Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter

 ,
,  , ,
, ,  and
and 
Abstract
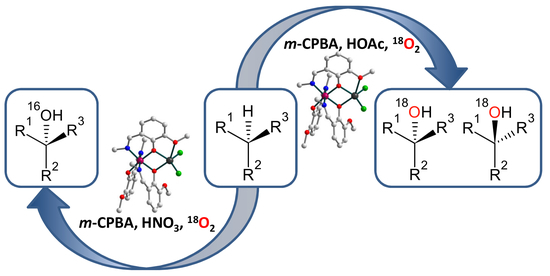
1. Introduction
2. Results and Discussion
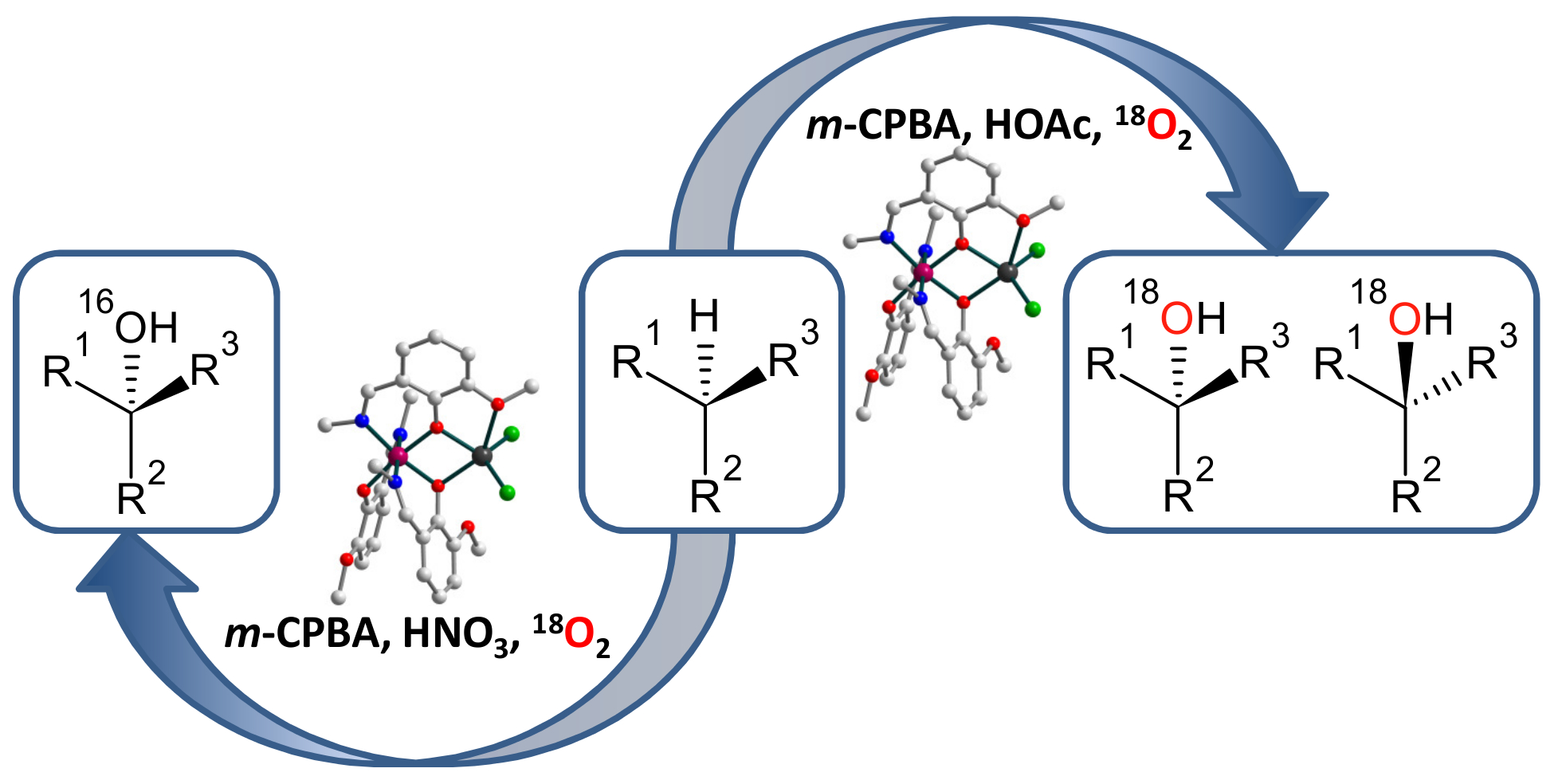
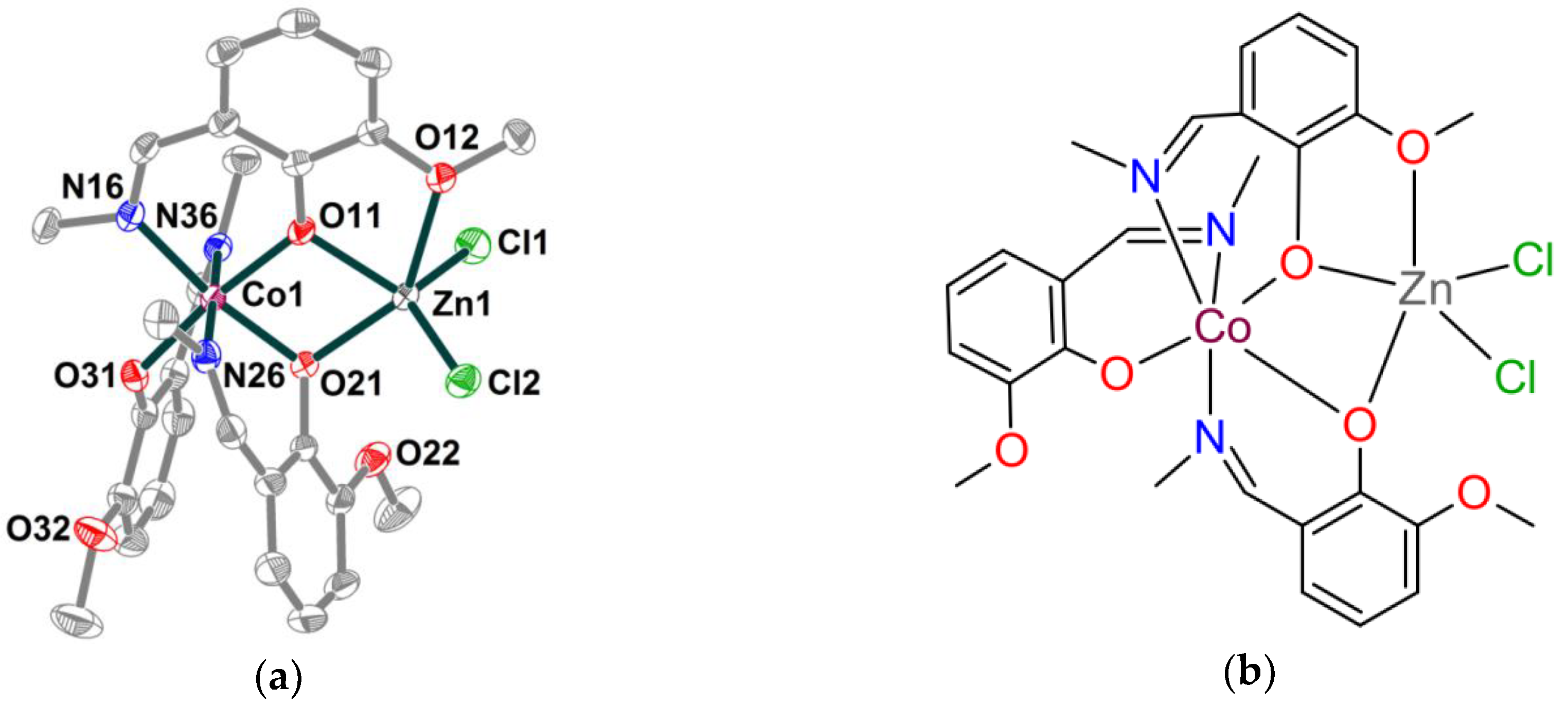
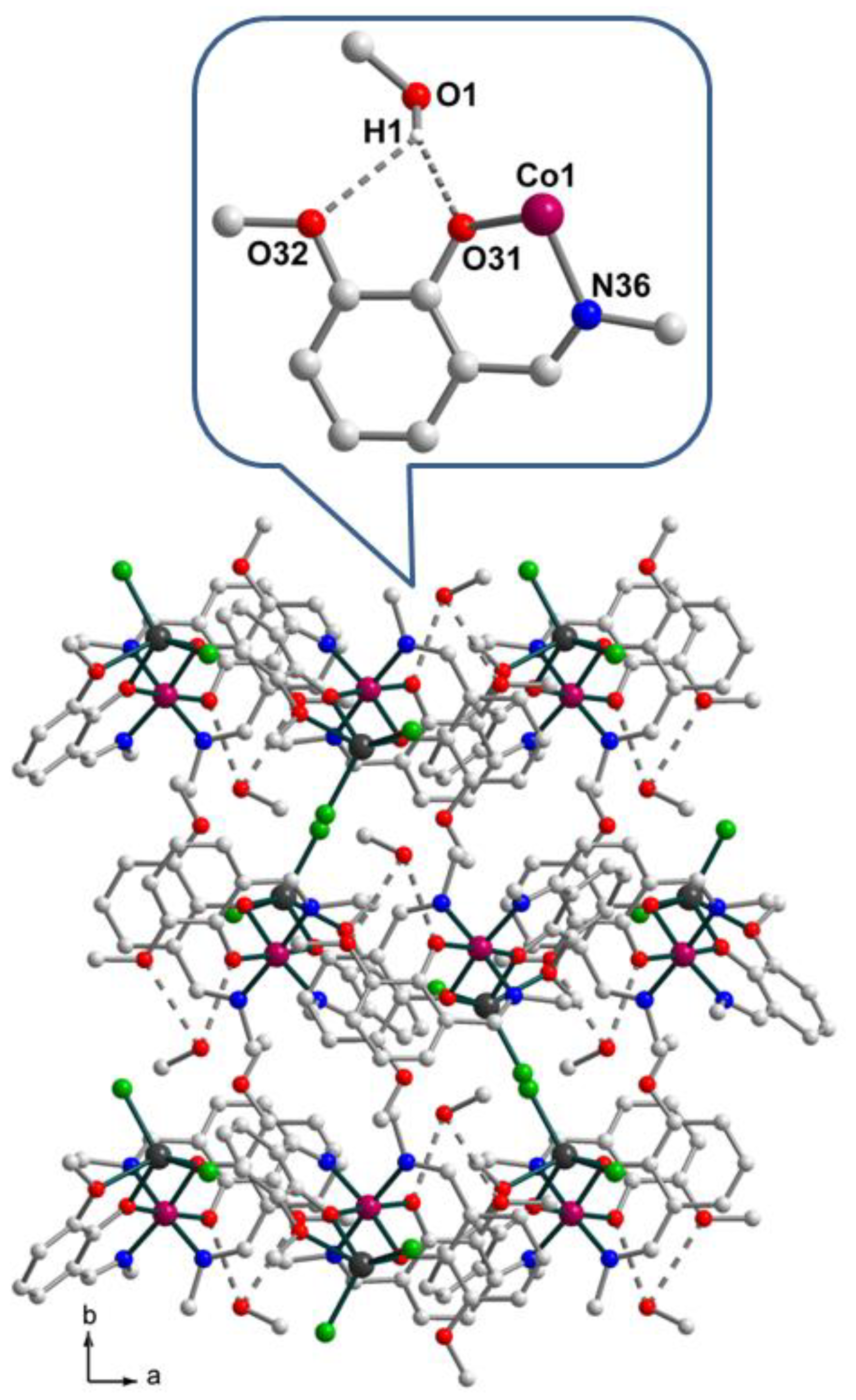
2.1. Synthesis and crystal structure
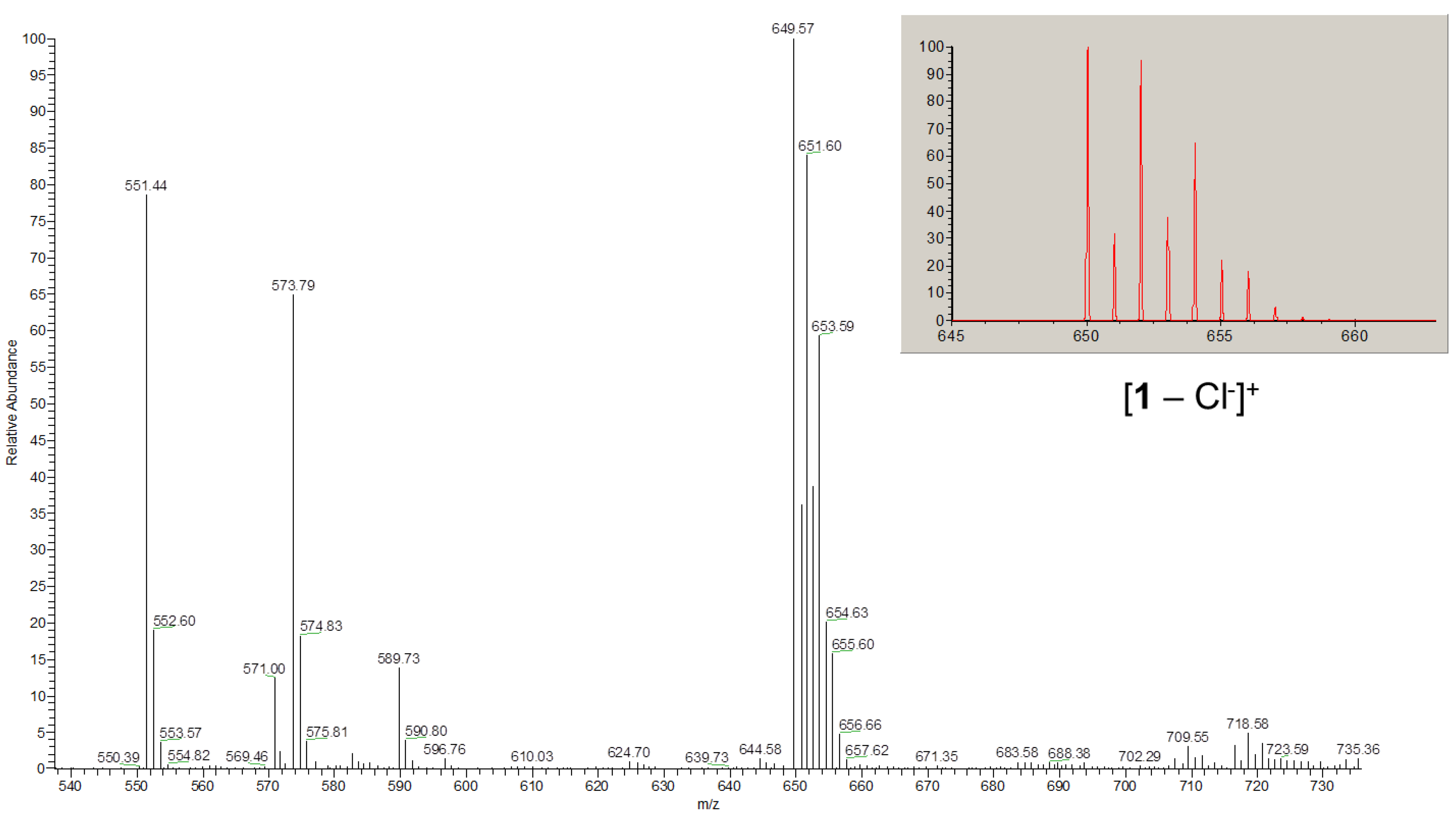
2.2. Spectroscopic characterization
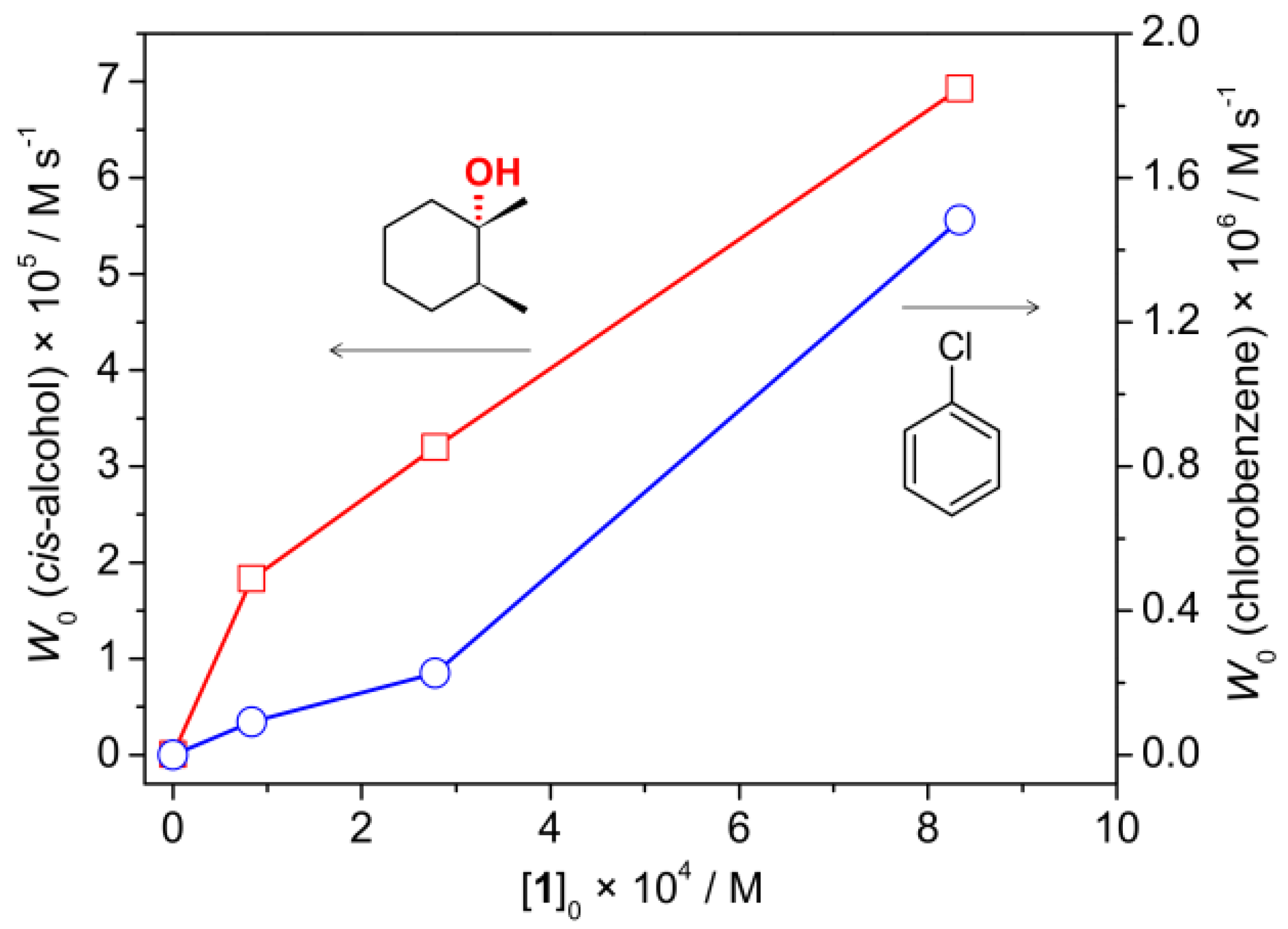
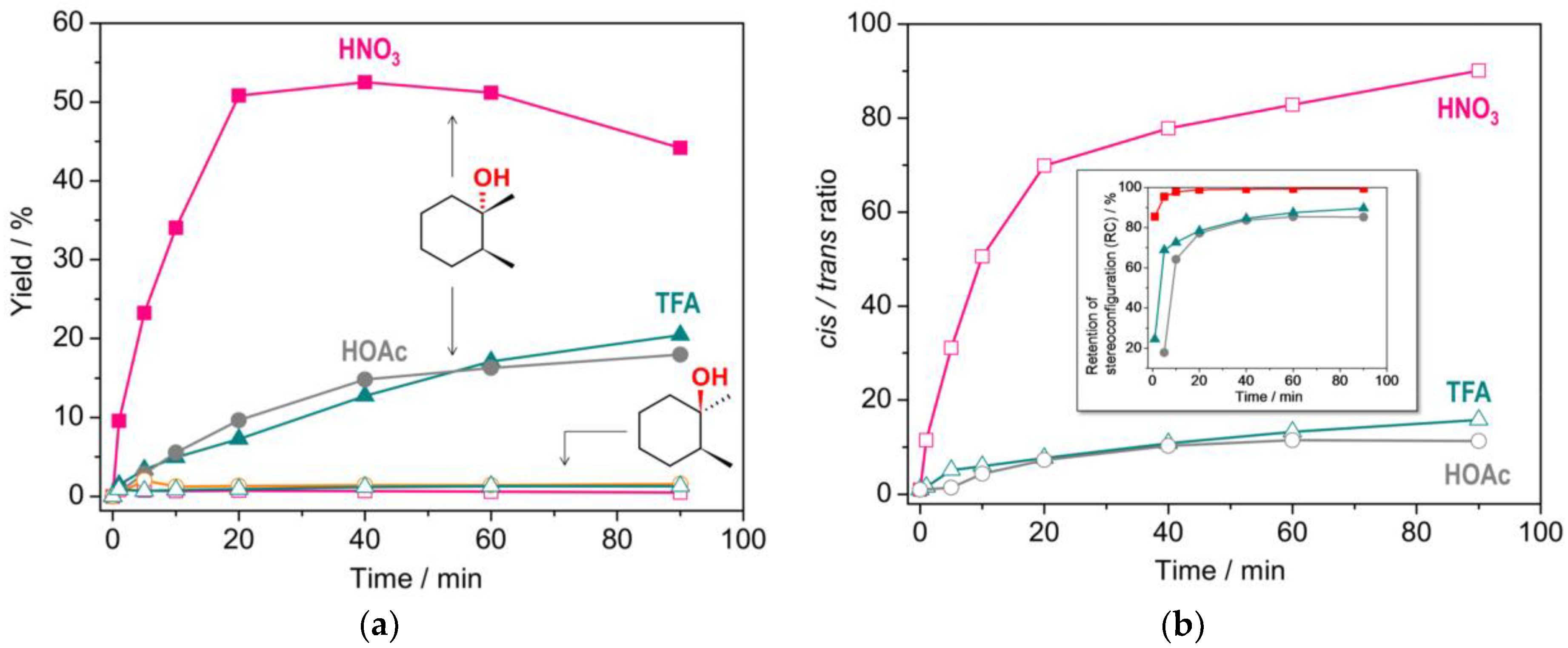
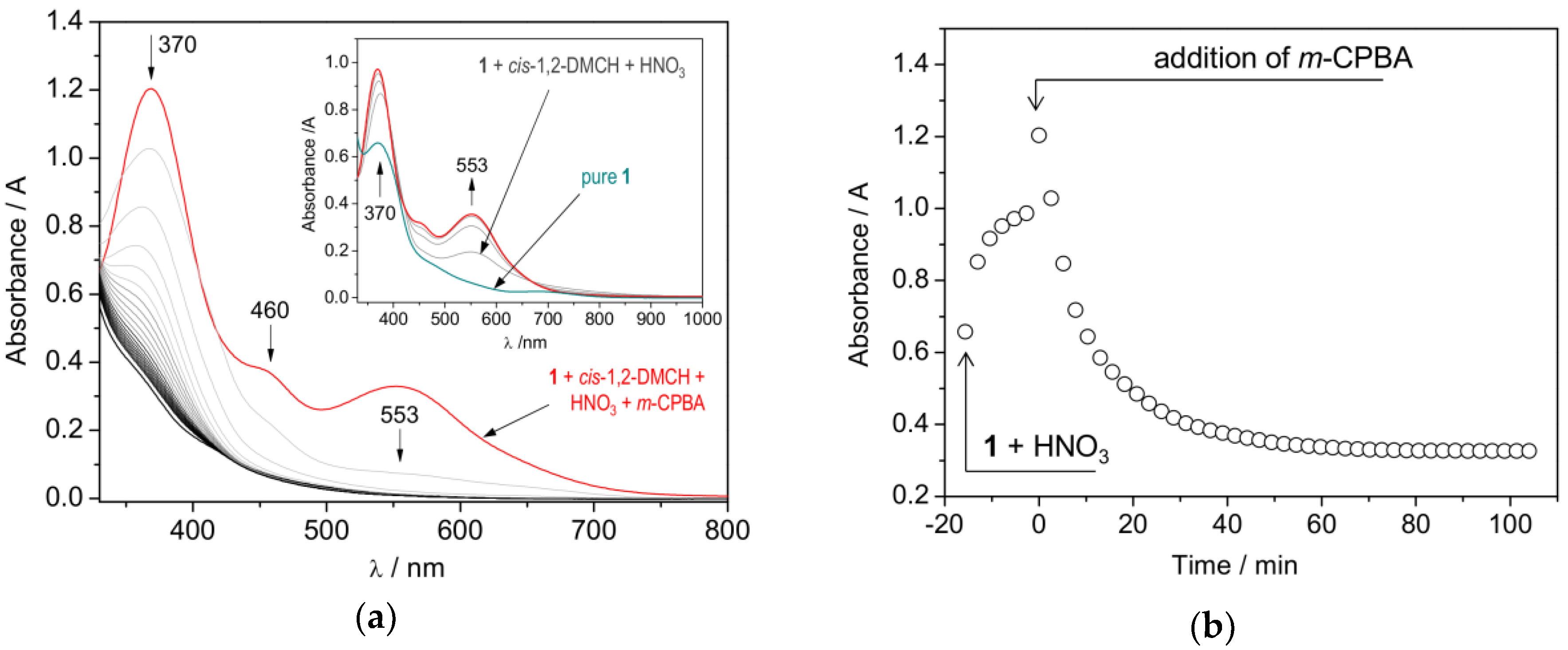
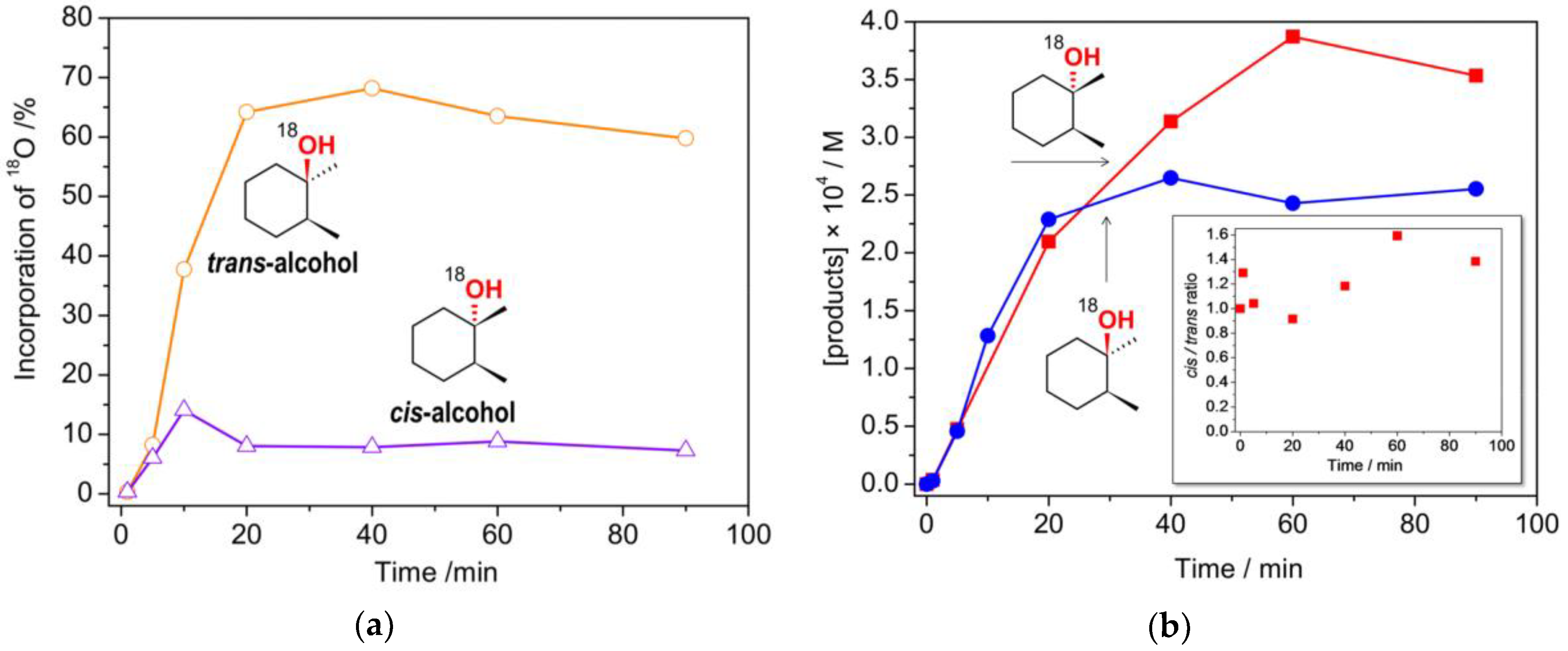
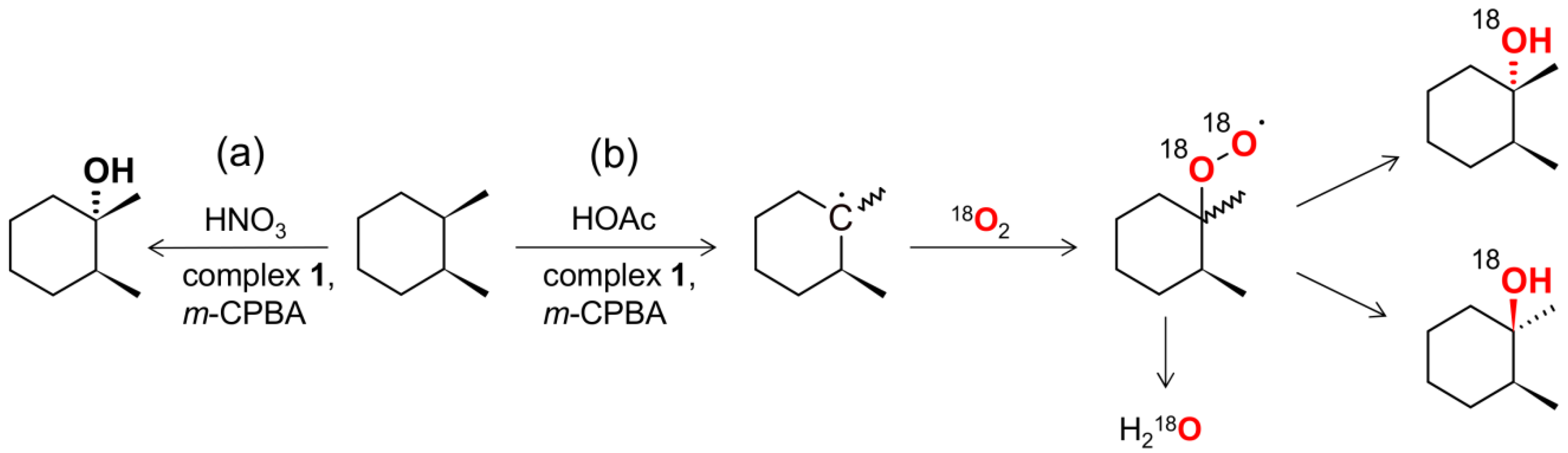
2.3. Catalytic activity
3. Materials and Methods
3.1. Reagents and General Procedures
3.2. Synthesis of [CoZnL3Cl2]·CH3OH (1)
3.3. Single-crystal X-ray diffraction
3.4. Catalytic oxidation of alkanes
3.5. Catalytic Reactions under 18O2 Atmosphere
3.6. Gas Chromatography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gensch, T.; James, M.J.; Dalton, T.; Glorius, F. Increasing Catalyst Efficiency in C–H Activation Catalysis. Angew. Chem. Int. Ed. 2018, 57, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C-H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Ping, L.; Chung, D.S.; Bouffard, J.; Lee, S.-G. Transition metal-catalyzed site- and regio-divergent C–H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [Google Scholar] [CrossRef] [PubMed]
- Tzouras, N.V.; Stamatopoulos, I.K.; Papastavrou, A.T.; Liori, A.A.; Vougioukalakis, G.C. Sustainable metal catalysis in C–H activation. Coord. Chem. Rev. 2017, 343, 25–138. [Google Scholar] [CrossRef]
- Hartwig, J.F.; Larsen, M.A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Central Sci. 2016, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon–Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef]
- Chu, J.C.K.; Rovis, T. Complementary Strategies for Directed C(sp3)-H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, O.; Yu, J.Q. Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.; Kohler, V.; Flitsch, S.L.; Turner, N.J. Cytochromes P450 as useful biocatalysts: addressing the limitations. Chem. Commun. 2011, 47, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Bryliakov, K.P.; Talsi, E.P. Active sites and mechanisms of bioinspired oxidation with H2O2, catalyzed by non-heme Fe and related Mn complexes. Coord. Chem. Rev. 2014, 276, 73–96. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Maji, S.; Chen, P.R.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Pfaff, F.F.; Wang, B.; Nam, W. Status of Reactive Non-Heme Metal-Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 13942–13958. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Cusso, O.; Borrell, M.; Costas, M. Oxidation of alkane and alkene moieties with biologically inspired nonheme iron catalysts and hydrogen peroxide: from free radicals to stereoselective transformations. J. Biol. Inorg. Chem. 2017, 22, 425–452. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Lee, Y.M.; Fukuzumi, S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, S.Y.; Yu, X.Q. C-H functionalization by high-valent Cp*Co(III) catalysis. Chem. Commun. 2017, 53, 3165–3180. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.W.; Mak, A.M.; Sullivan, M.B.; Dixon, D.J.; Seayad, J. Thioamide-Directed Cobalt(III)-Catalyzed Selective Amidation of C(sp3)-H Bonds. Angew. Chem. Int. Ed. 2017, 56, 16550–16554. [Google Scholar] [CrossRef] [PubMed]
- Kommagalla, Y.; Chatani, N. Cobalt(II)-catalyzed C-H functionalization using an N,N’-bidentate directing group. Coord. Chem. Rev. 2017, 350, 117–135. [Google Scholar] [CrossRef]
- Michigami, K.; Mita, T.; Sato, Y. Cobalt-Catalyzed Allylic C(sp3)-H Carboxylation with CO2. J. Am. Chem. Soc. 2017, 139, 6094–6097. [Google Scholar] [CrossRef] [PubMed]
- Barsu, N.; Bolli, S.K.; Sundararaju, B. Cobalt catalyzed carbonylation of unactivated C(sp3)-H bonds. Chem. Sci. 2017, 8, 2431–2435. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B.; Loginov, D.A.; Shul’pina, L.S.; Ikonnikov, N.S.; Idrisov, V.O.; Vinogradov, M.M.; Osipov, S.N.; Nelyubina, Y.V.; Tyubaeva, P.M. Stereoselective Alkane Oxidation with meta-Chloroperoxybenzoic Acid (MCPBA) Catalyzed by Organometallic Cobalt Complexes. Molecules 2016, 21, 1593. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, W.P.; Hu, R.J.; Lu, H.J.; Li, G.G. Cobalt-Catalyzed Cross-Dehydrogenative Coupling Reaction between Unactivated C(sp2)-H and C(sp3)-H Bonds. Org. Lett. 2017, 19, 4676–4679. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Nesterov, D.S. Polynuclear Cobalt Complexes as Catalysts for Light-Driven Water Oxidation: A Review of Recent Advances. Catalysts 2018, 8, 602. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. Stereoselective oxidation of alkanes with m-CPBA as an oxidant and cobalt complex with isoindole-based ligands as catalysts. RSC Adv. 2016, 6, 93756–93767. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Kokozay, V.N.; Pombeiro, A.J.L. Polynuclear Heterometallic Complexes from Metal Powders: The “Direct Synthesis” Approach. Eur. J. Inorg. Chem. 2014, 4496–4517. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Rocha, B.G.M.; Pombeiro, A.J.L.; Shul’pin, G.B. Oxidation of olefins with H2O2 catalysed by salts of group III metals (Ga, In, Sc, Y and La): epoxidation versus hydroperoxidation. Catal. Sci. Technol. 2016, 6, 1343–1356. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Rocha, B.G.M.; Pombeiro, A.J.L.; Shul’pin, G.B. Oxidation of Olefins with Hydrogen Peroxide Catalyzed by Bismuth Salts: A Mechanistic Study. ACS Catal. 2015, 5, 3823–3835. [Google Scholar] [CrossRef]
- Rocha, B.G.M.; Kuznetsov, M.L.; Kozlov, Y.N.; Pombeiro, A.J.L.; Shul’pin, G.B. Simple soluble Bi(III) salts as efficient catalysts for the oxidation of alkanes with H2O2. Catal. Sci. Technol. 2015, 5, 2174–2187. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Teixeira, F.A.; Bokach, N.A.; Pombeiro, A.J.L.; Shul’pin, G.B. Radical decomposition of hydrogen peroxide catalyzed by aqua complexes [M(H2O)n]2+ (M = Be, Zn, Cd). J. Catal. 2014, 313, 135–148. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Pombeiro, A.J.L.; Bokach, N.A.; Shul’pin, G.B. Generation of HO• Radical from Hydrogen Peroxide Catalyzed by Aqua Complexes of the Group III Metals [M(H2O)n]3+ (M = Ga, In, Sc, Y, or La): A Theoretical Study. ACS Catal. 2013, 3, 1195–1208. [Google Scholar] [CrossRef]
- Gamba, I.; Codola, Z.; Lloret-Fillol, J.; Costas, M. Making and breaking of the O-O bond at iron complexes. Coord. Chem. Rev. 2017, 334, 2–24. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kojima, T.; Lee, Y.M.; Nam, W. High-valent metal-oxo complexes generated in catalytic oxidation reactions using water as an oxygen source. Coord. Chem. Rev. 2017, 333, 44–56. [Google Scholar] [CrossRef]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Chen, K.; Que, L. Stereospecific alkane hydroxylation by non-heme iron catalysts: Mechanistic evidence for an Fe-V = O active species. J. Am. Chem. Soc. 2001, 123, 6327–6337. [Google Scholar] [CrossRef] [PubMed]
- Gryca, I.; Czerwinska, K.; Machura, B.; Chrobok, A.; Shul’pina, L.S.; Kuznetsov, M.L.; Nesterov, D.S.; Kozlov, Y.N.; Pombeiro, A.J.L.; Varyan, I.A.; et al. High Catalytic Activity of Vanadium Complexes in Alkane Oxidations with Hydrogen Peroxide: An Effect of 8-Hydroxyquinoline Derivatives as Noninnocent Ligands. Inorg. Chem. 2018, 57, 1824–1839. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L. A hydroperoxo-rebound mechanism of alkane oxidation with hydrogen peroxide catalyzed by binuclear manganese(IV) complex in the presence of an acid with involvement of atmospheric dioxygen. Inorg. Chim. Acta. 2017, 455, 666–676. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Levitsky, M.M.; Yalymov, A.I.; Korlyukov, A.A.; Vologzhanina, A.V.; Kozlov, Y.N.; Shul’pina, L.S.; Nesterov, D.S.; Pombeiro, A.J.L.; Lamaty, F.; et al. A heterometallic (Fe6Na8) cage-like silsesquioxane: synthesis, structure, spin glass behavior and high catalytic activity. RSC Adv. 2016, 6, 48165–48180. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Bilyachenko, A.N.; Nesterov, D.S.; Shul’pina, L.S.; Zubavichus, Y.V.; Pombeiro, A.J.L.; Levitsky, M.M.; Yalymov, A.I.; Shul’pin, G.B. Alkane oxidation with peroxides catalyzed by cage-like copper(II) silsesquioxanes. New J. Chem. 2015, 39, 187–199. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L.; Shul’pin, G.B. Oxidation of hydrocarbons with H2O2/O2 catalyzed by osmium complexes containing p-cymene ligands in acetonitrile. Catal. Sci. Technol. 2014, 4, 3214–3226. [Google Scholar] [CrossRef]
- Vikse, K.L.; Ahmadi, Z.; McIndoe, J.S. The application of electrospray ionization mass spectrometry to homogeneous catalysis. Coord. Chem. Rev. 2014, 279, 96–114. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; Garcia-Espana, E.; Basallote, M.G.; Munck, E.; Que, L.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C-H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef] [PubMed]
- Font, D.; Canta, M.; Milan, M.; Cusso, O.; Ribas, X.; Gebbink, R.J.M.K.; Costas, M. Readily Accessible Bulky Iron Catalysts exhibiting Site Selectivity in the Oxidation of Steroidal Substrates. Angew. Chem. Int. Ed. 2016, 55, 5776–5779. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Kasyanova, K.V.; Makhankova, V.G.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Stereospecific sp3 C-H oxidation with m-CPBA: A CoIII Schiff base complex as pre-catalyst vs. its CoIIICdII heterometallic derivative. Appl. Catal. A. 2018, 560, 171–184. [Google Scholar] [CrossRef]
- Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kuett, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; et al. Acidities of strong neutral Bronsted acids in different media. J. Phys. Org. Chem. 2013, 26, 162–170. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Skone, J.H.; Ning, M.; Wasada-Tsutsui, Y. Toward the accurate calculation of pK(a) values in water and acetonitrile. Bioenerg. 2013, 1827, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, A.M.; Shul’pin, G.B. Pyrazinecarboxylic acid and analogs: Highly efficient co-catalysts in the metal-complex-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2013, 257, 732–754. [Google Scholar] [CrossRef]
- Allard, M.M.; Xavier, F.R.; Heeg, M.J.; Schlegel, H.B.; Verani, C.N. Sequential Phenolate Oxidations in Octahedral Cobalt(III) Complexes with N2O3 Ligands. Eur. J. Inorg. Chem. 2012, 4622–4631. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Catalytic behaviour of a novel Fe(III) Schiff base complex in the mild oxidation of cyclohexane. Catal. Sci. Technol. 2015, 5, 1801–1812. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Kopylovich, M.N.; Pombeiro, A.J.L. Pronounced retention of stereoconfiguration upon sp3 C-H bonds hydroxylation of dimethylcyclohexanes and decahydronaphthalenes with m-CPBA oxidant and a Co-phthalocyanine catalyst. Mol. Catal. 2018, 459, 8–15. [Google Scholar] [CrossRef]
- Ghosh, M.; Pattanayak, S.; Dhar, B.B.; Singh, K.K.; Panda, C.; Sen Gupta, S. Selective C-H Bond Oxidation Catalyzed by the Fe-bTAML Complex: Mechanistic Implications. Inorg. Chem. 2017, 56, 10852–10860. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Chygorin, E.N.; Kokozay, V.N.; Bon, V.V.; Boca, R.; Kozlov, Y.N.; Shul’pina, L.S.; Jezierska, J.; Ozarowski, A.; Pombeiro, A.J.L. Heterometallic CoIII4FeIII2 Schiff Base Complex: Structure, Electron Paramagnetic Resonance, and Alkane Oxidation Catalytic Activity. Inorg. Chem. 2012, 51, 9110–9122. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: a review. J. Mol. Catal. A 2002, 189, 39–66. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599–1606. [Google Scholar] [CrossRef]
- Chatziefthimiou, S.D.; Lazarou, Y.G.; Hadjoudis, E.; Dziembowska, T.; Mavridis, I.M. Keto forms of salicylaldehyde Schiff bases: Structural and theoretical aspects. J. Phys. Chem. B 2006, 110, 23701–23709. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Mechanism of Selective C-H Hydroxylation Mediated by Manganese Aminopyridine Enzyme Models. ACS Catal. 2015, 5, 39–44. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Promoter | [Cat]0, mol%2 | Yield, %3 | Yield, %4 | cis/trans5 | TON6 | 3° : 2°7 |
| 1 | HNO3 | 0.08 | 17 | 61 | 82 | 198 | 38 : 1 |
| 2 | HNO3 | 0.8 | 11 | 40 | 82 | 13 | 38 : 1 |
| 3 | HNO3 | 0.3 | 14 | 50 | 90 | 48 | 38 : 1 |
| 48 | HNO3 | 0.3 | 37 | 37 | 29 | 56 | 37 : 1 |
| 5 | HOTf9 | 0.3 | < 1 | < 1 | > 40 | < 1 | ‒ |
| 6 | TFA10 | 0.3 | 7 | 26 | 16 | 25 | 24 : 1 |
| 7 | HOAc11 | 0.3 | 6 | 24 | 11 | 25 | 19 : 1 |
| 8 | PCA12 | 0.3 | < 1 | < 1 | 1 | < 1 | ‒ |
| 9 | Et3N | 0.3 | 2 | 6 | 1 | 2.2 | ‒ |
| 10 | Py13 | 0.3 | < 1 | 2 | 2 | 5.4 | ‒ |
| 1114 | HNO3 | 0.3 | 14 | 51 | 78 | 59 | 40 : 1 |
| 1215 | HNO3 | 0.3 | 10 | 36 | 59 | 36 | 32 : 1 |
| Entry | Substrate | Yield based on m-CPBA, %2 | cis/trans | 3° : 2° |
|---|---|---|---|---|
| 1 | cis-1,2-DMCH | 50 | 90 | 38 : 1 |
| 2 | cis-1,4-DMCH | 64 | 42 | 22 : 1 |
| 3 | trans-1,2-DMCH | 46 | 623 | 13 : 1 |
| 4 | trans-1,4-DMCH | 49 | 373 | 12 : 1 |
| 5 | Adamantane | 714 | – | 39 : 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterova, O.V.; Kasyanova, K.V.; Buvaylo, E.A.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter. Catalysts 2019, 9, 209. https://doi.org/10.3390/catal9030209
Nesterova OV, Kasyanova KV, Buvaylo EA, Vassilyeva OY, Skelton BW, Nesterov DS, Pombeiro AJL. Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter. Catalysts. 2019; 9(3):209. https://doi.org/10.3390/catal9030209
Chicago/Turabian StyleNesterova, Oksana V., Katerina V. Kasyanova, Elena A. Buvaylo, Olga Yu. Vassilyeva, Brian W. Skelton, Dmytro S. Nesterov, and Armando J.L. Pombeiro. 2019. "Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter" Catalysts 9, no. 3: 209. https://doi.org/10.3390/catal9030209
APA StyleNesterova, O. V., Kasyanova, K. V., Buvaylo, E. A., Vassilyeva, O. Y., Skelton, B. W., Nesterov, D. S., & Pombeiro, A. J. L. (2019). Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter. Catalysts, 9(3), 209. https://doi.org/10.3390/catal9030209