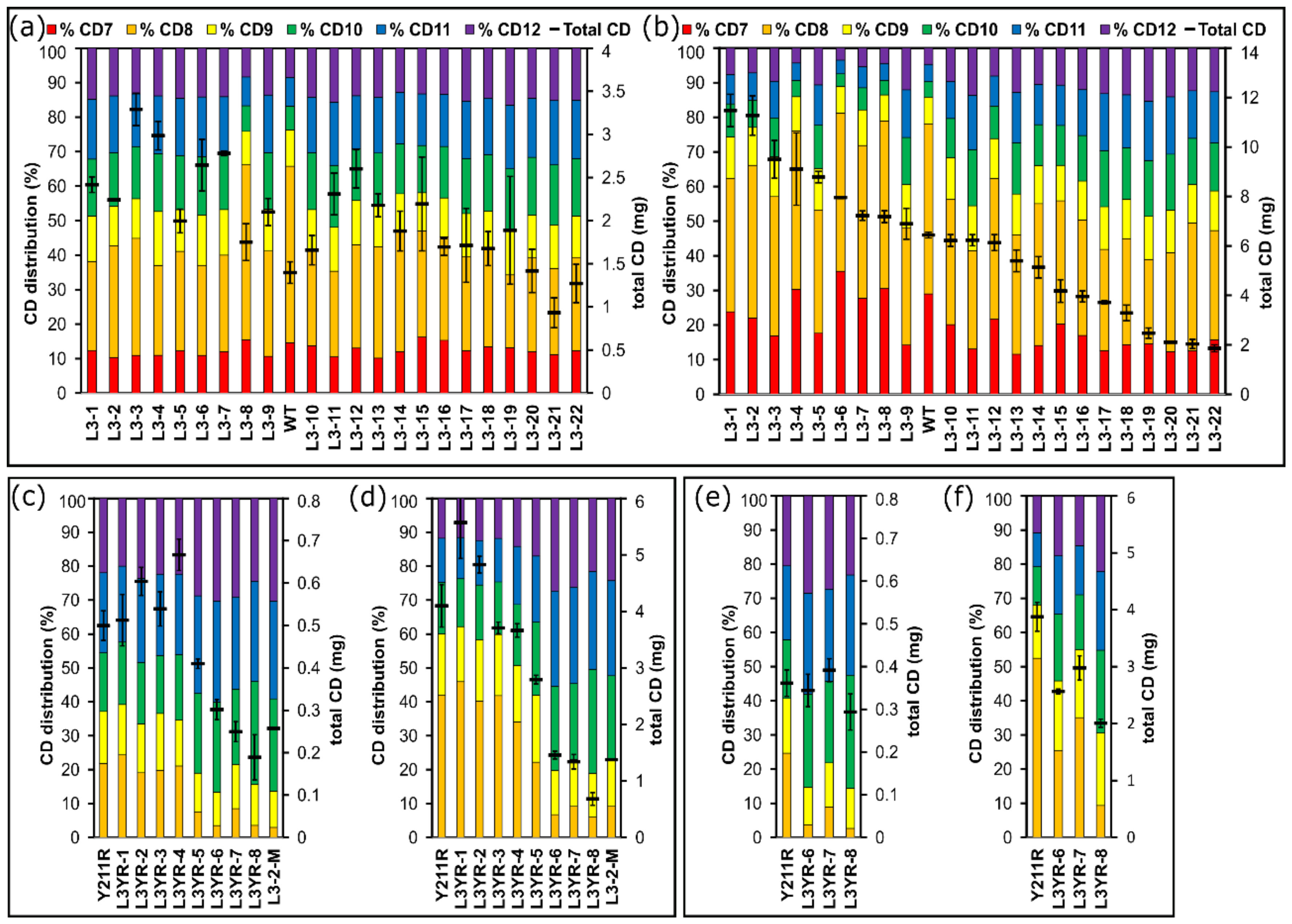
2.3. Comparison of the CD Synthesis by the CGTase Variants
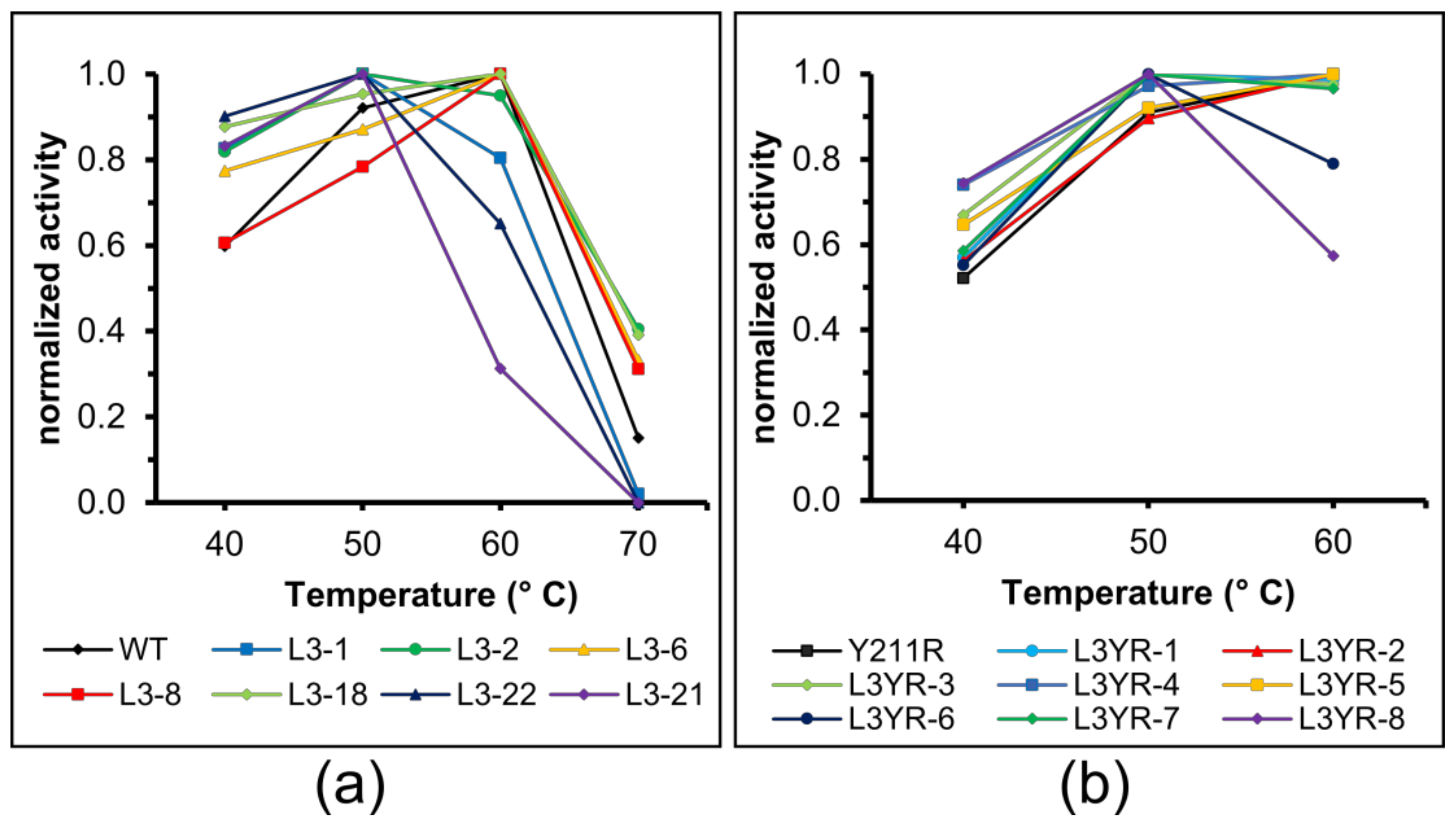
2.4. Thermostability of the CGTase Variants
3. Discussion
3.1. High Fitness of the CGTase Variant Library
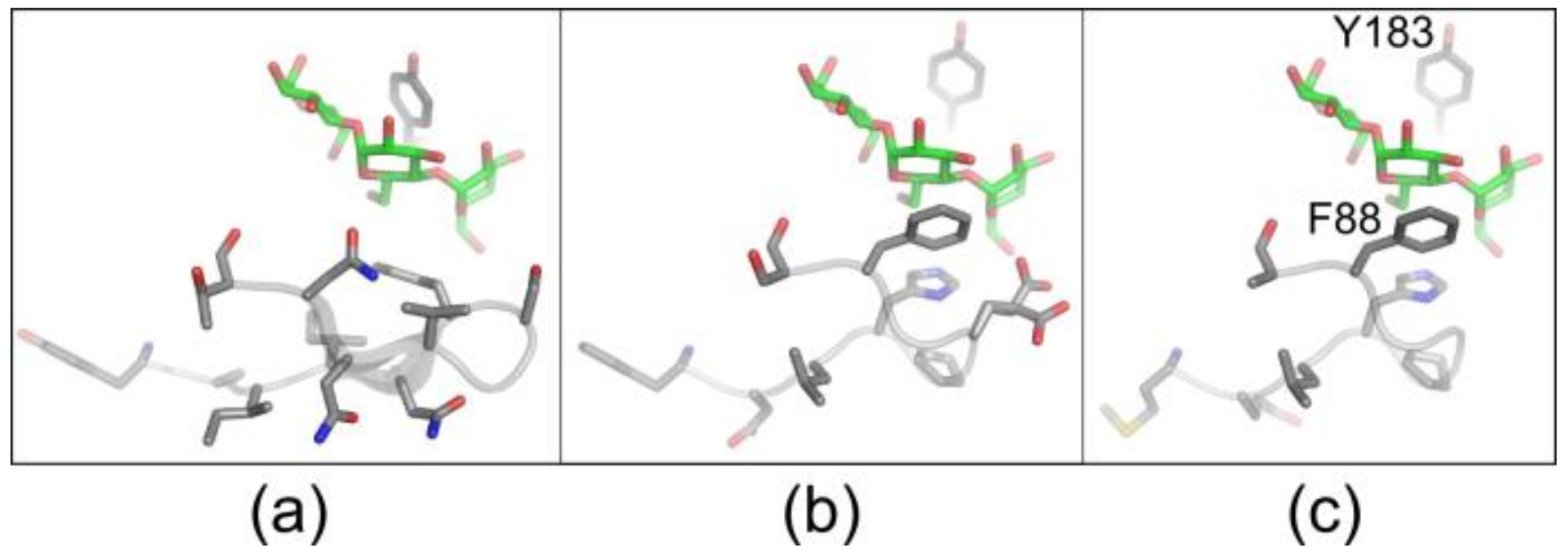
3.2. Selection of CGTase Variants with Changed CD Product Specificity
3.3. Variants Synthesizing High Yields of Large-Ring CD
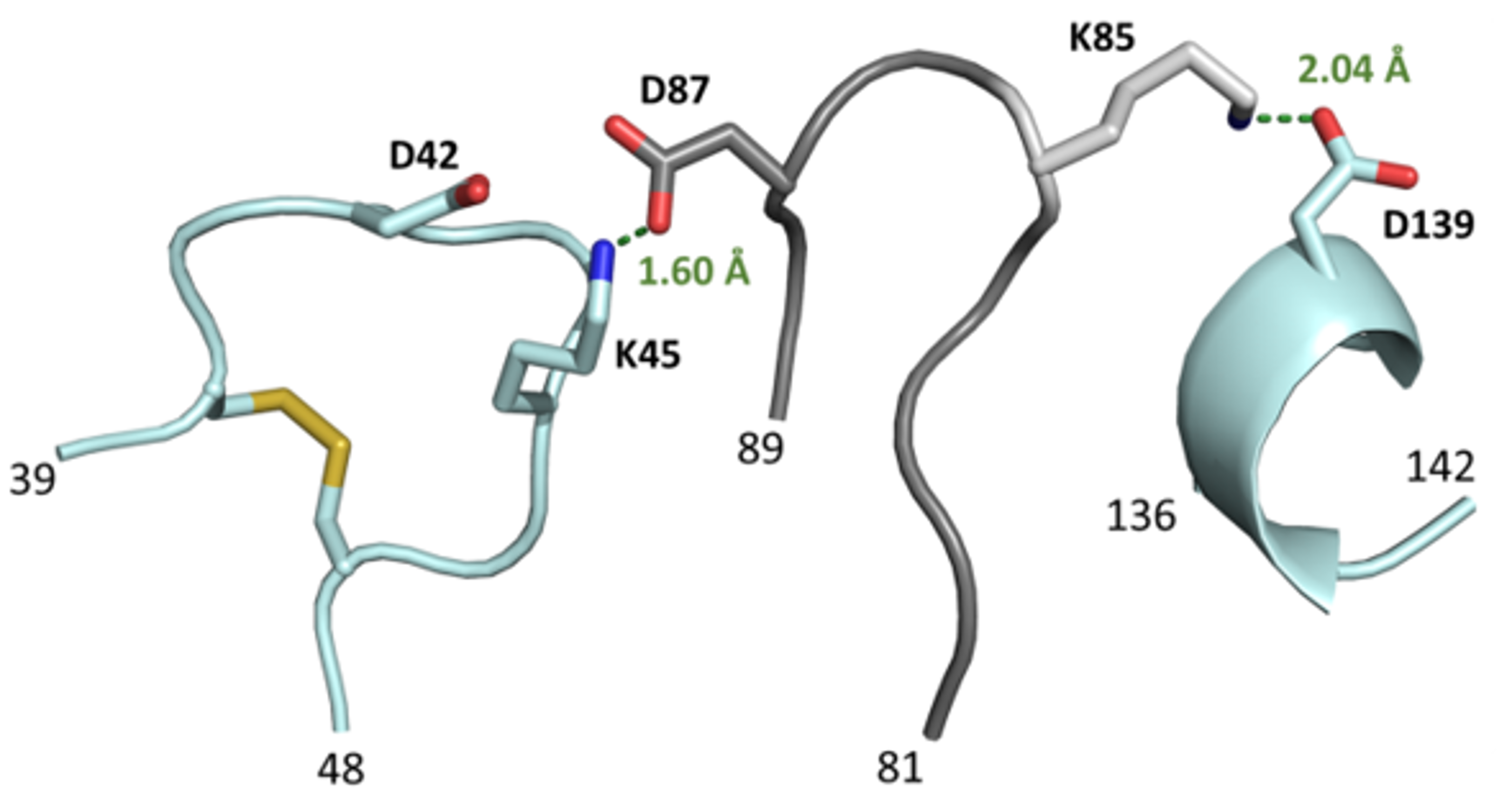
3.4. The L3 Loop Contributes to the Thermal Stability of the CGTase
4. Materials and Methods
4.1. Generation of the Vector Library
4.2. Agar Plate Screening
4.3. Recombinant Protein Production and Analysis
4.4. CD Synthesis and Analysis
4.5. Determination of Temperature Optimum and Thermostability of the Variants
4.6. Protein Sequence Alignments and Molecular Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cobb, R.E.; Chao, R.; Zhao, H. Directed evolution: Past, present and future. AIChE J. 2013, 59, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Dalby, P.A. Strategy and success for the directed evolution of enzymes. Curr. Opin. Struct. Biol. 2011, 21, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-C.; Vaisvila, R. Protein engineering: Single or multiple site-directed mutagenesis. Methods Mol. Biol. 2013, 978, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chica, R.A.; Doucet, N.; Pelletier, J.N. Semi-rational approaches to engineering enzyme activity: Combining the benefits of directed evolution and rational design. Curr. Opin. Biotechnol. 2005, 16, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Patrick, W.M.; Firth, A.E. Strategies and computational tools for improving randomized protein libraries. Biomol. Eng. 2005, 22, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S. Beyond directed evolution-semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.Y.; Snow, C.D.; Vizcarra, C.L.; Mayo, S.L.; Arnold, F.H. Comparison of random mutagenesis and semi-rational designed libraries for improved cytochrome P450 BM3-catalyzed hydroxylation of small alkanes. Protein Eng. Des. Sel. 2012, 25, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gao, H.; Zhu, X.; Wang, X.; Zhou, M.; Jiang, R. Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. BioTechniques 2012, 52, 149–158. [Google Scholar] [CrossRef] [PubMed]
- DePinto, J.A.; Campbell, L.L. Purification and properties of the amylase of Bacillus macerans. Biochemistry 1968, 7, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Zimmermann, W. Cyclodextrin glucanotransferase: From gene to applications. Appl. Microbiol. Biotechnol. 2005, 66, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Dijkstra, B.W. The cyclization mechanism of cyclodextrin glycosyltransferase (CGTase) as revealed by a gamma-cyclodextrin-CGTase complex at 1.8—A resolution. J. Biol. Chem. 1999, 274, 34868–34876. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, E. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Endo, T.; Nagase, H.; Ueda, H.; Kobayashi, S.; Nagai, T. Isolation, purification, and characterization of cyclomaltodecaose (.EPSILON.-Cyclodextrin), cyclomaltoundecaose (.ZETA.-Cyclodextrin) and cyclomaltotridecaose (.THETA.-Cyclodextrin). Chem. Pharm. Bull. 1997, 45, 532–536. [Google Scholar] [CrossRef]
- Larsen, K.L.; Endo, T.; Ueda, H.; Zimmermann, W. Inclusion complex formation constants of α-, β-, γ-, δ-, ε-, ζ-, η- and θ-cyclodextrins determined with capillary zone electrophoresis. Carbohydr. Res. 1998, 309, 153–159. [Google Scholar] [CrossRef]
- Ueda, H.; Endo, T. Large-ring cyclodextrins. In Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications; Dodziuk, H., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 370–380. ISBN 9783527608980. [Google Scholar]
- Akasaka, H.; Endo, T.; Nagase, H.; Ueda, H.; Kobayashi, S. Complex formation of cyclomaltononaose delta-cyclodextrin (delta-CD) with macrocyclic compounds. Chem. Pharm. Bull. 2000, 48, 1986–1989. [Google Scholar] [CrossRef] [PubMed]
- Dodziuk, H.; Ejchart, A.; Anczewski, W.; Ueda, H.; Krinichnaya, E.; Dolgonos, G.; Kutner, W. Water solubilization, determination of the number of different types of single-wall carbon nanotubes and their partial separation with respect to diameters by complexation with η-cyclodextrin. Chem. Commun. 2003, 9, 986–987. [Google Scholar] [CrossRef]
- Assaf, K.I.; Gabel, D.; Zimmermann, W.; Nau, W.M. High-affinity host-guest chemistry of large-ring cyclodextrins. Org. Biomol. Chem. 2016, 14, 7702–7706. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Yanase, M.; Takata, H.; Takaha, T.; Okada, S. Cyclodextrins are not the major cyclic alpha-1,4-glucans produced by the initial action of cyclodextrin glucanotransferase on amylose. J. Biol. Chem. 1997, 272, 15729–15733. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, M.; Wang, F.; Gu, Z.; Du, G.; Wu, J.; Chen, J. Gamma-cyclodextrin: A review on enzymatic production and applications. Appl. Microbiol. Biotechnol. 2007, 77, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Sanbe, H.; Takaha, T.; Kitahata, S.; Koizumi, K.; Okada, S. Comparative study of the cyclization reactions of three bacterial cyclomaltodextrin glucanotransferases. Appl. Environ. Microbiol. 2001, 67, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Li, J.; Shin, H.-D.; Chen, R.R.; Du, G.; Liu, L.; Chen, J. Recent advances in discovery, heterologous expression, and molecular engineering of cyclodextrin glycosyltransferase for versatile applications. Biotechnol. Adv. 2014, 32, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Sonnendecker, C.; Melzer, S.; Zimmermann, W. Engineered cyclodextrin glucanotransferases from Bacillus sp. G-825-6 produce large-ring cyclodextrins with high specificity. Microbiologyopen 2018, e757. [Google Scholar] [CrossRef] [PubMed]
- Sonnendecker, C.; Zimmermann, W. Domain shuffling of cyclodextrin glucanotransferases for tailored product specificity and thermal stability. FEBS Open Bio 2019, 9, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Bae, K.H.; Kim, T.J.; Park, K.H.; Lee, H.S.; Byun, S.M. Effect on product specificity of cyclodextrin glycosyltransferase by site-directed mutagenesis. Biochem. Mol. Biol. Int. 1997, 41, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.M.; Mahadi, N.M.; Hassan, O.; Rahman, R.N.Z.R.A.; Illias, R.M. A predominant β-CGTase G1 engineered to elucidate the relationship between protein structure and product specificity. J. Mol. Catal. B Enzym. 2009, 57, 270–277. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. ChemBioChem 2002, 3, 604. [Google Scholar] [CrossRef]
- Kaulpiboon, J.; Pongsawasdi, P.; Zimmermann, W. Molecular imprinting of cyclodextrin glycosyltransferases from Paenibacillus sp. A11 and Bacillus macerans with gamma-cyclodextrin. FEBS J. 2007, 274, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Kaulpiboon, J.; Pongsawasdi, P.; Zimmermann, W. Altered product specificity of a cyclodextrin glycosyltransferase by molecular imprinting with cyclomaltododecaose. J. Mol. Recognit. 2010, 23, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 47, 45–148. [Google Scholar] [CrossRef] [PubMed]
- Leemhuis, H.; Rozeboom, H.J.; Dijkstra, B.W.; Dijkhuizen, L. Improved thermostability of bacillus circulans cyclodextrin glycosyltransferase by the introduction of a salt bridge. Proteins 2004, 54, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.H. Molecular evolution over the mutational landscape. Evolution 1984, 38, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Hörtnagel, K.; Voloshin, O.N.; Kinal, H.H.; Ma, N.; Schaffer-Judge, C.; Camerini-Otero, R.D. Saturation mutagenesis of the E. coli RecA loop L2 homologous DNA pairing region reveals residues essential for recombination and recombinational repair. J. Mol. Biol. 1999, 286, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Decamp, M.; Jiang, L.; Bolon, D.; Clapham, P.R. Saturation mutagenesis of the HIV-1 envelope CD4 binding loop reveals residues controlling distinct trimer conformations. PLoS Pathog. 2016, 12, e1005988. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Wyss, M. Engineering proteins for thermostability: The use of sequence alignments versus rational design and directed evolution. Curr. Opin. Biotechnol. 2001, 12, 371–375. [Google Scholar] [CrossRef]
- Parsiegla, G.; Schmidt, A.K.; Schulz, G.E. Substrate binding to a cyclodextrin glycosyltransferase and mutations increasing the gamma-cyclodextrin production. Eur. J. Biochem. 1998, 255, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Leemhuis, H.; Kelly, R.M.; Dijkhuizen, L. Engineering of cyclodextrin glucanotransferases and the impact for biotechnological applications. Appl. Microbiol. Biotechnol. 2010, 85, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Sonnendecker, C.; Wei, R.; Kurze, E.; Wang, J.; Oeser, T.; Zimmermann, W. Efficient extracellular recombinant production and purification of a Bacillus cyclodextrin glucanotransferase in E. coli. Microb. Cell. Fact. 2017, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. The inoue method for preparation and transformation of competent e. Coli: "ultra-competent" cells. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Hamaker, K.; Tao, B.Y. Screening of Gamma Cyclodextrin-Producing Recombinant E. coli using congo red dye on solid complex media. Starch/Stärke 1993, 45, 181–182. [Google Scholar] [CrossRef]
- Park, C.S.; Park, K.H.; Kim, S.H. A rapid screening method for alkaline BETA-cyclodextrin glucanotransferase using phenolphthalein-methyl orange-containing-solid medium. Agric. Niol. Chem. 1989, 53, 1167–1169. [Google Scholar] [CrossRef]
- Oeser, T.; Wei, R.; Baumgarten, T.; Billig, S.; Föllner, C.; Zimmermann, W. High level expression of a hydrophobic poly (ethylene terephthalate)-hydrolyzing carboxylesterase from Thermobifida fusca KW3 in E. coli BL21(DE3). J. Biotechnol. 2010, 146, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Melzer, S.; Sonnendecker, C.; Föllner, C.; Zimmermann, W. Stepwise error-prone PCR and DNA shuffling changed the pH activity range and product specificity of the cyclodextrin glucanotransferase from an alkaliphilic Bacillus sp. FEBS Open Bio 2015, 5, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; Mosi, R.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Withers, S.G.; Dijkstra, B.W. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the alpha-amylase family. Nat. Struct. Biol. 1999, 6, 432–436. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CGTase | Tm (°C) |
|---|---|
| WT | 58.7 ± 0.19 |
| L3-1 | 59.7 ± 0.06 |
| L3-18 | 58.3 ± 0.08 |
| L3-21 | 52.5 ± 0.08 |
| Y211R | 67.3 ± 0.04 |
| L3YR-1 | 66.3 ± 0.08 |
| L3YR-6 | 64.8 ± 0.05 |
| L3YR-7 | 62.7 ± 0.05 |
| L3YR-8 | 63.5 ± 0.03 |
| L3-2M | 59.9 ± 0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonnendecker, C.; Zimmermann, W. Change of the Product Specificity of a Cyclodextrin Glucanotransferase by Semi-Rational Mutagenesis to Synthesize Large-Ring Cyclodextrins. Catalysts 2019, 9, 242. https://doi.org/10.3390/catal9030242
Sonnendecker C, Zimmermann W. Change of the Product Specificity of a Cyclodextrin Glucanotransferase by Semi-Rational Mutagenesis to Synthesize Large-Ring Cyclodextrins. Catalysts. 2019; 9(3):242. https://doi.org/10.3390/catal9030242
Chicago/Turabian StyleSonnendecker, Christian, and Wolfgang Zimmermann. 2019. "Change of the Product Specificity of a Cyclodextrin Glucanotransferase by Semi-Rational Mutagenesis to Synthesize Large-Ring Cyclodextrins" Catalysts 9, no. 3: 242. https://doi.org/10.3390/catal9030242
APA StyleSonnendecker, C., & Zimmermann, W. (2019). Change of the Product Specificity of a Cyclodextrin Glucanotransferase by Semi-Rational Mutagenesis to Synthesize Large-Ring Cyclodextrins. Catalysts, 9(3), 242. https://doi.org/10.3390/catal9030242