Abstract
Utilizing plant-based materials as a biofuel source is an increasingly popular attempt to redesign the global energy cycle. This endeavour underlines the potential of cellulase enzymes for green energy production and requires the structural and functional engineering of natural enzymes to enhance their utilization. In this work, we aimed to engineer enzymatic and functional properties of Endoglucanase I (EGI) by swapping the Ala43-Gly83 region of Cellobiohydrolase I (CBHI) from Trichoderma reesei. Herein, we report the enhanced enzymatic activity and improved thermal stability of the engineered enzyme, called EGI_swapped, compared to EGI. The difference in the enzymatic activity profile of EGI_swapped and the EGI enzymes became more pronounced upon increasing metal-ion concentrations in the reaction media. Notably, the engineered enzyme retained a considerable level of enzymatic activity after thermal incubation for 90 min at 70 °C while EGI completely lost its enzymatic activity. Circular Dichroism spectroscopy studies revealed distinctive conformational and thermal susceptibility differences between EGI_swapped and EGI enzymes, confirming the improved structural integrity of the swapped enzyme. This study highlights the importance of swapping the metal-ion coordination region in the engineering of EGI enzyme for enhanced structural and thermal stability.
1. Introduction
Lignocellulose is considered the most abundant and sustainable energy resource in the world [1,2]. Due to the growing demand on green alternative energy sources, a substantial global effort is being exerted to redesign the global carbon cycle [3] in an effort to provide sustainabile fuels from plant-based cellulosic materials [4]. Plant-based cellulosic materials as a biofuel resource offer significant advantages over fossil fuel sources, e.g., decreasing global emission, providing a long-term rural development [5,6], the particular challenges still have to be surmounted in the deconstruction processes of cellulose to cellulosic biomass. The naturally endowed complexity of cellulose requires the synergistic action of cellulase enzymes for the effective breaking down of cellulosic biomass [7,8]. Apart from efforts in different pre-treatment techniques for a more efficient breaking down of cellulose, researchers focused on the development of new cellulase enzymes with improved efficiency via protein engineering [9,10].
In protein engineering, the combinations of experimental and computational methods are frequently employed to study the structure-function relationships of enzymes and to reveal the impacts of structural variations on their stability. Domain swapping is one of the prominent tools in the protein engineering arsenal that aims to provide the alterations on the scaffold of proteins for attaining the improvements in existing catalytic and enzymatic activities [11]. So far, numerous studies were reported on domain swapping (DS), ranging from cysteine proteinase inhibitors [12], exploring SH2 and SH3 domains [13], L-histidonol dehydrogenase proteins [14], and prion proteins [15].
Naturally occurring cellulase enzymes usually underperform in biotechnological and industrial applications, and hence this limitation intrigues researchers to develop new cellulase enzymes that can efficiently perform under less restricted conditions [16]. Understanding the molecular basis of how enzymes maintain their stability at harsh conditions is a recurring topic biochemistry [17]. Thermostability could be affected by numerous factors; e.g., hydrogen bond networks, hydrophobic and packing interactions, and charge clusters [18]. The presence of metal ions could directly alter thermostability [18,19,20] via the enhancement of hydrogen networking and packing interactions with the aliphatic residues’ tendency to interact with metal ions via unpaired electrons [21].
The aim of this study is to create a suitable environment for metal-ion coordination in EGI enzyme via swapping a region from CBHI enzyme, being isolated from Trichoderma reseei [22,23]. The findings indicate that swapping the metal-ion coordination region from the CBHI enhanced structural integrity and resulted in a novel endoglucanase enzyme, EGI_swapped, which displayed unique structural and functional properties and is a promising catalyst candidate for industrial applications. To the best of our knowledge, the work presented here is the first study in terms of engineering the structural and functional properties of an EGI enzyme from Trichoderma reesei via swapping in the literature.
2. Results & Discussion
2.1. Construction of EGI_Swapped and Cloning Approach
In this study, we aim to alter the existing structural features of EGI enzyme via domain swapping from CBHI enzyme. These enzymes are both isolated from Trichoderma reesei by sharing the certain level of structural similarity but differentiated in the mode of action (Figure S1). First, we perform the structural alignment between EGI and CBHI (PDB id: 1EG1 and 1DY4) to find appropriate regions for domain swapping. We find that choosing the Ala43-Gly83 region (CBHI numbering) is promising due to its closeness to the Co2+ metal ion in the crystal structure of CBHI, which does not exist in the crystal structure of EGI [24] (see Figure S2). Among many metal ions, Co2+ ion is reported as a cellulase activator [25].
In the crystal structure of CBHI, two Co2+ ions are reported as one is linked to His206 and Glu239, and another one is linked to Glu295 and Glu325. We just focused on a Co2+ ion linked to His206 and Glu239 [24], based on the structural alignment results. Around this particular Co2+ ion, there is a network of negatively charged and polar residues [24], e.g., Asp63, Asn64, and Glu65, which can provide support for the coordination of Co2+. Before deciding on a swapped region, we measure the distances of the OD1 atom of Asp63, ND2 atom of Asn64 and OE2 atom of Glu65 to Co2+ ion, in CBHI, as ~5.6 Å, ~11 Å, and ~9 Å, respectively. Even these distances are out of range for minimum direct metal-ion binding to residues (Asp/Glu) [26], yet these residues are in the range of coordination as they are close enough to create a “high hydrophobicity” contrast [26,27]. Mainly, metal ions are classified into three according to their polarizability; (i) hard, (ii) soft, and (iii) borderline. Hard means a “low-polarizability” atom or ion that is deformed only by difficulty, and soft means a “high-polarizability” atom or ion that is easily deformed [27,28]. The Co2+ ion is reported as borderline based on its polarizability. Due to the nature of not being easily deformed, these particular distances would be enough to keep Co2+ ion in true geometry with His residue [28,29].
Specifically, a fixed aromatic residue or negatively charged one provides an appropriate environment for effective metal ion binding by creating new H-bond networking or contributing to the existing one in advance [21]. Hence, swapping of Ala43-Gly83 region of CBHI to EGI has satisfied these constraints by covering the residues of Asp63 to Glu65, close to fixed Histidine in EGI’s location in order to create a favorable neighbour pocket for Co2+ ion coordination. With Ala to Glu replacement 64th position (EGI numbering) upon swapping, this favorable environment has been also supported in EGI_swapped for Co2+ ion coordination, in terms of donating more electrons and thus creating an advanced H-bond network, with respect to its native counterpart (see Figure S3). In Figure 1, the domain swapping is described by indicating Co-linked His and Glu residues.
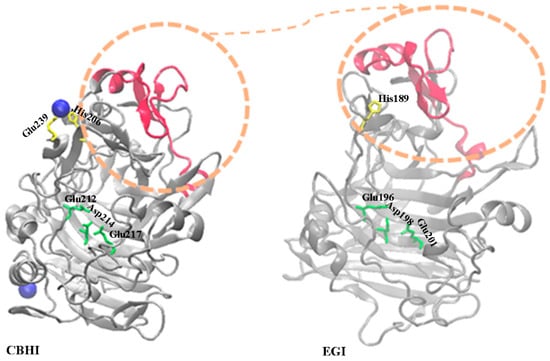
Figure 1.
The rationality of the swapping approach. The crystal structures of CBHI (PDB id: 1DY4) and EGI (PDB id: 1EG1) are drawn by colouring the swapped-domain in red. The catalytic residues of CBHI, Glu212, Asp214 and Glu217, and EGI, Glu196, Asp198 and Glu201, are depicted in ‘licorice’ format and coloured as ‘green’. The His206 and Glu239 residues directly linked to Co2+ in CBHI’s structure is drawn in ‘licorice’ format and colored as ‘yellow’. To perform distance measurement and visualization of the structure, the Visual molecular dynamic is used [34].
Stahlberg et al. reported that divalent metal ions, as Ni2+, Co2+, Ca2+, Mn2+, and Mg2+, seem to be required by crystallization [24]. There are several CBHI structures containing the different ions, e.g., Sm3+ and Gd3+, than divalent ones [30,31]. Before deciding on the swapped region for Co2+ coordination, the crystal structures displaying more than 95% sequence similarity and also the high level of structural similarity within our reference structure (CBHI enzyme) are carefully examined. Among 26 reported crystal structures, only 7 ones contain different metal ions than Co2+, e.g., Sm3+ in PDB id: 4P1H, Gd3+ in PDB id: 5TC9, and Ca2+ in PDB id: 1CEL, 2CEL, 3CEL and 4CEL [30,31,32,33]. To make a true decision for swapping, we also perform the structural alignments between EGI (PDB id: 1EG1) with them. These alignment results suggest that the ion coordination regions of CBHI’s structures are not well aligned in EGI’s structure with the existence of structural breaks, and thus they are not suitable to perform swapping (Figure S4). Actually, Ca2+, Gd3+, and Sm3+ ions are coordinated by CBHI in the region, covering Glu295 and Glu325, as similar to another Co2+ ion in our reference CBHI enzyme. This particular region is too far from our swapped-region, Ala43-Gly83, as displayed in Figure S4.
It is stated in the literature that tunnel forming loops existed in the structure of CBHI is not observed in EGI [24,35] due to the loss of particular genes via horizontal gene transfer during the evolutionary process [36]. Even a certain level of structural similarity has been preserved between the two, this particular gene loss results in the differentiation in their mode of action as depicted in Figure S1. Instead of swapping just a few residues in close contact, the swapping of the whole region eliminates the possibility of generation of the low- or non-functional enzyme due to misfolding or unfolding (Figure S2). Further, this swapped region is rich, for Gly content that provides additional conformational flexibility in the swapped enzyme to adopt Co2+ coordination (Figure S3).
Before transforming the swapped gene into P.pastoris expression vector, the positive clones were double-checked by EcoR I single digestion (Figure S5a,b). As displayed in Figure S3, there is a high level of similarity and identity between EGI_swapped and EGI enzymes, 94.3% and 96.4% respectively. In parallel to prediction (Table S1), the molecular weight of the swapped enzyme is reported around ~50 kDa, including a 6xHis tag in the expression vector used for the purification step, see Figure S6.
2.2. Enzymatic Activities
First, we find the optimal pH-buffer combinations for both EGI_swapped and EGI enzymes. The optimal pH-buffer combination is found by scanning pH 4–5 and pH 6–7 ranges in 50 mM sodium acetate and potassium phosphate buffers toward 0.5% w/v CMC at 50 °C for 3 h, respectively. For EGI_swapped and EGI enzymes, the highest enzymatic activity is recorded at 50 mM sodium acetate buffer pH 4.7 as in line with the reported PI value (Table S1). Then, the rest of all enzyme activity and thermal stability tests are performed in 50 mM sodium acetate pH 4.7 buffer. Just for the circular dichroism (CD) spectroscopy measurement, the concentration of buffer is decreased to 20 mM at pH 4.7.
As depicted in Figure 1, the swapped region is not close to the catalytic triad. This particular distance from the swapped region to the catalytic triad creates an expectation that alterations in enzyme activity are most probably due to a change in the thermal stability of EGI_swapped. Among 1 h results, the first notable difference in enzymatic activities is captured at the highest temperature, 50 °C (see Figure S9). Upon extending the reaction time to 3 h, the notable difference is again recorded at 50 °C (Figure 2). Indeed, the difference in activities of EGI and EGI_swapped become more visible, around ~20% difference, at 6 h (Figure 2). These results point out the alterations in thermal stability of EGI_swapped with respect to the EGI enzyme.
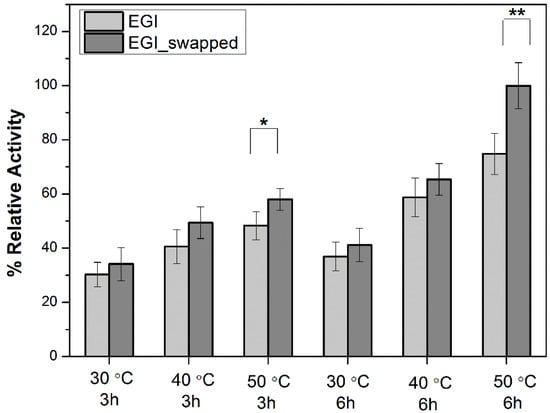
Figure 2.
The enzyme activity profiles of EGI and EGI_swapped enzymes toward CMC for 3 h and 6 h at 30–50 °C (/10 °C). Data are express as mean ± SD, n = 3. p-values between 0.1 and 0.05, and p-value smaller than 0.05 are indicated with ‘*’ and ‘**’ on plots, respectively.
Moreover, we aim to investigate either there is any impact of Co2+ addition on enzymatic activities of EGI and EGI_swapped, or not. To reveal this, we performed activity tests on enzymes toward 0.5% w/v CMC by following two paths; (i) pre-incubating enzymes with Co2+ (Figure 3) and (ii) pre-incubating CMC with Co2+ (Figure S7) prior to the hydrolysis reaction. We report that the relative activities of EGI are 48% without any Co2+, and 47% for 0.05 mM and 0.1 mM Co2+ ion additions. As Co2+ ion addition increases to 0.2 mM and 0.5 mM, its enzymatic activity dramatically decreases to 28% and 24%, respectively. This particular trend could be explained as the accumulated Co2+ ions randomly bind to the tertiary structure of EGI due to the lack of a favorable environment for ion binding. This result is in line with the literature search that there is no reported crystal structure in PDB, displaying more than a 70% sequence similarity with EGI (PDB id: 1EG1) to be considered as a homolog, with the presence of Co2+ or any relevant metal ions [35].
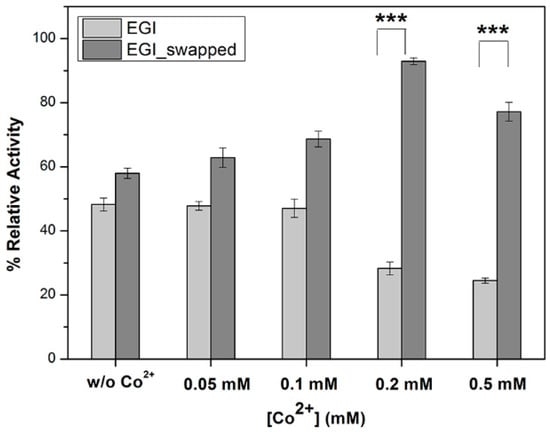
Figure 3.
The effects of Co2+-ion pre-incubation with enzymes prior to hydrolysis reaction on their enzymatic activity profiles. Through enzyme assays toward 0.5% w/v CMC, the impacts of Co2+ ion pre-incubation with enzymes were investigated for different Co2+ ion concentrations, 0 mM, 0.05 mM, 0.1 mM, 0.2 mM and 0.5 mM, in final. The highest data among all activity tests, EGI_swapped at 50 °C for 6 h, is taken as 100% to calculate the rest. Data are express as mean ± SD, n = 3. To indicate the significant differences in enzymatic activities of EGI and EGI_swapped enzymes, p-value smaller than 0.001 are indicated with ‘***’ on plots.
For EGI_swapped, we report that the relative activities of EGI_swapped are 58% without any Co2+, 63% with 0.05 mM Co2+ and 69% with 0.1 mM Co2+ (Figure 3). The most notable result is recorded as 92% with 0.2 mM and 77% with 0.5 mM Co2+ ion concentrations (Figure 3). Herein, we conclude that Co2+ ion is coordinated better in EGI_swapped with 0.2 mM concentration. Due to the addition of more Co2+ ion, its enzymatic activity decreases to 77% but still it is better than the enzymatic activity of EGI_swapped with no Co2+ ion addition, at 57%. When we compare the enzymatic activities of EGI_swapped with respect to native, Co2+ ion coordination is significantly different in EGI_swapped with a p-value smaller than 0.001 for 0.2 mM and 0.5 mM Co2+ ion concentrations, see Figure 3.
As previously stated, two different paths [(i) and (ii)] are followed to investigate whether Co2+ ions inhibit enzymatic activity by binding to alternative sites in their tertiary structure or if they occupy any side of the CMC, long polymeric chain, to prevent the proper attack of enzymes to CMC. Based on the results of path (ii) in Figure S7, we conclude that Co2+ ion incubating with CMC does not alter the results.
2.3. Thermal Stabilities of EGI and EGI_Swapped
Thermal stabilities of EGI and EGI_swapped enzymes are evaluated by measuring their remaining activities toward 0.5% w/v CMC at 30 °C for 1 h after subjecting them to thermal incubation at 30–70 °C/10 °C for 30 min, 60 min, and 90 min. An enzymatic activity without any thermal incubation 30°C for 1 h is taken as a reference, 100%, to calculate the rest. In parallel to the enzymatic activity profile in Figure 2, the notable differences in enzymatic activities are captured upon the increase in incubation temperature and extension in incubation time.
As displayed in Figure 4, the first notable difference among 30 min incubation results is reported at 70 °C, e.g., EGI_swapped displays ~1.5-fold better activity than EGI. Among 60 min incubation results, the notable differences are at 60 °C and 70 °C, e.g., EGI_swapped displays ~1.6-fold and ~2.8-fold higher activity than EGI at 60 °C and 70 °C, respectively. Upon extending the incubation time to 90 min, ~1.8-fold at 50 °C and ~2-fold at 60 °C higher enzymatic activities are reported for EGI_swapped with respect to EGI. Among all data sets, the most striking thermal stability result is reported for 90 min thermal incubation at 70 °C. Notably, swapped enzyme still shows enzymatic activity after thermal incubation for 90 min at 70 °C, e.g., 14% relative activity, while the enzymatic activity of EGI is completely lost. These thermal stability assay results together with the enzyme activity assay results performed with and without Co2+ ions (Figure 2 and Figure 3) prove to us that EGI_swapped is a more promising catalyst than its native counterpart by meeting the demands of the harsh conditions of biotechnological application.
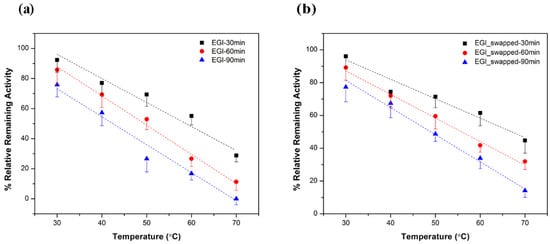
Figure 4.
The thermal stability results; (a) EGI and (b) EGI_swapped enzymes toward CMC. The remaining relative activities of enzymes toward 0.5% w/v CMC were measured after incubating enzymes at different temperatures (30–70 °C/10 °C) for 30, 60 and 90 minutes. For each EGI and EGI_swapped enzymes, the enzymatic activities at 30 °C without any thermal incubation were set to 100% in order to calculate the rest. Data are express as mean ± SD, n = 3.
The thermal stability of the enzyme is controlled by numerous factors; e.g., hydrogen bond networks, hydrophobic and packing interactions, and charge clusters [18]. It is stated for metal ions that they can provide a geometrically defined scaffold, evolving into the reorganization of the local interactions [26]. In some cases, this defined scaffold can modulate the active site of enzymes through long-range interactions to some extent that are dependent on the position of metal in the protein, its interaction with solvent, and the accessibility of counter-ions, etc. [26] Based on the findings in Figure 4, we could conclude that swapping the Ala43-Gly83 region of CBHI to EGI has resulted in an alteration to the enzyme’s structure; e.g., the enhancement of hydrogen networking and packing interactions, [18,19,20]. Hence, we evidently end up with a novel endoglucanase enzyme and displayed altered thermal stability with respect to its native counterpart. The existence of local reorganizations are expected in proteins’ scaffold upon the metal ion binding [26]. Consequently, observing the significant differences in thermal stabilities of EGI_swapped and EGI would imply the existence of these local reorganizations in the scaffold of EGI_swapped.
2.4. Analysis of Coordinated Co2+ Ions in the Structures of EGI and EGI_Swapped Enzymes
As already demonstrated, EGI_swapped displays altered enzymatic activity and thermal stability with respect to its native counterpart, see Figure 2, Figure 3 and Figure 4. These findings strongly suggest that the favorable environment for Co2+ ion coordination is created in EGI_swapped but still additional findings are required for further demonstrations. We perform ICP-OES measurements with EGI_swapped and EGI enzymes. Immediately after 20 min heat denaturation of EGI_swapped and EGI_swapped at 95 °C, the amount of released Co2+ ion is measured as 2.2 µM and 8 μM in EGI_swapped and EGI enzymes, respectively. Herein, ~3.6-fold stronger Co2+ ion coordination in EGI_swapped with respect to EGI could be accounted as another strong implication for the existence of Co2+ ion coordination in the EGI_swapped enzyme. In ICP-OES analysis, we detected a minute amount of Co2+ ions in expression buffer and thus free Co2+ ions alternatively bind to EGI’s structure and they are released upon heating at 95 °C.
Stahlberg et al. suggested that divalent metal ions are required for the crystallization of the CBHI (PDB id: 1DY4) enzyme [24]. This suggestion seems to be suffered since there are several CBHI structures in literature that are crystallized with metal ion different than divalent ones; e.g., 4P1H and 5TC9 with Sm3+ and Gd3+ [30,31,32,33] (see Figure S4). It has been already suggested in the literature that there is a selective binding mode of metal ions to proteins, and it is controlled by several factors, e.g., metal ion radii, number of unpaired electrons, coordination number and preferred coordination geometry of metal ion, the availability of His, and Glu and Asp residues in proteins’ structure, etc. [27]. With the well-combination of all these factors, proteins bind to one appropriate metal ion much more tightly than others [26]. To investigate whether EGI_swapped displays any specific binding to other divalent atoms (Ca2+, Mg2+, and Zn2+) or not, we measured the amount of released Ca2+, Mg2+ and Zn2+ ions upon the thermal denaturation of EGI_swapped and EGI enzymes at 95°C for 20 min through ICP-OES method. We report the ratio of released ions from EGI_swapped to EGI as ~1.0 for Ca2+, ~1.1 for both Mg2+ and Zn2+. Hence, these particular differences in the ratio of released Co2+ and other divalent atoms from EGI_swapped to EGI enzymes, ~3.6 and ~1.0/~1.1, could be accounted as strong evidence for specificity toward Co2+, but not Ca2+, Mg2+, and Zn2+. For Sm3+ and Gd3+ ions, there is no proper detector in our ICP-OES instrument. That is why we could not report the amount of released Sm3+ and Gd3+ ions from EGI_swapped and EGI enzymes.
There is no set of rules for metal ions’ binding to proteins as similar to their binding to inorganic molecules, several constrains should be satisfied for metal ion binding to protein molecules. For instance, the existence of true geometry in the protein structure, the presence of His and Asp/Glu residues in proper geometry, and the overcoming size and shape limitations are several constraints that originate from the proteins’ structures [26,27]. The polarizability, radii of metal ion, and the number of unpaired electrons are other constraints’ coming from metal ions’ site [26,27]. Upon the meeting of these constraints both from proteins’ and metal ions’ sites, the specific metal binding to protein has occurred. In the light of enzyme assay, thermal stability and ICP-OES measurement results, we draw a conclusion that swapping of Ala43-Gly83 region of CBHI to EGI enzyme enabled EGI_swapped enzyme to provide the several constrains mentioned above that are actually lacking in EGI enzyme (see Figure 2 and Figure 3).
2.5. The Change in Secondary Structures Upon Swapping
The altered enzymatic activity and thermal stability results in the swapped enzyme have already pointed out that domain swapping should transform the secondary structure of the EGI significantly. Now we use Circular Dichroism (CD) spectroscopy approach in order to evaluate the possible structural variations of EGI_swapped and EGI enzymes. The temperature sweep of CD spectra between 25 °C and 70 °C reveals distinct conformational and thermal susceptibility between the secondary structure of EGI and EGI_swapped enzymes, see Figure 5a,b. EGI enzyme shows antiparallel β-sheet features in the spectra region of 210–240 nm with a distinctive peak ca. 215 nm. In contrast, EGI_swapped demonstrates the superposition of an antiparallel β-sheet at ca. 215 nm and α-helix like features at ca. 225 nm [37]. Peculiarly, temperature dependent CD study reveals that the structure of EGI enzyme significantly changes with increasing temperature. Although the maxima of ca. 215 is increased slightly, the structural changes exhibit a significant broadening at ca. 230 nm. These structural changes correlate with the temperature dependent activity assays of EGI enzyme. Increasing temperature induces critical alterations in the secondary structure of EGI enzyme, and prolonged temperature exposures could render the enzyme completely inert as shown in Figure 4a.
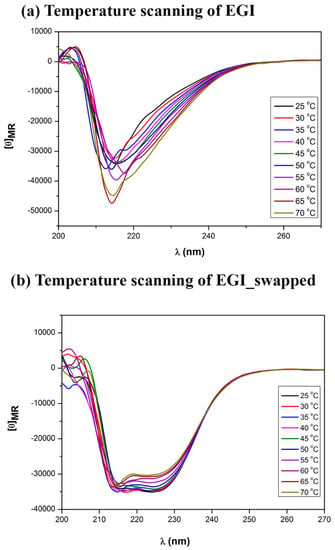
Figure 5.
Temperature scanning of EGI (a) and EGI_swapped enzymes (b). Through Circular Dichroism, the change in the structure of endoglucanase I enzyme upon domain swapping has been displayed. The enzyme samples were prepared in 20 mM sodium acetate buffer (pH 4.7) that was corrected for the backgrounds. The changes in secondary structures of native and swapped enzymes upon an increase in temperature (25–70 °C/5 °C) were captured. The secondary structure changes of native and domain-swapped enzymes are carefully tracked within 210–220 nm and 215–230 nm, respectively. All results are displayed in mean residue ellipticity ([θ]).
Unlike to EGI, the EGI_swapped enzyme exhibits subtle changes in temperature sweep. Antiparallel β-sheet dominant CD feature ca. 215 nm does not show changes in increased temperature. Meanwhile, α-helix associated CD absorption at ca. 225 remains highly stable until the temperature of 50 °C then is slightly reduced but remains prominent as it correlates with the temperature dependent activity assays of EGI_swapped, see Figure 4b. Here, we deduce that the temperature-dependent activity variation between EGI and EGI_swapped enzymes originates from the structural changes and confirms that domain swapping has altered the conformation of the enzyme, unequivocally leading to a novel endoglucanase enzyme, EGI_swapped.
Furthermore, we also investigate the changes in secondary structures of EGI and EGI_swapped enzymes upon thermal denaturation, for the duration of an hour at 95 °C. Conforming to the thermal stability profiles (Figure 4), EGI_swapped contains a considerable level of its secondary structure while antiparallel β-sheet structures of EGI is drastically degraded (Figure 6). Evidently, swapping of the Ala43-Gly83 region of CBHI to EGI enzymes improves the structural integrity and thermal stability significantly (Figure S10).
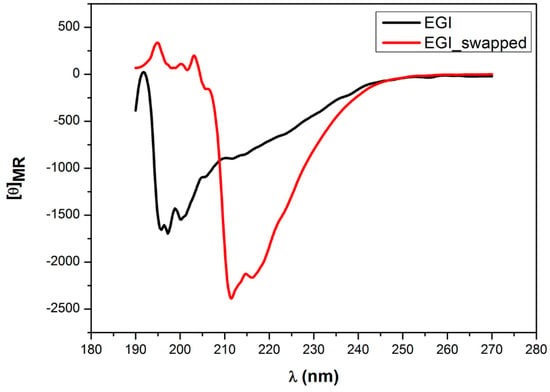
Figure 6.
The CD spectroscopy measurement of EGI and EGI_swapped enzymes after 1 h denaturation at 95 °C. Through Circular Dichroism, the change in structures of EGI and EGI_swapped was carefully traced between 195 nm and 230 nm after 1-h heat denaturation at 95 °C and 3-h incubation at room temperature. The CD measurement was performed at room temperature with samples prepared in 20 mM sodium acetate buffer (pH 4.7) that was corrected for the backgrounds. For EGI_swapped, the peaks at 211 nm and 216 nm are indicated. All results are displayed in mean residue ellipticity ([θ]).
To elucidate the impact of Co2+ presence on secondary structures enzymes, we track the secondary structure of these enzymes at various Co2+ concentrations (Figure 7). In the case of the EGI enzyme, increasing Co2+ concentration results in narrowing and the red-shift of the antiparallel β-sheet dominant CD profile. Upon the addition of 0.05 mM Co2+, two negative maxima in the CD pattern emerges at a wavelength of 216 nm and 220 nm, as is similar to EGI_swapped but in a narrower range (Figure 7a). With the addition of higher concentrations of Co2+ to EGI enzyme, the sharper peaks are observed. The sharpened peaks remain to be linked with the presence of Co2+ while they are not correlated with the concentration of Co2+ suggesting nonspecific interactions with Co2+. The enzyme assay results performed with various Co2 ion concentrations (Figure 3a) and the release of ~2.2 µM Co2+ ion from the EGI enzyme upon heat denaturation are also verified by these non-specific interactions. Contrary to EGI, the impact of Co2+ is more stable and obvious in EGI_swapped. Increasing Co2+ concentrations exhibit the correlation with a peak intensity ca. 211 nm. The newly formed peak could be interpreted as the higher amount of stable antiparallel β-sheet formation, contributing the improved enzymatic activity and higher thermal stability, as previously showed in Figure 4.
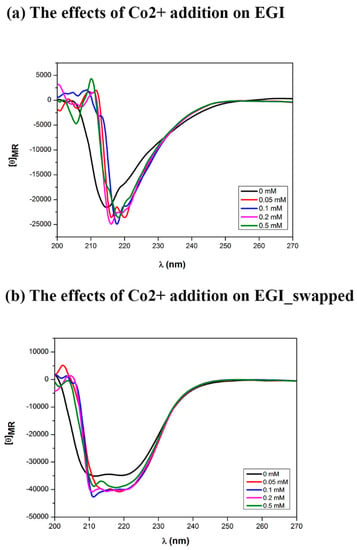
Figure 7.
The effects of Co2+ ion addition on secondary structures of EGI and EGI_swapped enzymes. Through Circular Dichroism, the differences in secondary structures of EGI (a) and EGI_swapped (b) upon Co2+ ion addition have been captured within 210–220 nm and 215–230 nm, respectively. Protein samples were prepared in 50 mM sodium acetate buffer (pH 4.7) that was corrected for background at room temperature. All results are displayed in mean residue ellipticity ([θ]).
3. Material & Methods
3.1. The Construction of EGI_Swapped and Cloning Approach
A foresight for swapped domain between EGI (pdb id: 1EG1) [35] and CBHI (PDB id: 1DY4) [24] was provided with the structural alignment, performed via TM-Align web-server [38]. Based on the structural alignment results, EGI_swapped was constructed, and the finalized nucleotide sequence of the swapped enzyme was synthesized by Microsynth AG® (Balgach, Switzerland) in the pUC57 plasmid.
To clone this swapped gene into pPICZαA vector, its initial amplification was performed with PCR by using forward (FEGI) and reverse (REGI) primers. The reaction conditions were set to 96 °C for 120 s, 94 °C for 30 s, 55 °C for 30 s, 72 °C for 4 min with 35 cycles and 72 °C for 7 min. After digesting the amplified gene and pPICZαA vector with EcoRI and XbaI restriction enzymes (Thermo Scientific, USA), the rapid ligation kit (Thermo Scientific, Waltham, MA, USA) was used to ligate the digested EGI_swapped gene into the digested sites of a pPICZαA expression vector (Invitrogen, San Diego, CA, USA). Finally, 20 µL of ligation mixture was transformed into Escherichia Coli XL-1 blue cells, cultured on low-salt Luria-Bertani (LB) broth containing 25 µg∙mL−1 of Zeocin [16]. The selection of positive colonies from LB plates was done with colony PCR by priming the AOX site of pPICZαA vector and they were isolated with QIAprep miniprep spin kit (QIAGEN). For double verification, these isolated plasmids were subjected to single digestion with EcoRI restriction enzyme. Prior to transformation of selected plasmids to Pichia pastoris cells, they were linearized with SacI to enable its integration to the AOX1 chromosomal locus of P. pastoris KM71H (AOX: ARG4, arg4) (Invitrogen). The competent P. pastoris KM71H cells with slow methanol utilizing phenotype were prepared according to a procedure of Wu and Letchworth [39]. Approximately, ~2 µg of linearized plasmid was transformed into KM71H P. pastoris cells by using 1.5 kV charging voltage, 25 F capacitance and 200 Ω resistance. The transformation product was spread onto YPD plates containing 100 µg∙mL−1 of Zeocin. After 48 h incubation at 30 °C, the positive ones were also selected with colony PCR from YPD-zeo plates by priming the AOX site of pPICZαA vector. Eventually, they were transferred to YPD Broth medium at 30 °C for 48 h with 250 rpm shakings.
3.2. P. pastoris Expression & Purification
Previously, EGI enzyme was cloned into pPICZαA vector and expressed in P.pastoris [40]. By following a similar procedure, EGI and EGI_swapped genes in pPICZαA vector were initially inoculated into 50 mL YPD-Broth medium for 48 h at 30 °C with 250 rpm shaking. As the optical density of cultures was reached to ~30 arbitrary units at 600 nm, the cells were collected by centrifugation (4200 rpm for 10 min at 4 °C). The pellets were suspended into expression buffer, containing 100 mM potassium phosphate buffer (pH 6.0), 1.34% Yeast Nitrogen Base (YNB), 4 × 10−5 % biotin and 2% (v/v) methanol in final. They were expressed at 30 °C for 72 h with 250 rpm shaking by inducing expression cultures with 2% v/v methanol in final, in every 24 h. At the end of the expression, proteins were collected by centrifugation at 4200 rpm for 25 min at 4 °C. After concentration of collected products with Sartocon Micro and Ultrafiltration system with 10 and 100 kDa cutoffs (Sartorius Stedim), they were purified with nickel-coated beads. During binding and elution steps, 20 mM sodium phosphate buffers with 50 mM imidazole and 500 mM imidazole were used.
3.3. Gel Electrophoresis
Followed by purification, the presence of swapped enzyme was checked with sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), prepared as 5% (w/v) stacking and 12% (w/v) separation gel. Enzyme samples were boiled at 95 °C for 5 min before loading them to SDS-PAGE. The gel was stained with Coomassie Brillant Blue Dye R-250 (Thermo Scientific, Waltham, MA, USA).
3.4. Bioinformatic Analysis of EGI and EGI_Swapped
Several bioinformatics tools were used to indicate changes in EGI enzyme upon swapping. First, the missing parts of EGI, deposited up to Thr371 (PDB id: 1EG1) [35], were predicted with I-TASSER [41,42] before performing its structural alignment with CBHI enzyme (PDB id: 1DY4) [24] via TM-Align web-server [38]. For visualization purpose, Visual Molecular Dynamics (VMD) was used [34]. By providing default parameters, the multiple alignments between EGI_swapped and EGI enzymes was performed [43]. The change in molecular weight and PI value of enzyme was predicted via ExPaSy-PI/MW tool [44,45].
3.5. Enzyme Assays
First of all, the purification buffer of enzymes was exchanged with reaction buffer through Sartocon Micro and Ultrafiltration system with 10 kDa cutoffs (Sartorius Stedim, Göttingen, Germany), and enzyme concentrations were determined via Bradford Assay [46]. The activities of these enzymes were determined by the 3,5-dinitrosalicyclic acid (DNS) against Carboxymethyl Cellulase (CMC) (Megazyme®, Bray, Ireland) as described by Gusakov et al [47]. The first step was to check the optimal buffer and pH combinations for both. For this purpose, activity tests were performed in 50 mM sodium acetate buffer (pH 4–5) and in 50 mM potassium phosphate buffer (pH 6–7) by narrowing pH ranges as 4, 4.3, 4.7, and 5 for sodium acetate buffer and 6, 6.3, 6.7, and 7 for potassium phosphate buffer. The activities of enzymes were measured against 0.5% w/v CMC, prepared separately for each buffer-pH combination, for 3 h at 50 °C by keeping the amount of enzyme as 20 µg in final.
After determination of optimal pH-buffer combination for enzymes, the following enzyme assays were performed in 50 mM sodium acetate buffer (pH 4.7) for 1 h, 3 h and 6 h at 30–50 °C (/10 °C) against 0.5% w/v CMC. Again, the amount of enzymes was kept as 20 µg in final. Prior to all assays, enzymes and substrate were separately pre-incubated at the designed temperature for 10 min. To terminate the reaction, DNS reagent was added to the reaction mixture that was boiled at 95 °C for 5 min. Upon cooling down the reaction mixture to room temperature, the absorbance of the reduced sugar was measured at a wavelength of 540 nm. The amount of reduced sugar was calculated from standard calibration curve after subtracting A540 background of enzymes and substrate. To test the reproducibility of data, three samples from two independent protein expression and purification batches of EGI and EGI_swapped enzymes, were taken. Since the reaction volume and the amount of enzyme were kept constant in all reactions, mean values were represented as relative values, obtained by setting the highest value to 100% for calculating the rest, with standard deviations below 10%.
Moreover, the enzyme activity tests were performed with the presence of Co2+ ions at several final concentrations, 0 mM, 0.05 mM, 0.1 mM, 0.2 mM and 0.5 mM. Two different paths were followed to reveal the impacts of Co2+ ions on activity; (i) incubating EGI_swapped and EGI enzymes with Co2+ ions or (ii) incubating 0.5% w/v CMC with Co2+ ions for 10 min at room temperature just before performing enzymatic assays described by Gusakov et al [47]. As outlined in above, the reaction was terminated by adding DNS reagent to reaction mixture that was further boiled at 95 °C for 5 min. The absorbance of reduced sugar was measured at a wavelength of 540 nm after cooling the reaction mixture to room temperature. Followed by proper subtraction of enzymes’ and substrate’ absorbance at 540 nm (A540), the amount of reduced sugar was calculated from the calibration curve. Three samples from two independent protein expressions and purification batches were taken to test the reproducibility of data. To follow the impact of Co2+ ion addition on enzymes’ activities, the highest data among all activity test results, e.g., EGI_swapped at 50°C for 6 h in our case, was taken as 100% to calculate the rest.
3.6. Thermal Stability Assay
To delve into the effects of swapping on thermal stability, EGI_swapped and EGI enzymes were initially incubated at 30–70 °C (/10 °C) for 30 min, 60 min and 90 min in 50 mM sodium acetate buffer (pH 4.7) by keeping the amount of enzymes as 20 µg in final. Followed by thermal incubation, they were chilled on ice for 20 min to allow structural recovery. Then, their remaining enzymatic activities were determined by DNS against 0.5% w/v CMC at 30 °C for 1 h in the same way as described above [47]. Similarly, the absorbance of reduced sugar was measured at a wavelength of 540 nm upon cooling of the reaction mixture to room temperature, and the amount of reduced sugar was calculated from standard calibration curve after subtracting A540 background of enzymes and substrate. To test the reproducibility of data, three samples from two independent expressions and purification batches of swapped and native enzymes were taken. The relative remaining activities were calculated by setting the enzymatic activity value of each enzyme, at 30 °C for 1 h, without any thermal incubation to 100%.
As mentioned above, all enzyme activity and thermal stability tests are performed by the 3,5-Dinitrosalicylic acid (DNS) method against 0.5% w/v CMC as outlined by Gusakov et al [47]. Among all available methods, colorimetric methods are most commonly used for measuring the amount of reduced sugar released during the enzymatic reaction [48]. 3,5-Dinitrosalicylic acid (DNS) is a recently used colorimetric assay [49]. Compared to the Nelson–Somogyi method, it is reported as being faster, less toxic and more convenient compared to the Nelson–Somogyi method [50]. To obtain the realistic results in DNS assay, we aimed to improve the sensitivity and precision of the method, DNS assay for our case as much as possible. For this particular purpose, we initially satisfied it by working on the linearization of enzyme assay with trying different enzyme-substrate combinations, incubation time and temperatures. The reproducibility of our data was also tested before reporting by increasing the number of samples coming from two independent purification and expression batches.
3.7. Measurement of Co(II) Ion Concentration in EGI and EGI_Swapped Enzymes
To assess whether suitable environment for Co2+ ion coordination was successfully integrated into EGI_swapped or not, we performed inductively coupled optical emission spectrometry measurement (ICP-OES) (Vista-Pro Axial; Varian Pty Ltd., Mulgrave, Australia). Followed by expression and purification steps, enzyme samples of 0.5 mg/ml concentration were boiled at 95 °C for 20 min, and the amount of released Co2+ upon denaturation was detected with an ICP-OES measurement. In addition to the Co2+ ion, the amount of released Ca2+, Mg2+ and Zn2+ ions were also measured through ICP-OES method after 20 min of thermal denaturation at 95 °C.
3.8. Circular Dichroism (CD) Spectroscopy Measurements
CD spectrums of enzymes were collected by using a J-815 Circular Dichroism Spectropolarimeter (Jasco International Co., Tokyo, Japon) under a N2 stream purge equipped with a thermostatically controlled cuvette. The measurement was performed with a quartz cuvette (Hellma Analytics, 1.0 mm path length) and the scanning speed was 100 nm/min [51]. Temperature scanning was performed from 25 °C to 70 °C as 5 °C increments. In each temperature, ten scans were averaged to obtain the full spectra of samples prepared in 20 mM of sodium acetate buffer (pH 4.7) that were corrected for the background.
To explore more about the effects of Co2+ addition on the structures of EGI and EGI_swapped enzymes, Co2+ ions at various concentrations (0.05 mM, 0.1 mM, 0.2 mM and 0.5 mM) were added to the enzyme samples. Prior to the collection of full spectra, they were incubated for 10 min at room temperature, in 20 mM sodium acetate buffer (pH 4.7). The full spectra of samples were collected at room temperature with scanning speeds as 100 nm/min with ten scans that were averaged to obtain the full spectra of samples.
Further, the effects of thermal incubation on enzymes’ structures were investigated with CD spectroscopy measurement after they were subjected to thermal incubation at 95 °C.
Enzymes were subjected to thermal incubation at 95 °C for 1 h and then were incubated at room temperature for 3 h. Again; CD spectra samples were collected as ten averages with scanning speed as 100 nm/min at room temperature. There was no Co2+ ion addition on enzyme samples that subjected to thermal denaturation.
By using the following equation, the mean residue ellipticity was calculated;
where θ is the observed ellipticity mmDeg, c is the molar concentration, l is the cell length in centimetres.
4. Conclusions
In this study, we present how swapping is a strong protein-engineering tool in order to create an endoglucanase enzyme, called as EGI_swapped, with novel structural and functional properties. Basically, we aimed to create the favorable environment for providing a Co2+ ion coordination in the endoglucanase enzyme by swapping the Ala43-Gly83 region of CBHI to EGI.
In the first part of this study, we described the impacts of swapping on enzyme activities with improvements in EGI_swapped. The difference in enzymatic activities of EGI_swapped and EGI become more prominent upon increasing Co2+ ion concentrations. This particular result could be considered as a strong implication for the existence of Co2+ ion coordination in EGI_swapped. As being different than EGI, EGI_swapped displays improved thermal stability that becomes more apparent with the extension of reaction time and temperature. Specifically, the EGI_swapped enzyme shows quantifiable activity against CMC while EGI enzyme completely loses its activity after 90 min thermal incubation at 70 °C. ICP-OES measurements suggest that the existence of Co2+ ion in EGI_swapped is significantly different than EGI; e.g.~3.6-fold stronger Co2+ ion coordination in EGI_swapped with respect to the EGI enzyme. This particular difference could provide a basis to explain improvements in enzymatic activity and thermal stability of EGI_swapped with respect to its native counterpart.
In the second part of the study, we identify the structural differences in EGI_swapped and native enzymes via Circular Dichroism (CD) spectroscopy. Swapping the Ala43-Gly83 region of CBHI to EGI leads to a transformation of the secondary structure, and results in distinct conformational and thermal susceptibility. In EGI_swapped enzyme, the newly formed peaks are observed upon Co2+ ion addition via CD spectroscopy. Unlike EGI, the considerable level of secondary structure is reported in CD spectroscopy of EGI_swapped after heat denaturation at 95 °C for 1 h. This result verifies the thermal stability results by indicating that swapping improves the structural integrity of the enzyme and contributes significantly to its thermal stability without disrupting its activity. Indeed, we end up with a novel endoglucanase enzyme with more promising enzymatic and functional properties, in terms of meeting the demands of harsh conditions of industrial applications, compared to its native counterpart.
To the best of our knowledge, this study is the first attempt in literature to alter the functional and structural properties of endoglucanase enzyme from Trichoderma reesei via swapping from cellobiohydrolase I enzyme. The computational and experimental tools of protein-engineering approaches have been perfectly combined to end up with a creation of new endoglucanase enzyme with altered thermal and structural properties. In literature, this study can be considered as the first successful attempt to understand the impacts of divalent metal ion coordination in endoglucanase enzyme from Trichoderma reseei. We believe that this study will be used as a basis and motivation for others, aiming to fulfill the gaps in cellulase research about the structural and functional importance of metal ion coordination.
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4344/9/2/130/s1, Figure S1: The depiction of cellobiohydrolase and endoglucanase mode of action; Figure S2: The structural alignment results between EGI and CBHI enzymes (a) TM-alignment results between enzymes are displayed. ‘:’ donates aligned residue pairs whose distances are smaller than 5 Å. The aligned region, Ala43-Gly83 (CBHI numbering) is indicated with *. The structural break happened around His189 (EGI numbering) is indicated with **. (b) The structural presentation of alignment results are provided in ‘cartoon’ format for overall, and ‘surf’ format just for swapped region. His206 in CBHI and His189 in EGI are drawn in ‘licorice’ format as yellow and red, respectively. In ‘surf’ presentation, the end points of swapped domain (Ala43 and Gly 83 as CBHI numbering) are indicated; Figure S3: The multiple protein alignment results between EGI_swapped and EGI; Figure S4: The crystal structures of cellobiohydrolase I enzymes. These cellobiohydrolase I enzymes have displayed more than 95% sequence similarity to CBHI (pdb id: 1DY4). These structures are crystallized with metal ions different than Co2+ ion. On the left side, the crystal structures are displayed with metal ions. Structures and metal ions are drawn in ‘cartoon’ and ‘VDW’ format, respectively, with Visual Molecular dynamics. The corresponding parts of our swapped region, Ala43-Gly83, in cellobiohydrolase enzymes are displayed by indicating the distances of Ala43 and Gly83 to metal ions. On the right side, the structural alignment results of cellobiohydrolase enzymes, pdb id: 1CEL, 4P1H and 5TC9, with EGI enzyme (pdb id: 1EG1) are displayed by indicating Glu residues involved in metal ions coordination; Figure S5: The cloning results of EGI_swapped; (a) the colony PCR results and (b) the double verification of isolated plasmid with EcoR I, and (c) Raw files of colony PCR results and single digest verification with EcoR I. After selection of positive clones through colony PCR, the isolated plasmids (last four ones) were double checked with single EcoR I digestion as displayed in (b). The colony PCR’s products and linearized plasmid were visualized in 1.2% w/v Agarose gel by ethidium bromide staining. M indicates a GeneRuler Ladder Mix (Fermentas); Figure S6. SDS-PAGE results of EGI_swapped; Figure S7: The effects of Co2+-ion pre-incubation with CMC prior to hydrolysis reaction on their enzymatic activity profiles; Figure S8. The construction of pH profiles for native and domain swapped enzymes; Figure S9: The enzyme activity profiles of EGI and EGI_swapped enzymes toward CMC for 1 h at 30–50 °C(/10 °C); Figure S10: The temperature path of EGI_swapped and EGI enzymes.
Author Contributions
O.U.S. initiated the research. A.Y. and O.U.S. designed the study. A.Y. performed and analyzed all computational and experimental work excluding circular dichroism (CD) spectroscopy study under the supervision of O.U.S. H.K. performed the CD spectroscopy measurements. H.K. and A.Y. analyzed the CD results. A.Y. wrote the manuscript, and H.K. and O.U.S. revised the manuscript. All authors read and approved the manuscript.
Acknowledgments
The Scientific and Technological Research Council of Turkey supported this work with grant number 112T901. The authors have special thanks to Yusuf Tutuş (Sabanci University) for contribution to ICP-OES readings.
Conflicts of Interest
The authors declare that they have no competing interest.
Abbreviations
| Cellobiohydrolase I | CBHI |
| CD | Circular Dichroism |
| Carboxymethyl cellulase | CMC |
| 3,5-Dinitrosalicylic acid | DNS |
| Endoglucanese I | EGI |
| Endoglucanese I swapped | EGI_swapped |
| PDB | Protein Data Bank |
References
- Lin, L.L.; Yan, R.; Liu, Y.Q.; Jiang, W.J. In-depth investigation of enzymatic hydrolysis of biomass wastes based on three major components: Cellulose, hemicellulose and lignin. Bioresour. Technol. 2010, 101, 8217–8223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Lynd, L.R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems. Biotechnol. Bioeng. 2004, 88, 797–824. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.; Muñoz-Dorado, J.; de la Rubia, T.; Martínez, J. Biodegradation and biological treatments of cellulose, hemicellulose and lignin: An overview. Int. Microbial. 2002, 5, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Eibinger, M.; Sattelkow, J.; Ganner, T.; Plank, H.; Nidetzky, B. Single-molecule study of oxidative enzymatic deconstruction of cellulose. Nat. Commun. 2017, 8, 894. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef]
- Donohoe, B.S.; Resch, M.G. Mechanisms employed by cellulase systems to gain access through the complex architecture of lignocellulosic substrates. Curr. Opin. Chem. Biol. 2015, 29, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Percival Zhang, Y.H.; Himmel, M.E.; Mielenz, J.R. Outlook for cellulase improvement: Screening and selection strategies. Biotechnol. Adv. 2006, 24, 452–481. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Cellulases and biofuels. Curr. Opin. Biotechnol. 2009, 20, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Chanzy, H.; Lamed, R.; Shoham, Y. Cellulose, cellulases and cellulosomes. Curr. Opin. Struct. Biol. 1998, 8, 548–557. [Google Scholar] [CrossRef]
- Bennett, M.; Choe, S.; Eisenberg, D. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Abrahamson, M.; Grubb, A.; Jaskolski, M. Domain swapping in N-truncated human cystation. Chem. J. Mol. Biol. 2004, 341, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Schering, N.; Casale, E.; Caccia, P.; Giordano, P.; Battistini, C. Dimer formation through domain swapping in the crystal structure of the Grb2-SH2-Ac-pYVNV complex. Biochemistry 2000, 39, 13376–13382. [Google Scholar] [CrossRef]
- Barbosa, J.; Sivarman, J.; Li, Y.; Larocque, R.; Matte, A. Mechanism of action and NAD+ binding mode revealed by the crystal structure of L-histidinol dehydrogenase. Proc. Natl. Acad. Sci. USA 2002, 99, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Knaus, K.; Morillas, M.; Swietnicki, W.; Malone, M.; Surewicz, WK.; Yee, V.C. Crystal structure of the human prion protein reveals a mechanism for oligomrerization. Nat. Struct. Biol. 2001, 8, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Yenenler, A.; Sezerman, O.U. Design and characterizations of two novel cellulases through single-gene shuffling of Cel12A (EG3) gene from Trichoderma reseei. Protein Eng. Des. Sel. 2016, 29, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Timucin, E.; Sezerman, O.U. The conserved lid tryptophan, W211, potentiates thermostability and thermoactivity in bacterial thermoalkalophilic lipases. PLoS ONE 2013, 8, e85186. [Google Scholar] [CrossRef]
- Jaenicke, R.; Bohm, G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998, 8, 738–748. [Google Scholar] [CrossRef]
- Tanaka, S.; Igarashi, S.; Ferri, S.; Sode, K. Increasing stability of water-soluble PQQ glucose dehydrogenase by increasing hydrophobic interaction at dimeric interface. BMC Biochem. 2005, 6, 1. [Google Scholar] [CrossRef]
- Kirino, H.; Aoki, M.; Aoshima, M.; Hayashi, Y.; Ohba, M. Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur. J. Biochem. 1994, 220, 275–281. [Google Scholar] [CrossRef]
- Keeling, A.A. Understanding Humic Substances: Advanced Methods, Properties and Applications; Wiley: Cambridge, UK, 2000. [Google Scholar]
- Hakansson, U.; Fagerstam, L.G.; Pettersson, G.; Andersson, L. 1,4-β-glucan glucanohydrolase from the cellulolytic fungus Trichoderma viride QM 9414. Biochem. J. 1979, 179, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, S.; Schweickart, V.; Ladner, M.; Gelfand, D.; Kwok, S.; Myambo, K.; Innis, M. Molecular cloning of exo-cellobiohydrolase from Trichoderma reesei strain L27. Nat. Biotechnol. 1983, 1, 691–696. [Google Scholar] [CrossRef]
- Stahlberg, J.; Henriksson, H.; Divne, C.; Isaksson, R.; Pettersson, G.; Johansson, G.; Jones, T.A. Structural basis for enantiomer binding and separation of a common beta-blocker: Crystal structure of cellobiohydrolase Cel7A with bound (S)-propranolol at 1.9 A resolution. J. Mol. Biol. 2001, 305, 79–93. [Google Scholar] [CrossRef]
- Cassia Pereira, J.; Giese, E.C.; Souza Moretti, M.M.; Santos Gomes, A.C.; Perrone, O.M.; Boscolo, M.; Silva, R.; Gomes, E.; Bocchini Martins, D.A. Effect of Metal Ions, Chemical Agents and Organic Compounds on Lignocellulolytic Enzymes Activities. In Enzyme Inhibitors and Activators; Senturk, M., Ed.; IntechOpen: London, UK, 2017. [Google Scholar]
- Glusker, J.P. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 1991, 42, 1–76. [Google Scholar]
- Sigel, R.K.; Pyle, A.M. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A. YETI: An interactive molecular mechanics program for small-molecule protein complexes. J. Comput. Chem. 1986, 9, 269–280. [Google Scholar] [CrossRef]
- Vedani, A.; Huhta, D.W. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990, 112, 4759–4767. [Google Scholar] [CrossRef]
- Bodenheimer, A.M.; Meilleur, F. Crystal structures of wild-type Trichoderma reesei Cel7A catalytic domain in open and closed states. FEBS Lett. 2016, 590, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Bodenheimer, A.M.; Cuneo, M.J.; Swartz, P.D.; Myles, D.A.; Meilleur, F. Crystal Structure of Wild Type Hypocrea Jecorina Cel7a in a Monoclinic Crystal Form. Available online: https://www.rcsb.org/structure/4p1h (accessed on 29 January 2019).
- Divne, C.; Stahlberg, J.; Reinikainen, T.; Ruohonen, L.; Pettersson, G.; Knowles, J.K.; Teeri, T.T.; Jones, T.A. The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 1994, 265, 524–528. [Google Scholar] [CrossRef]
- Stahlberg, J.; Divne, C.; Koivula, A.; Piens, K.; Claeyssens, M.; Teeri, T.T.; Jones, T.A. Activity studies and crystal structures of catalytically deficient mutants of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 1996, 264, 337–349. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Zou, J.Y.; Divne, C.; Davies, G.J.; Sinning, I.; Stahlberg, J.; Reinikainen, T.; Srisodsuk, M.; Teeri, T.T.; Jones, T.A. The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 A resolution, and a comparison with related enzymes. J. Mol. Biol. 1997, 272, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Rouvinen, J.; Bergfors, T.; Teeri, T.; Knowles, J.K.; Jones, T.A. Three-dimensional structure of Pcellobiohydrolase II from Trichoderma reesei. Science 1990, 249, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2007, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Letchworth, G.J. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques 2004, 36, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Akcapinar, G.B.; Gul, O.; Sezerman, U. Effect of codon optimization on the expression of Trichoderma reesei endoglucanase 1 in Pichia pastoris. Biotechnol. Prog. 2011, 27, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gusakov, A.V.; Kondratyeva, E.G.; Sinitsyn, A.P. Comparison of two methods for assaying reducing sugars in the determination of carbohydrase activities. Int. J. Anal. Chem. 2011, 2011, 283658. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K.; Hazlewood, G.P. Enzymology and Other Characteristics of Cellulases and Xylanases; CABI Publishing: Oxford, UK, 2001; pp. 11–60. [Google Scholar]
- Chen, X.; Li, W.; Ji, P.; Zhao, Y.; Hua, C.; Han, C. Engineering the conserved and noncatalytic residues of a thermostable beta-1,4-endoglucanase to improve specific activity and thermostability. Sci. Rep. 2018, 8, 2954. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Biely, P.; Poutanen, K. Interlaboratory testing of methods for assay of xylanase activity. J. Biotechnol. 1992, 23, 257–270. [Google Scholar] [CrossRef]
- Kurt, H.; Yuce, M.; Hussain, B.; Budak, H. Dual-excitation upconverting nanoparticle and quantum dot aptasensor for multiplexed food pathogen detection. Biosens. Bioelectron. 2016, 81, 280–286. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).