Influence of Base-Catalyzed Organosolv Fractionation of Larch Wood Sawdust on Fraction Yields and Lignin Properties

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Feedstock composition
2.2. Interpretation of results from experimental design
2.2.1. Pulp fraction
Higher fractionation efficiency in comparison to catalyzed ethanol organosolv pulping
Higher fractionation efficiency in comparison to catalyzed ethanol organosolv pulping
2.2.2. Lignin fraction
Higher purity compared to Kraft pulping
Higher yields compared to catalyzed ethanol organosolv pulping
2.3. Influence of pulping conditions on lignin properties
2.3.1. Ultimate analysis
2.3.2. Molecular weight analysis
2.3.3. Quantitative 31P-NMR analysis
2.3.4. Thioacidolysis GC/MS analysis
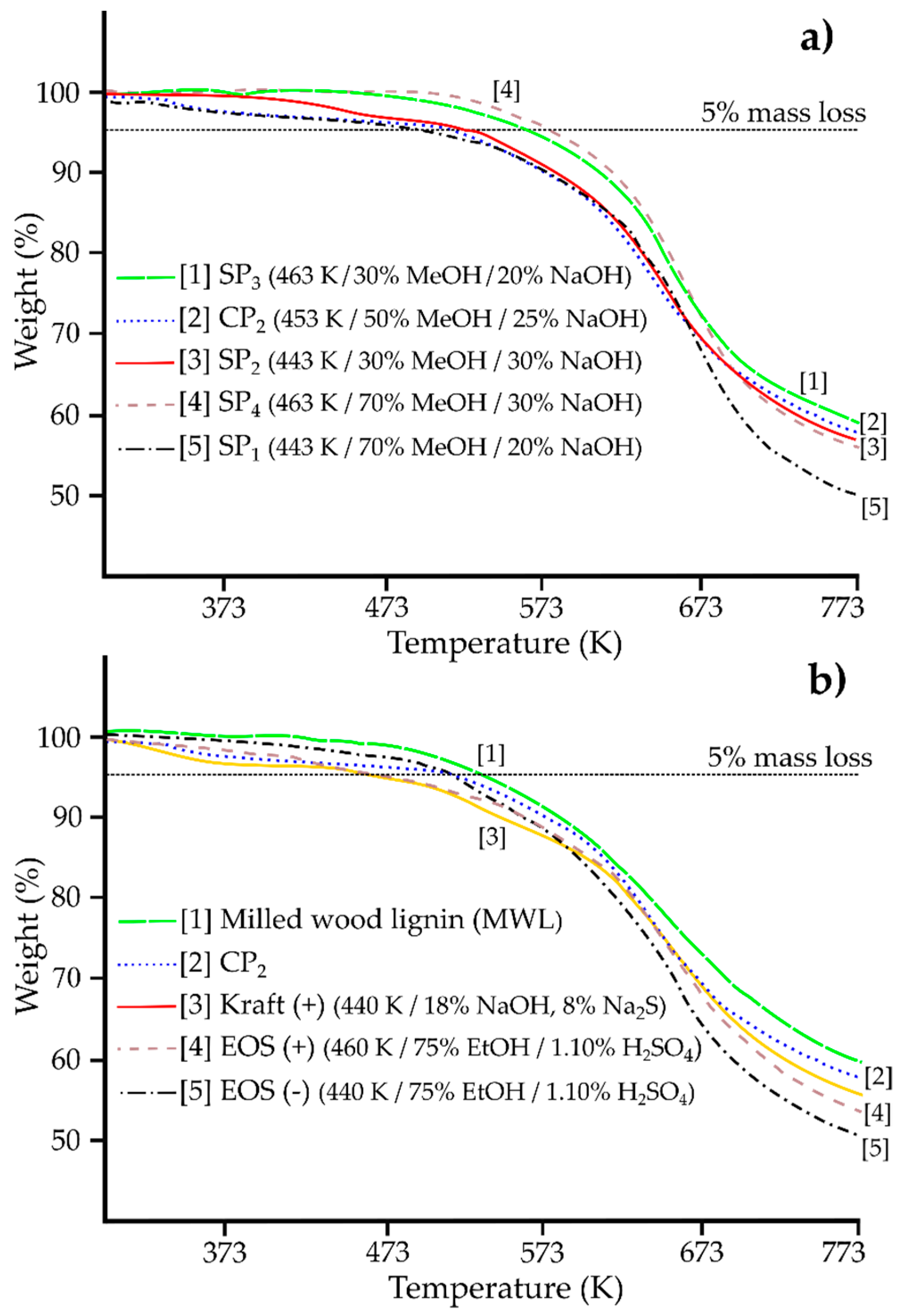
2.3.5. Thermal analysis
3. Materials and Methods
3.1. Base-catalyzed organosolv fractionation
3.2. Data Analysis
3.3. Lab-scale Kraft pulping
3.4. Crude milled wood lignin (MWLc) preparation
3.5. Milled wood lignin purification
3.6. Lignin purification
3.7. Analysis of chemical composition
3.8. Thermal analysis
3.9. Further analysis techniques
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery; Mussatto, S.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780128023235. [Google Scholar]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the way for lignin valorisation: Recent advances in bioengineering, biorefining and catalysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [PubMed]
- Nitsos, C.; Rova, U.; Christakopoulos, P. Organosolv fractionation of softwood biomass for biofuel and biorefinery applications. Energies 2018, 11, 50. [Google Scholar] [CrossRef]
- Kleinert, T.; Tayenthal, K.v. Über neuere Versuche zur Trennung von Cellulose und Inkrusten verschiedener Hölzer. Angew. Chem. 1931, 44, 788–791. [Google Scholar] [CrossRef]
- Rodríguez, A.; Espinosa, E.; Domínguez-Robles, J.; Sánchez, R.; Bascón, I.; Rosal, A. Different solvents for organosolv pulping. In Pulp and Paper Processing; Kazi, S.N., Ed.; InTechOpen: London, United Kingdom, 2018; ISBN 978-1-78923-847-1. [Google Scholar]
- Sixta, H. Handbook of Pulp; Wiley-VCH: Weinheim, Germany, 2006; ISBN 9783527309979. [Google Scholar]
- Løhre, C.; Kleinert, M.; Barth, T. Organosolv extraction of softwood combined with lignin-to-liquid-solvolysis as a semi-continuous percolation reactor. Biomass Bioenergy 2017, 99, 147–155. [Google Scholar] [CrossRef]
- Rossberg, C.; Janzon, R.; Saake, B.; Leschinsky, M. Effect of process parameters in pilot scale operation on properties of organosolv lignin. Bioresources 2019, 14. [Google Scholar] [CrossRef]
- Nitsos, C.; Stoklosa, R.; Karnaouri, A.; Vörös, D.; Lange, H.; Hodge, D.; Crestini, C.; Rova, U.; Christakopoulos, P. Isolation and characterization of organosolv and alkaline lignins from hardwood and softwood biomass. ACS Sustain. Chem. Eng. 2016, 4, 5181–5193. [Google Scholar] [CrossRef]
- Imlauer-Vedoya, C.M.; Vergara-Alarcón, P.; Area, M.C.; Revilla, E.; Felissia, F.E.; Villar, J.C. Fractionation of Pinus radiata wood by combination of steam explosion and organosolv delignification. Maderas. Cienc. Tecnol. 2019, 21, 587–598. [Google Scholar] [CrossRef]
- Lesar, B.; Humar, M.; Hora, G.; Hachmeister, P.; Schmiedl, D.; Pindel, E.; Siika-aho, M.; Liitiä, T. Utilization of recycled wood in biorefineries: Preliminary results of steam explosion and ethanol/water organosolv pulping without a catalyst. Eur. J. Wood Prod. 2016, 74, 711–723. [Google Scholar] [CrossRef]
- Baumeister, M.; Edel, E. Äthanol-Wasser-Aufschluss. Das Papier 1980, 34, V9–V18. [Google Scholar]
- Schröter, M.C. Possible lignin reaction in the organocell pulping process. Tappi J. 1991, 73, 197–200. [Google Scholar]
- Fullerton, T.J. Soda-anthraquinone pulping. The advantages of using oxygen-free conditions. Tappi J. 1979, 62, 55–57. [Google Scholar]
- Lindner, A.; Wegener, G. Characterization of lignins from organosolv pulping according to the organocell process. Part 1. Elemental analysis, nonlignin portions and functional groups. J. Wood Chem. Technol. 1988, 8, 323–340. [Google Scholar] [CrossRef]
- Lindner, A.; Wegener, G. Characterization of lignins from organosolv pulping according to the organocell process. Part 2. Residual lignins. J. Wood Chem. Technol. 1989, 9, 443–465. [Google Scholar] [CrossRef]
- Lindner, A.; Wegener, G. Characterization of lignins from organosolv pulping according to the organocell process. Part 3. Permanganate oxidation and thioacidolysis. J. Wood Chem. Technol. 1990, 10, 331–350. [Google Scholar] [CrossRef]
- Lindner, A.; Wegener, G. Characterization of lignins from organosolv pulping according to the organocell process. Part 4. Molecular weight determination and investigation of fractions isolated by GPC. J. Wood Chem. Technol. 1990, 10, 351–363. [Google Scholar] [CrossRef]
- Da Ronch, F.; Caudullo, G.; Tinner, W.; de Rigo, D. Larix decidua and other larches in Europe: Distribution, habitat, usage and threats. In European Atlas of Forest Tree Species, 2016th ed.; San-Miguel-Ayanz, J., de Rigo, D., Caudullo, G., Durrant, T.H., Mauri, A., Eds.; Publication Office of the European Union: Luxembourg, 2016; ISBN 978-92-79-36740-3. [Google Scholar]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood Kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Hochegger, M.; Cottyn-Boitte, B.; Cézard, L.; Schober, S.; Mittelbach, M. Influence of ethanol organosolv pulping conditions on physicochemical lignin properties of European larch. Int. J. Chem. Eng. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass. Technical Report NREL/TP-510-42618; 2012. Available online: https://www.nrel.gov/docs/gen/fy13/42618.pdf (accessed on 26 November 2019).
- Sjöström, E. Wood Chemistry. Fundamentals and Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 1993; ISBN 9780126474817. [Google Scholar]
- Carlson, R.; Carlson, J.E. Design and Optimization in Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 9780080455273. [Google Scholar]
- Tran, A. Effect of cooking temperature on Kraft pulping of hardwood. Tappi J. 2002, 1, 13–19. [Google Scholar]
- Yang, R.; Lucia, L.; Ragauskas, A.; Jameel, H. Oxygen delignification chemistry and its impact on pulp fibers. J. Wood Chem. Technol. 2003, 23, 13–29. [Google Scholar] [CrossRef]
- Borrega, M.; Larsson, P.T.; Ahvenainen, P.; Ceccherini, S.; Maloney, T.; Rautkari, L.; Sixta, H. Birch wood pre-hydrolysis vs pulp post-hydrolysis for the production of xylan-based compounds and cellulose for viscose application. Carbohydr. Polym. 2018, 190, 212–221. [Google Scholar] [CrossRef]
- Ehman, N.; Tarrés, Q.; Delgado-Aguilar, M.; Vallejos, M.; Felissia, F.; Area, M.; Mutjé, P. From pine sawdust to cellulose nanofibres. Cell Chem. Technol. 2016, 50, 361–367. [Google Scholar]
- Liu, J.; Korpinen, R.; Mikkonen, K.S.; Willför, S.; Xu, C. Nanofibrillated cellulose originated from birch sawdust after sequential extractions: A promising polymeric material from waste to films. Cellulose 2014, 21, 2587–2598. [Google Scholar] [CrossRef]
- Hörhammer, H. A Larch Biorefinery Producing Pulp and Lactic Acid. Ph.D. Thesis, Aalto University, Espoo, Finland, 2014. [Google Scholar]
- McDonough, T.J. The chemistry of organosolv delignification. Tappi J. 1993, 76, 186–193. [Google Scholar]
- Hu, F.; Jung, S.; Ragauskas, A. Pseudo-lignin formation and its impact on enzymatic hydrolysis. Bioresour. Technol. 2012, 117, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.D.; Meng, X.; Kumar, R.; Ragauskas, A.J. Recent advances in understanding the pseudo-lignin formation in a lignocellulosic biorefinery. Green Chem. 2018, 20, 2192–2205. [Google Scholar] [CrossRef]
- Burkhardt, S.; Kumar, L.; Chandra, R.; Saddler, J. How effective are traditional methods of compositional analysis in providing an accurate material balance for a range of softwood derived residues? Biotechnol. Biofuels 2013, 6, 90. [Google Scholar] [CrossRef]
- Baumberger, S.; Abaecherli, A.; Fasching, M.; Gellerstedt, G.; Gosselink, R.; Hortling, B.; Li, J.; Saake, B.; de Jong, E. Molar mass determination of lignins by size-exclusion chromatography: Towards standardisation of the method. Holzforschung 2007, 61, 205. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Abächerli, A.; Semke, H.; Malherbe, R.; Käuper, P.; Nadif, A.; van Dam, J.E.G. Analytical protocols for characterisation of sulphur-free lignin. Ind. Crops Prod. 2004, 19, 271–281. [Google Scholar] [CrossRef]
- Bauer, S.; Sorek, H.; Mitchell, V.D.; Ibáñez, A.B.; Wemmer, D.E. Characterization of miscanthus giganteus lignin isolated by ethanol organosolv process under reflux condition. J. Agric. Food Chem. 2012, 60, 8203–8212. [Google Scholar] [CrossRef]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; Peinder, P.D.; Boelens, R.; van Es, D.S.; Grisel, R.J.H.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef]
- Zhu, M.-Q.; Wen, J.-L.; Su, Y.-Q.; Wei, Q.; Sun, R.-C. Effect of structural changes of lignin during the autohydrolysis and organosolv pretreatment on Eucommia ulmoides Oliver for an effective enzymatic hydrolysis. Bioresour. Technol. 2015, 185, 378–385. [Google Scholar] [CrossRef]
- Gilarranz, M.A.; Rodríguez, F.; Oliet, M. Lignin behavior during the autocatalyzed methanol pulping of eucalyptus globulus changes in molecular weight and functionality. Holzforschung 2000, 54, 373–380. [Google Scholar] [CrossRef]
- Pan, X.; Kadla, J.F.; Ehara, K.; Gilkes, N.; Saddler, J.N. Organosolv ethanol lignin from hybrid poplar as a radical scavenger: Relationship between lignin structure, extraction conditions, and antioxidant activity. J. Agric. Food Chem. 2006, 54, 5806–5813. [Google Scholar] [CrossRef] [PubMed]
- Gordobil, O.; Moriana, R.; Zhang, L.; Labidi, J.; Sevastyanova, O. Assesment of technical lignins for uses in biofuels and biomaterials: Structure-related properties, proximate analysis and chemical modification. Ind. Crops Prod. 2016, 83, 155–165. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. 31P NMR in wood chemistry: A review of recent progress. Res. Chem. Intermed. 1995, 21, 373. [Google Scholar] [CrossRef]
- Balakshin, M.; Capanema, E. On the quantification of lignin hydroxyl groups with 31P and 13C NMR spectroscopy. J. Wood Chem. Technol. 2015, 35, 220–237. [Google Scholar] [CrossRef]
- Yang, X.; Li, N.; Lin, X.; Pan, X.; Zhou, Y. Selective cleavage of the aryl ether bonds in lignin for depolymerization by acidic lithium bromide molten salt hydrate under mild conditions. J. Agric. Food Chem. 2016, 64, 8379–8387. [Google Scholar] [CrossRef]
- Lai, C.; Tu, M.; Shi, Z.; Zheng, K.; Olmos, L.G.; Yu, S. Contrasting effects of hardwood and softwood organosolv lignins on enzymatic hydrolysis of lignocellulose. Bioresour. Technol. 2014, 163, 320–327. [Google Scholar] [CrossRef]
- Zhao, Y.; Tagami, A.; Dobele, G.; Lindström, M.E.; Sevastyanova, O. The impact of lignin structural diversity on performance of cellulose nanofiber (CNF)-starch composite films. Polymers 2019, 11, 538. [Google Scholar] [CrossRef]
- Koumba-Yoya, G.; Stevanovic, T. Study of organosolv lignins as adhesives in wood panel production. Polymers 2017, 9, 46. [Google Scholar] [CrossRef]
- Huang, F.; Singh, P.M.; Ragauskas, A.J. Characterization of milled wood lignin (MWL) in loblolly pine stem wood, residue, and bark. J. Agric. Food Chem. 2011, 59, 12910–12916. [Google Scholar] [CrossRef] [PubMed]
- Toloue Farrokh, N.; Suopajärvi, H.; Mattila, O.; Umeki, K.; Phounglamcheik, A.; Romar, H.; Sulasalmi, P.; Fabritius, T. Slow pyrolysis of by-product lignin from wood-based ethanol production—A detailed analysis of the produced chars. Energy 2018, 164, 112–123. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Poly(ethylene oxide)/organosolv lignin blends: Relationship between thermal properties, chemical structure, and blend behavior. Macromolecules 2004, 37, 6904–6911. [Google Scholar] [CrossRef]
- Vroom, K.E. The H factor: A means of expressing cooking times and temperatures as a single variable. Pulp Paper Mag. Can. 1957, 58, 228–231. [Google Scholar]
- Zinovyev, G.; Sumerskii, I.; Rosenau, T.; Balakshin, M.; Potthast, A. Ball milling’s effect on pine milled wood lignin’s structure and molar mass. Molecules 2018, 23, 2223. [Google Scholar] [CrossRef]
- Björkman, A. Studies on finely divided wood. Part 1. Extraction of lignin with neutral solvents. Sven Papperstidn 1956, 59, 477–485. [Google Scholar]
- Janson, J. Calculation of the polysaccharide composition of wood and pulp. Paperi Ja Puu 1970, 52, 323–329. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose | Hemicellulose a | Total Lignin b | Extractives c | Ash [21] | |
|---|---|---|---|---|---|
| Content % w/w | 42.3 ± 0.4 | 29.6 ± 0.2 | 26.4 ± 0.1 | 2.70 ± 0.09 | 0.17 ± 0.02 |
| Parameters a | Pulp Fraction b | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| T | S | C | Total | KL c | ASL | TL | Cellulose | Hemicellulose | |
| Sawdust | 100 | 26.07 | 0.35 | 26.42 | 42.34 | 29.56 | |||
| SP1 (−/+/−) | 443 | 70 | 20 | 54.00 | 4.28 | 0.35 | 4.63 | 37.61 | 11.76 |
| SP2 (−/−/+) | 443 | 30 | 30 | 52.46 | 2.12 | 0.37 | 2.49 | 37.73 | 12.25 |
| SP3 (+/−/−) | 463 | 30 | 20 | 44.69 | 0.35 | 0.20 | 0.55 | 35.12 | 9.02 |
| SP4 (+/+/+) | 463 | 70 | 30 | 34.42 | 0.62 | 0.19 | 0.81 | 26.50 | 7.10 |
| CP1 (0/0/0) | 453 | 50 | 25 | 45.79 | 0.96 | 0.26 | 1.22 | 34.07 | 10.50 |
| CP2 (0/0/0) | 453 | 50 | 25 | 46.19 | 0.88 | 0.31 | 1.20 | 34.51 | 10.74 |
| CP3 (0/0/0) | 453 | 50 | 25 | 46.12 | 0.92 | 0.26 | 1.18 | 34.27 | 10.67 |
| CP1-3 | 46.0 | 0.92 | 0.28 | 1.20 | 34.3 | 10.6 | |||
| Avg ± SD | 0.2 | 0.03 | 0.02 | 0.02 | 0.2 | 0.1 | |||
| Pulp Fraction a | Lignin Fraction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | KL b | ASL | Cell | Hcell | Total | KL | ASL | TL | CH | Ash | |
| Sawdust | 100 | 26.07 | 0.35 | 42.34 | 29.56 | - | - | - | - | - | - |
| Kraft (−) | 52.08 | 4.84 | 0.72 | 36.86 | 9.66 | 23.27 | 19.14 | 0.35 | 19.50 | 0.51 | 0.56 |
| Kraft (+) | 42.83 | 1.98 | 0.29 | 33.05 | 7.51 | 26.90 | 22.76 | 0.55 | 23.31 | 0.46 | 0.68 |
| EOS (−) c | 47.79 | 8.45 | 0.18 | 36.98 | 2.23 | 16.36 | 15.48 | 0.07 | 15.55 | 0.09 | >0.05 |
| EOS (0) | 38.56 | 4.91 | 0.15 | 32.84 | 0.67 | 19.42 | 18.12 | 0.15 | 18.27 | 0.08 | >0.05 |
| EOS (+) | 29.74 | 5.31 | 0.12 | 24.05 | 0.27 | 20.83 | 19.52 | 0.24 | 19.78 | 0.18 | >0.05 |
| Lignin Fraction a | Aqueous Fraction | ||||||
|---|---|---|---|---|---|---|---|
| Total | KL | ASL | TL | CH | Ash | ASL | |
| SP1 (−/+/−) | 18.44 | 16.78 | 0.56 | 17.34 | 0.40 | 0.10 | 4.56 |
| SP2 (−/−/+) | 21.53 | 19.57 | 0.62 | 20.19 | 0.64 | 0.27 | 3.94 |
| SP3 (+/−/−) | 24.82 | 22.38 | 0.87 | 23.24 | 0.31 | 0.19 | 3.61 |
| SP4 (+/+/+) | 23.08 | 21.07 | 0.52 | 21.59 | 0.18 | 0.28 | 7.06 |
| CP1 (0/0/0) | 22.28 | 20.01 | 0.65 | 20.66 | 0.28 | 0.23 | 5.63 |
| CP2 (0/0/0) | 22.16 | 20.24 | 0.58 | 20.82 | 0.22 | 0.24 | 5.62 |
| CP3 (0/0/0) | 22.57 | 20.42 | 0.67 | 21.10 | 0.25 | 0.25 | 5.62 |
| CP1-3 | 22.3 | 20.2 | 0.64 | 20.9 | 0.25 | 0.24 | 5.62 |
| Avg ± SD | 0.2 | 0.1 | 0.05 | 0.2 | 0.03 | 0.01 | 0.01 |
| Pulp Fraction a | Lignin Fraction | Aqueous Fraction | Lignin Recovery | |||
|---|---|---|---|---|---|---|
| KL | ASL | KL | ASL | ASL | % | |
| SP1 (−/+/−) | 4.28 | 0.35 | 16.78 | 0.56 | 4.56 | 100.4 |
| SP2 (−/−/+) | 2.12 | 0.37 | 19.57 | 0.62 | 3.94 | 100.8 |
| SP3 (+/−/−) | 0.35 | 0.20 | 22.38 | 0.87 | 3.61 | 103.8 |
| SP4 (+/+/+) | 0.62 | 0.19 | 21.07 | 0.52 | 7.06 | 111.5 |
| CP1 (0/0/0) | 0.96 | 0.26 | 20.01 | 0.65 | 5.63 | 104.1 |
| CP2 (0/0/0) | 0.88 | 0.31 | 20.24 | 0.58 | 5.62 | 104.6 |
| CP3 (0/0/0) | 0.92 | 0.26 | 20.42 | 0.67 | 5.62 | 105.6 |
| CP1-3 | 0.92 | 0.28 | 20.2 | 0.64 | 5.62 | 104.8 |
| Avg ± SD | 0.03 | 0.02 | 0.1 | 0.05 | 0.01 | 0.5 |
| C % w/w | H % w/w | O % w/w | S % w/w | H/C mol/mol | O/C mol/mol | Mn a kg/mol | Mw kg/mol | |
|---|---|---|---|---|---|---|---|---|
| MWL | 60.78 | 5.71 | 33.51 | - b | 1.04 | 0.50 | 1.76 | 2.61 |
| SP1 (−/+/−) | 66.13 | 5.78 | 28.09 | - | 1.05 | 0.32 | 1.89 | 2.94 |
| SP2 (−/−/+) | 65.32 | 5.78 | 28.90 | - | 1.06 | 0.33 | 1.80 | 2.88 |
| SP3 (+/−/−) | 66.75 | 5.77 | 27.48 | - | 1.04 | 0.31 | 1.68 | 2.77 |
| SP4 (+/+/+) | 67.27 | 5.85 | 26.88 | - | 1.04 | 0.30 | 1.75 | 2.56 |
| CP1 (0/0/0) | 66.43 | 5.79 | 27.78 | - | 1.05 | 0.31 | 1.65 | 2.58 |
| CP2 (0/0/0) | 66.36 | 5.78 | 27.86 | - | 1.05 | 0.31 | 1.62 | 2.52 |
| CP3 (0/0/0) | 66.27 | 5.82 | 27.91 | - | 1.05 | 0.32 | 1.62 | 2.55 |
| CP1-3 | 66.35 | 5.80 | 27.85 | - | 1.05 | 0.31 | 1.63 | 2.55 |
| Avg ± SD | 0.08 | 0.02 | 0.07 | - | <0.01 | <0.01 | 0.02 | 0.03 |
| Kraft (−) | 61.78 | 5.33 | 30.23 | 2.66 | 0.99 | 0.44 | 1.81 | 3.05 |
| Kraft (+) | 61.57 | 5.44 | 30.26 | 2.73 | 0.97 | 0.43 | 1.78 | 2.92 |
| EOS (−) | 66.42 | 6.22 | 27.36 | - | 1.12 | 0.31 | 1.06 | 2.50 |
| EOS (0) | 66.83 | 5.86 | 27.32 | - | 1.05 | 0.31 | 1.12 | 2.40 |
| EOS (+) | 67.18 | 6.04 | 26.78 | - | 1.08 | 0.30 | 0.97 | 2.02 |
| 31P-NMR Analysis | Thermal Analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| OHAlk mmol/g | OHPh mmol/g | OHtotal mmol/g | Ph/Alk ratio | COOH mmol/g | β-O-4 % (µmol/g) | Tg K | T5% K | |
| MWLa | 3.67 | 1.84 | 5.51 | 0.5 | 0.11 | 100 (947) | 432 | 563 |
| SP1 (−/+/−) | 1.54 | 3.47 | 5.01 | 2.3 | 0.43 | 8.2 (78) | 433 | 496 |
| SP2 (−/−/+) | 1.52 | 3.44 | 4.96 | 2.3 | 0.52 | 4.1 (39) | 433 | 528 |
| SP3 (+/−/−) | 1.18 | 3.60 | 4.78 | 3.1 | 0.51 | 2.6 (25) | 429 | 567 |
| SP4 (+/+/+) | 1.38 | 3.83 | 5.21 | 2.8 | 0.47 | 2.7 (26) | 426 | 578 |
| CP1 (0/0/0) | 1.51 | 3.68 | 5.19 | 2.4 | 0.50 | 4.6 (44) | 424 | 497 |
| CP2 (0/0/0) | 1.54 | 3.69 | 5.23 | 2.4 | 0.50 | 5.0 (47) | 425 | 512 |
| CP3 (0/0/0) | 1.59 | 3.78 | 5.37 | 2.4 | 0.52 | 4.8 (45) | 425 | 483 |
| CP1-3 | 1.55 | 3.72 | 5.27 | 2.4 | 0.51 | 4.8 (45) | 425 | 497 |
| Avg ± SD | 0.04 | 0.06 | 0.09 | >0.1 | 0.01 | 0.2 (2) | 1 | 14 |
| Kraft (−) | 1.48 | 3.56 | 5.04 | 2.41 | 0.61 | 7.4 (70) | - b | 473 |
| Kraft (+) | 1.44 | 4.01 | 5.45 | 2.78 | 0.68 | 3.7 (35) | - b | 412 |
| EOS (−) | 1.92 | 1.78 | 3.70 | 0.9 | 0.09 | 45.8 (443) | 393 | 512 |
| EOS (0) | 1.61 | 2.35 | 3.96 | 1.5 | 0.07 | 36.8 (365) | 402 | 516 |
| EOS (+) | 1.37 | 2.5 | 3.87 | 1.8 | 0.09 | 24.6 (233) | 401 | 471 |
| Variable | Assignment | (−) | (0) | (+) |
|---|---|---|---|---|
| X1 | Temperature (K) | 443 | 453 | 463 |
| X2 | Methanol (MeOH)/H2O (v/v) | 30:70 | 50:50 | 70:30 |
| X3 | NaOH (w/w odw a) | 20 | 25 | 30 |
| Parameter | Low | High |
|---|---|---|
| Reaction time, min | 58 | 134 |
| H-factor, h | 850 | 1650 |
| NaOH, % (odw) | 15.0 | 17.7 |
| Na2S, % (odw) | 5.76 | 7.61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochegger, M.; Trimmel, G.; Cottyn-Boitte, B.; Cézard, L.; Majira, A.; Schober, S.; Mittelbach, M. Influence of Base-Catalyzed Organosolv Fractionation of Larch Wood Sawdust on Fraction Yields and Lignin Properties. Catalysts 2019, 9, 996. https://doi.org/10.3390/catal9120996
Hochegger M, Trimmel G, Cottyn-Boitte B, Cézard L, Majira A, Schober S, Mittelbach M. Influence of Base-Catalyzed Organosolv Fractionation of Larch Wood Sawdust on Fraction Yields and Lignin Properties. Catalysts. 2019; 9(12):996. https://doi.org/10.3390/catal9120996
Chicago/Turabian StyleHochegger, Markus, Gregor Trimmel, Betty Cottyn-Boitte, Laurent Cézard, Amel Majira, Sigurd Schober, and Martin Mittelbach. 2019. "Influence of Base-Catalyzed Organosolv Fractionation of Larch Wood Sawdust on Fraction Yields and Lignin Properties" Catalysts 9, no. 12: 996. https://doi.org/10.3390/catal9120996
APA StyleHochegger, M., Trimmel, G., Cottyn-Boitte, B., Cézard, L., Majira, A., Schober, S., & Mittelbach, M. (2019). Influence of Base-Catalyzed Organosolv Fractionation of Larch Wood Sawdust on Fraction Yields and Lignin Properties. Catalysts, 9(12), 996. https://doi.org/10.3390/catal9120996