Enhancing Enzymatic Properties of Endoglucanase I Enzyme from Trichoderma Reesei via Swapping from Cellobiohydrolase I Enzyme


Abstract
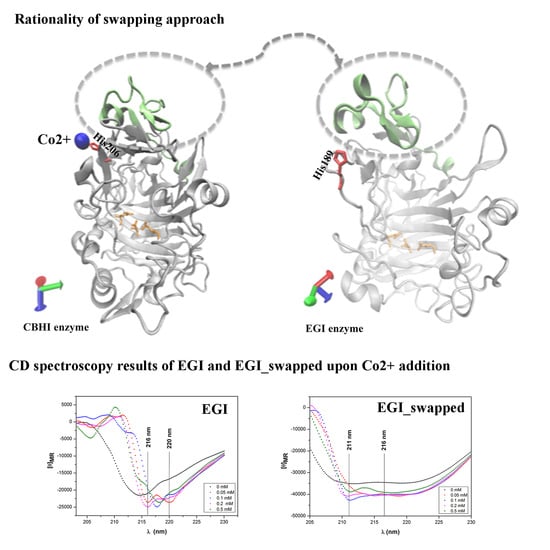
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results & Discussion
2.1. Construction of EGI_Swapped and Cloning Approach
2.2. Enzymatic Activities
2.3. Thermal Stabilities of EGI and EGI_Swapped
2.4. Analysis of Coordinated Co2+ Ions in the Structures of EGI and EGI_Swapped Enzymes
2.5. The Change in Secondary Structures Upon Swapping
3. Material & Methods
3.1. The Construction of EGI_Swapped and Cloning Approach
3.2. P. pastoris Expression & Purification
3.3. Gel Electrophoresis
3.4. Bioinformatic Analysis of EGI and EGI_Swapped
3.5. Enzyme Assays
3.6. Thermal Stability Assay
3.7. Measurement of Co(II) Ion Concentration in EGI and EGI_Swapped Enzymes
3.8. Circular Dichroism (CD) Spectroscopy Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Cellobiohydrolase I | CBHI |
| CD | Circular Dichroism |
| Carboxymethyl cellulase | CMC |
| 3,5-Dinitrosalicylic acid | DNS |
| Endoglucanese I | EGI |
| Endoglucanese I swapped | EGI_swapped |
| PDB | Protein Data Bank |
References
- Lin, L.L.; Yan, R.; Liu, Y.Q.; Jiang, W.J. In-depth investigation of enzymatic hydrolysis of biomass wastes based on three major components: Cellulose, hemicellulose and lignin. Bioresour. Technol. 2010, 101, 8217–8223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Lynd, L.R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems. Biotechnol. Bioeng. 2004, 88, 797–824. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.; Muñoz-Dorado, J.; de la Rubia, T.; Martínez, J. Biodegradation and biological treatments of cellulose, hemicellulose and lignin: An overview. Int. Microbial. 2002, 5, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Eibinger, M.; Sattelkow, J.; Ganner, T.; Plank, H.; Nidetzky, B. Single-molecule study of oxidative enzymatic deconstruction of cellulose. Nat. Commun. 2017, 8, 894. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef]
- Donohoe, B.S.; Resch, M.G. Mechanisms employed by cellulase systems to gain access through the complex architecture of lignocellulosic substrates. Curr. Opin. Chem. Biol. 2015, 29, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Percival Zhang, Y.H.; Himmel, M.E.; Mielenz, J.R. Outlook for cellulase improvement: Screening and selection strategies. Biotechnol. Adv. 2006, 24, 452–481. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Cellulases and biofuels. Curr. Opin. Biotechnol. 2009, 20, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Chanzy, H.; Lamed, R.; Shoham, Y. Cellulose, cellulases and cellulosomes. Curr. Opin. Struct. Biol. 1998, 8, 548–557. [Google Scholar] [CrossRef]
- Bennett, M.; Choe, S.; Eisenberg, D. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Abrahamson, M.; Grubb, A.; Jaskolski, M. Domain swapping in N-truncated human cystation. Chem. J. Mol. Biol. 2004, 341, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Schering, N.; Casale, E.; Caccia, P.; Giordano, P.; Battistini, C. Dimer formation through domain swapping in the crystal structure of the Grb2-SH2-Ac-pYVNV complex. Biochemistry 2000, 39, 13376–13382. [Google Scholar] [CrossRef]
- Barbosa, J.; Sivarman, J.; Li, Y.; Larocque, R.; Matte, A. Mechanism of action and NAD+ binding mode revealed by the crystal structure of L-histidinol dehydrogenase. Proc. Natl. Acad. Sci. USA 2002, 99, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Knaus, K.; Morillas, M.; Swietnicki, W.; Malone, M.; Surewicz, WK.; Yee, V.C. Crystal structure of the human prion protein reveals a mechanism for oligomrerization. Nat. Struct. Biol. 2001, 8, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Yenenler, A.; Sezerman, O.U. Design and characterizations of two novel cellulases through single-gene shuffling of Cel12A (EG3) gene from Trichoderma reseei. Protein Eng. Des. Sel. 2016, 29, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Timucin, E.; Sezerman, O.U. The conserved lid tryptophan, W211, potentiates thermostability and thermoactivity in bacterial thermoalkalophilic lipases. PLoS ONE 2013, 8, e85186. [Google Scholar] [CrossRef]
- Jaenicke, R.; Bohm, G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998, 8, 738–748. [Google Scholar] [CrossRef]
- Tanaka, S.; Igarashi, S.; Ferri, S.; Sode, K. Increasing stability of water-soluble PQQ glucose dehydrogenase by increasing hydrophobic interaction at dimeric interface. BMC Biochem. 2005, 6, 1. [Google Scholar] [CrossRef]
- Kirino, H.; Aoki, M.; Aoshima, M.; Hayashi, Y.; Ohba, M. Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur. J. Biochem. 1994, 220, 275–281. [Google Scholar] [CrossRef]
- Keeling, A.A. Understanding Humic Substances: Advanced Methods, Properties and Applications; Wiley: Cambridge, UK, 2000. [Google Scholar]
- Hakansson, U.; Fagerstam, L.G.; Pettersson, G.; Andersson, L. 1,4-β-glucan glucanohydrolase from the cellulolytic fungus Trichoderma viride QM 9414. Biochem. J. 1979, 179, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, S.; Schweickart, V.; Ladner, M.; Gelfand, D.; Kwok, S.; Myambo, K.; Innis, M. Molecular cloning of exo-cellobiohydrolase from Trichoderma reesei strain L27. Nat. Biotechnol. 1983, 1, 691–696. [Google Scholar] [CrossRef]
- Stahlberg, J.; Henriksson, H.; Divne, C.; Isaksson, R.; Pettersson, G.; Johansson, G.; Jones, T.A. Structural basis for enantiomer binding and separation of a common beta-blocker: Crystal structure of cellobiohydrolase Cel7A with bound (S)-propranolol at 1.9 A resolution. J. Mol. Biol. 2001, 305, 79–93. [Google Scholar] [CrossRef]
- Cassia Pereira, J.; Giese, E.C.; Souza Moretti, M.M.; Santos Gomes, A.C.; Perrone, O.M.; Boscolo, M.; Silva, R.; Gomes, E.; Bocchini Martins, D.A. Effect of Metal Ions, Chemical Agents and Organic Compounds on Lignocellulolytic Enzymes Activities. In Enzyme Inhibitors and Activators; Senturk, M., Ed.; IntechOpen: London, UK, 2017. [Google Scholar]
- Glusker, J.P. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 1991, 42, 1–76. [Google Scholar]
- Sigel, R.K.; Pyle, A.M. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A. YETI: An interactive molecular mechanics program for small-molecule protein complexes. J. Comput. Chem. 1986, 9, 269–280. [Google Scholar] [CrossRef]
- Vedani, A.; Huhta, D.W. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990, 112, 4759–4767. [Google Scholar] [CrossRef]
- Bodenheimer, A.M.; Meilleur, F. Crystal structures of wild-type Trichoderma reesei Cel7A catalytic domain in open and closed states. FEBS Lett. 2016, 590, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Bodenheimer, A.M.; Cuneo, M.J.; Swartz, P.D.; Myles, D.A.; Meilleur, F. Crystal Structure of Wild Type Hypocrea Jecorina Cel7a in a Monoclinic Crystal Form. Available online: https://www.rcsb.org/structure/4p1h (accessed on 29 January 2019).
- Divne, C.; Stahlberg, J.; Reinikainen, T.; Ruohonen, L.; Pettersson, G.; Knowles, J.K.; Teeri, T.T.; Jones, T.A. The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 1994, 265, 524–528. [Google Scholar] [CrossRef]
- Stahlberg, J.; Divne, C.; Koivula, A.; Piens, K.; Claeyssens, M.; Teeri, T.T.; Jones, T.A. Activity studies and crystal structures of catalytically deficient mutants of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 1996, 264, 337–349. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Zou, J.Y.; Divne, C.; Davies, G.J.; Sinning, I.; Stahlberg, J.; Reinikainen, T.; Srisodsuk, M.; Teeri, T.T.; Jones, T.A. The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 A resolution, and a comparison with related enzymes. J. Mol. Biol. 1997, 272, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Rouvinen, J.; Bergfors, T.; Teeri, T.; Knowles, J.K.; Jones, T.A. Three-dimensional structure of Pcellobiohydrolase II from Trichoderma reesei. Science 1990, 249, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2007, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Letchworth, G.J. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques 2004, 36, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Akcapinar, G.B.; Gul, O.; Sezerman, U. Effect of codon optimization on the expression of Trichoderma reesei endoglucanase 1 in Pichia pastoris. Biotechnol. Prog. 2011, 27, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gusakov, A.V.; Kondratyeva, E.G.; Sinitsyn, A.P. Comparison of two methods for assaying reducing sugars in the determination of carbohydrase activities. Int. J. Anal. Chem. 2011, 2011, 283658. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K.; Hazlewood, G.P. Enzymology and Other Characteristics of Cellulases and Xylanases; CABI Publishing: Oxford, UK, 2001; pp. 11–60. [Google Scholar]
- Chen, X.; Li, W.; Ji, P.; Zhao, Y.; Hua, C.; Han, C. Engineering the conserved and noncatalytic residues of a thermostable beta-1,4-endoglucanase to improve specific activity and thermostability. Sci. Rep. 2018, 8, 2954. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Biely, P.; Poutanen, K. Interlaboratory testing of methods for assay of xylanase activity. J. Biotechnol. 1992, 23, 257–270. [Google Scholar] [CrossRef]
- Kurt, H.; Yuce, M.; Hussain, B.; Budak, H. Dual-excitation upconverting nanoparticle and quantum dot aptasensor for multiplexed food pathogen detection. Biosens. Bioelectron. 2016, 81, 280–286. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yenenler, A.; Kurt, H.; Sezerman, O.U. Enhancing Enzymatic Properties of Endoglucanase I Enzyme from Trichoderma Reesei via Swapping from Cellobiohydrolase I Enzyme. Catalysts 2019, 9, 130. https://doi.org/10.3390/catal9020130
Yenenler A, Kurt H, Sezerman OU. Enhancing Enzymatic Properties of Endoglucanase I Enzyme from Trichoderma Reesei via Swapping from Cellobiohydrolase I Enzyme. Catalysts. 2019; 9(2):130. https://doi.org/10.3390/catal9020130
Chicago/Turabian StyleYenenler, Aslı, Hasan Kurt, and Osman Uğur Sezerman. 2019. "Enhancing Enzymatic Properties of Endoglucanase I Enzyme from Trichoderma Reesei via Swapping from Cellobiohydrolase I Enzyme" Catalysts 9, no. 2: 130. https://doi.org/10.3390/catal9020130
APA StyleYenenler, A., Kurt, H., & Sezerman, O. U. (2019). Enhancing Enzymatic Properties of Endoglucanase I Enzyme from Trichoderma Reesei via Swapping from Cellobiohydrolase I Enzyme. Catalysts, 9(2), 130. https://doi.org/10.3390/catal9020130