Highly Selective pH-Dependent Ozonation of Cyclohexane over Mn/γ-Al2O3 Catalysts at Ambient Reaction Conditions


Abstract
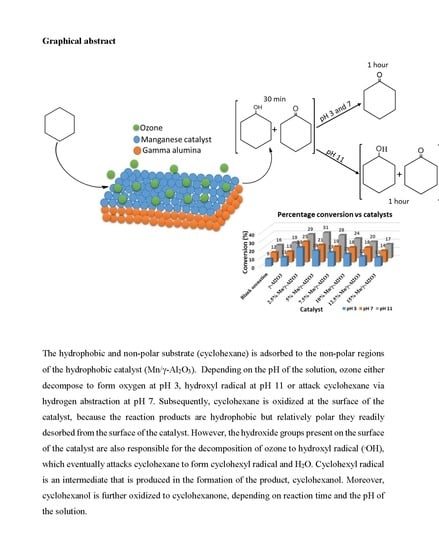
:
1. Introduction
2. Results and Discussion
2.1. Catalysts Characterisation
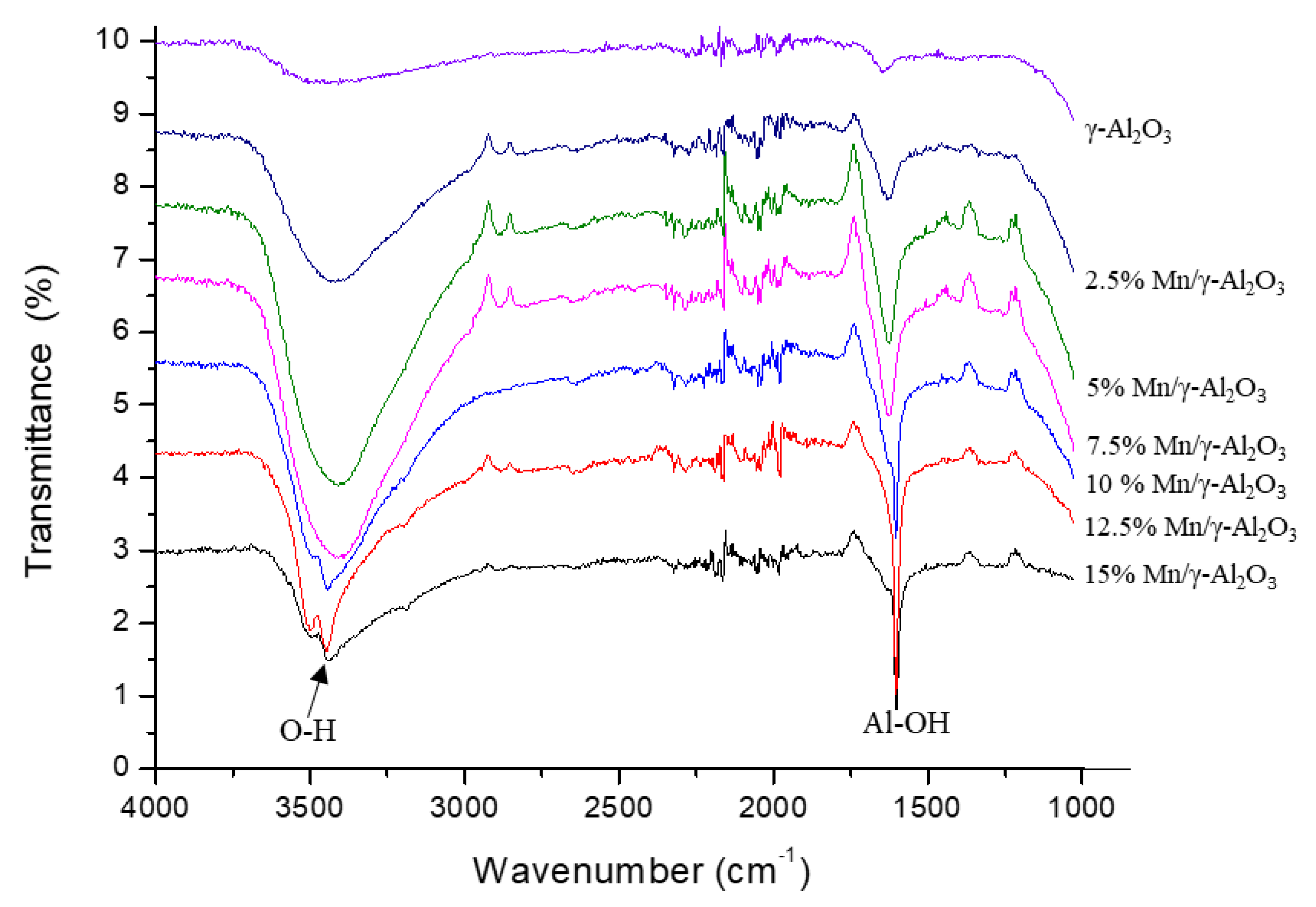
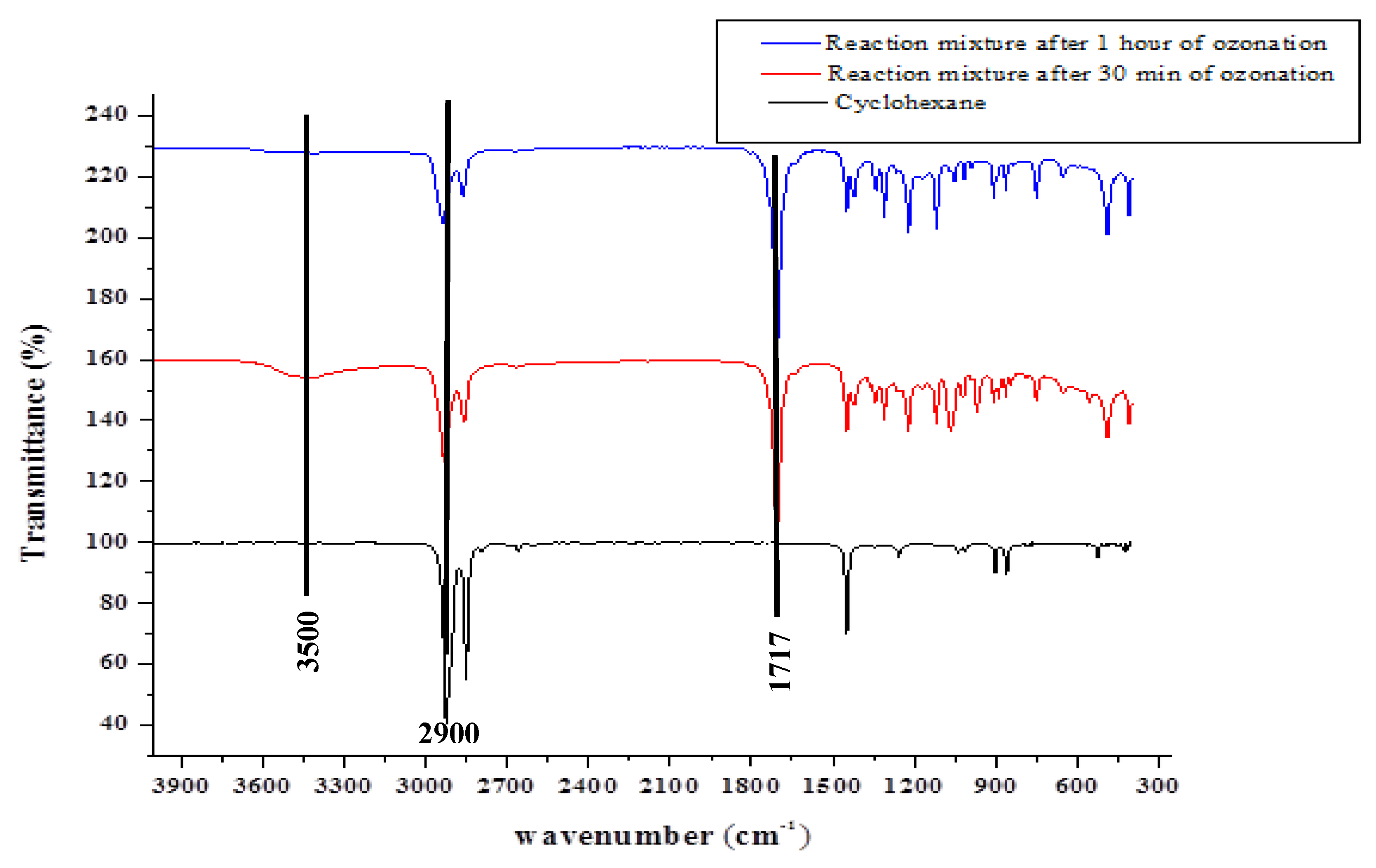
2.1.1. Fourier Transform Infrared (FT-IR) Spectroscopy
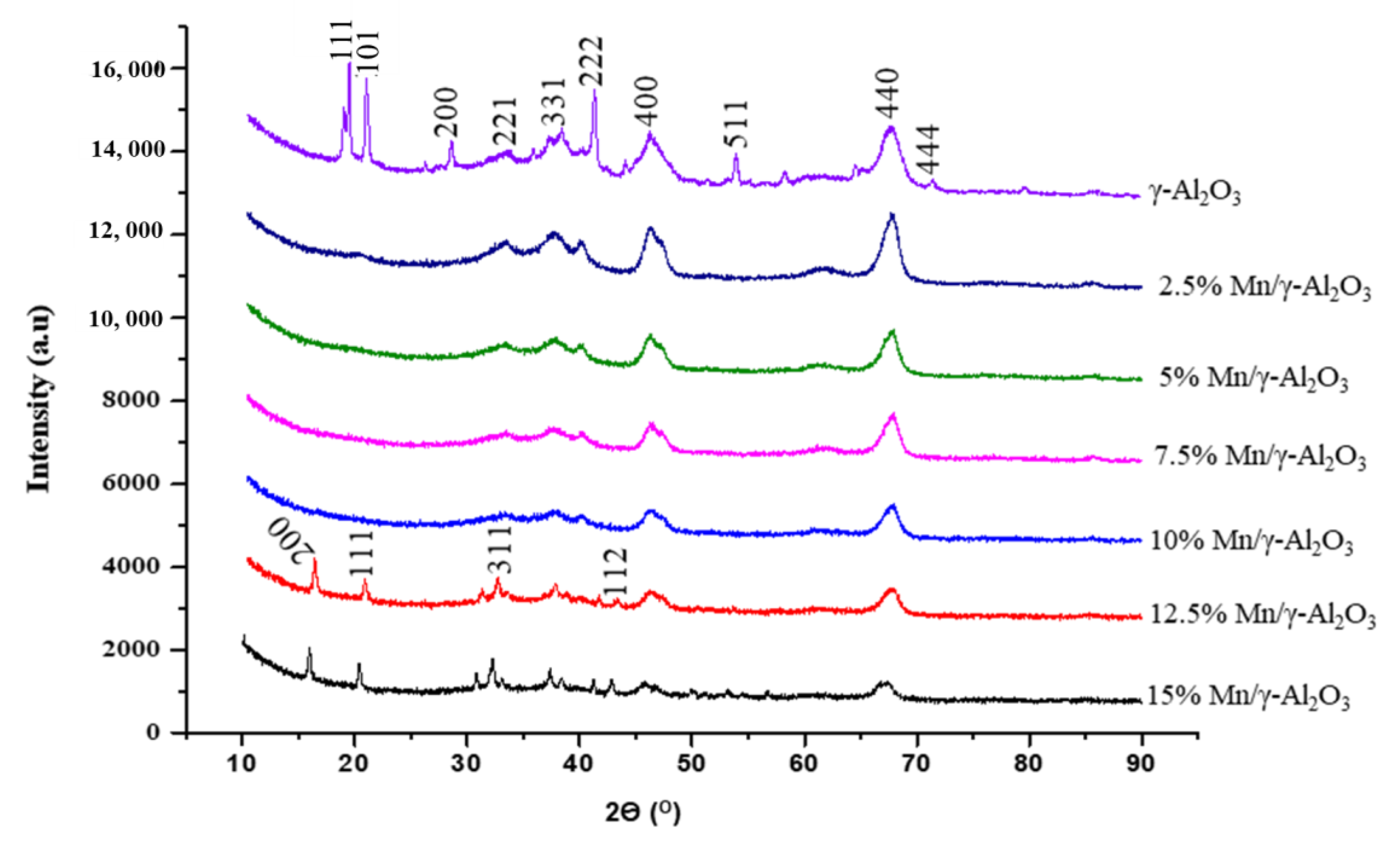
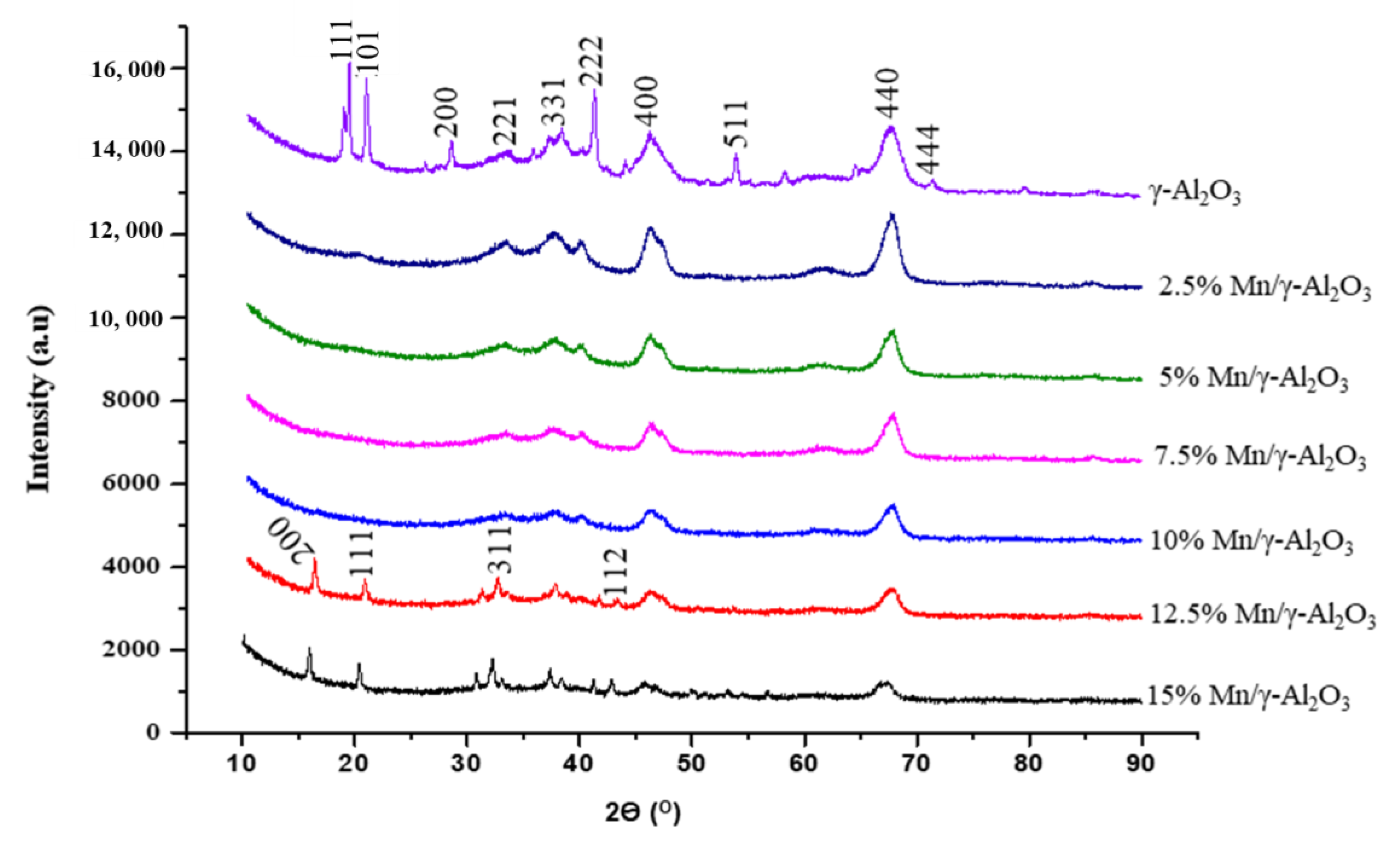
2.1.2. X-Ray Diffraction
2.1.3. Scanning Electron Microscopy (SEM)
2.1.4. Transmission Electron Microscopy (TEM)
2.1.5. Inductively Coupled Plasma (ICP) and Energy Dispersed X-Ray (EDX) Spectroscopy
2.1.6. Brunauer-Emmet-Teller (BET) Surface Area
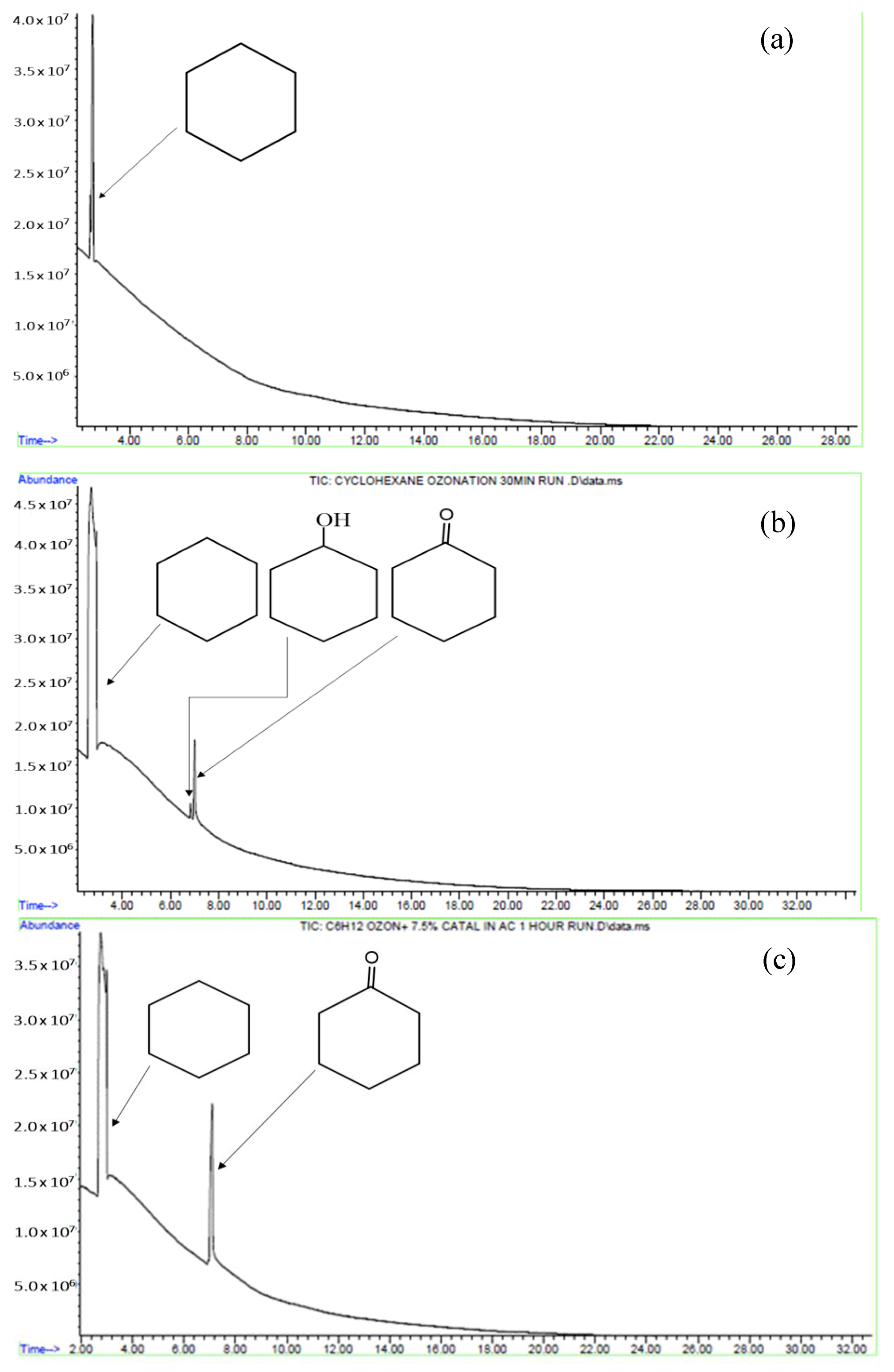
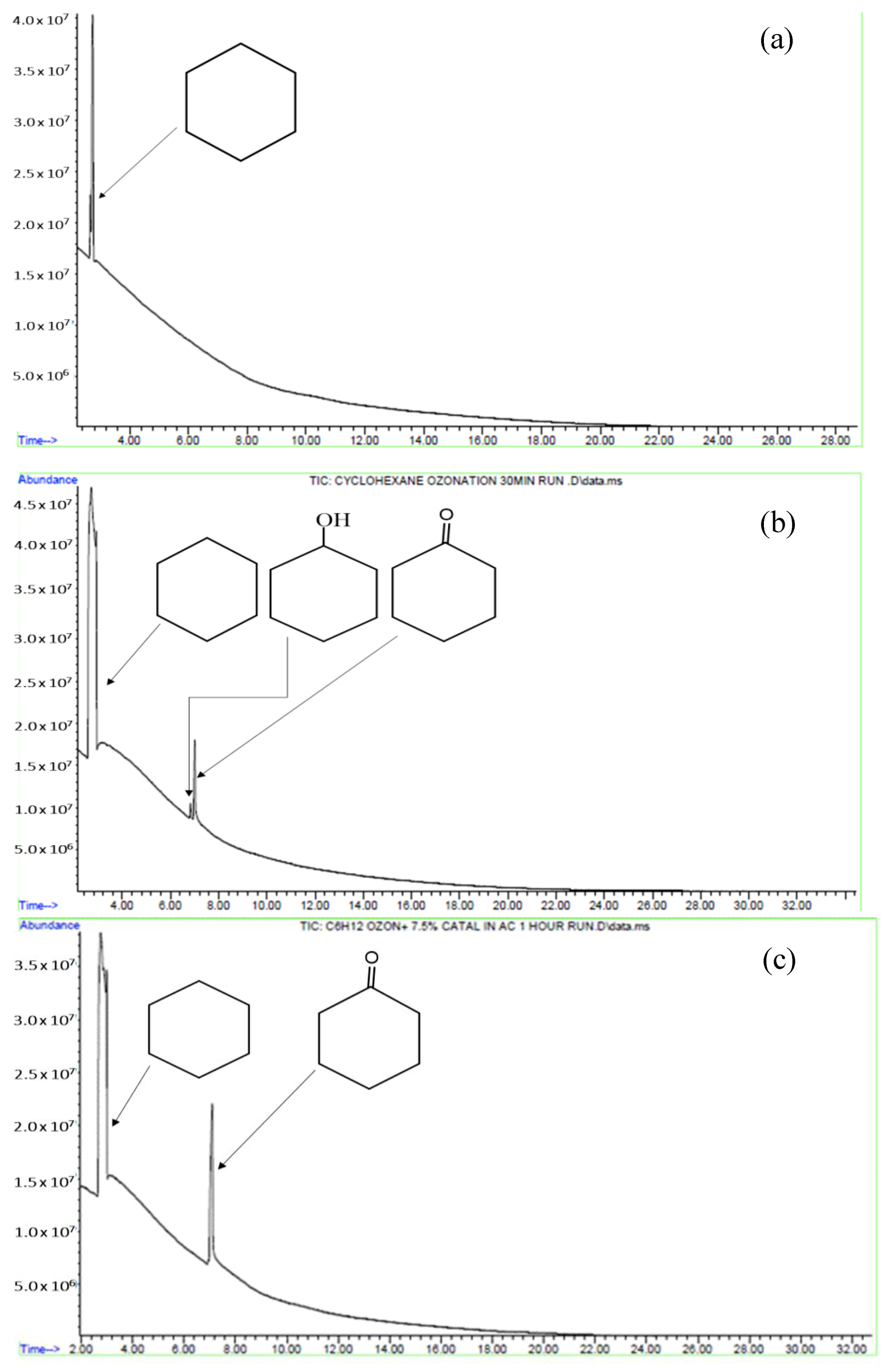
2.2. Characterisation of the Reaction Mixture
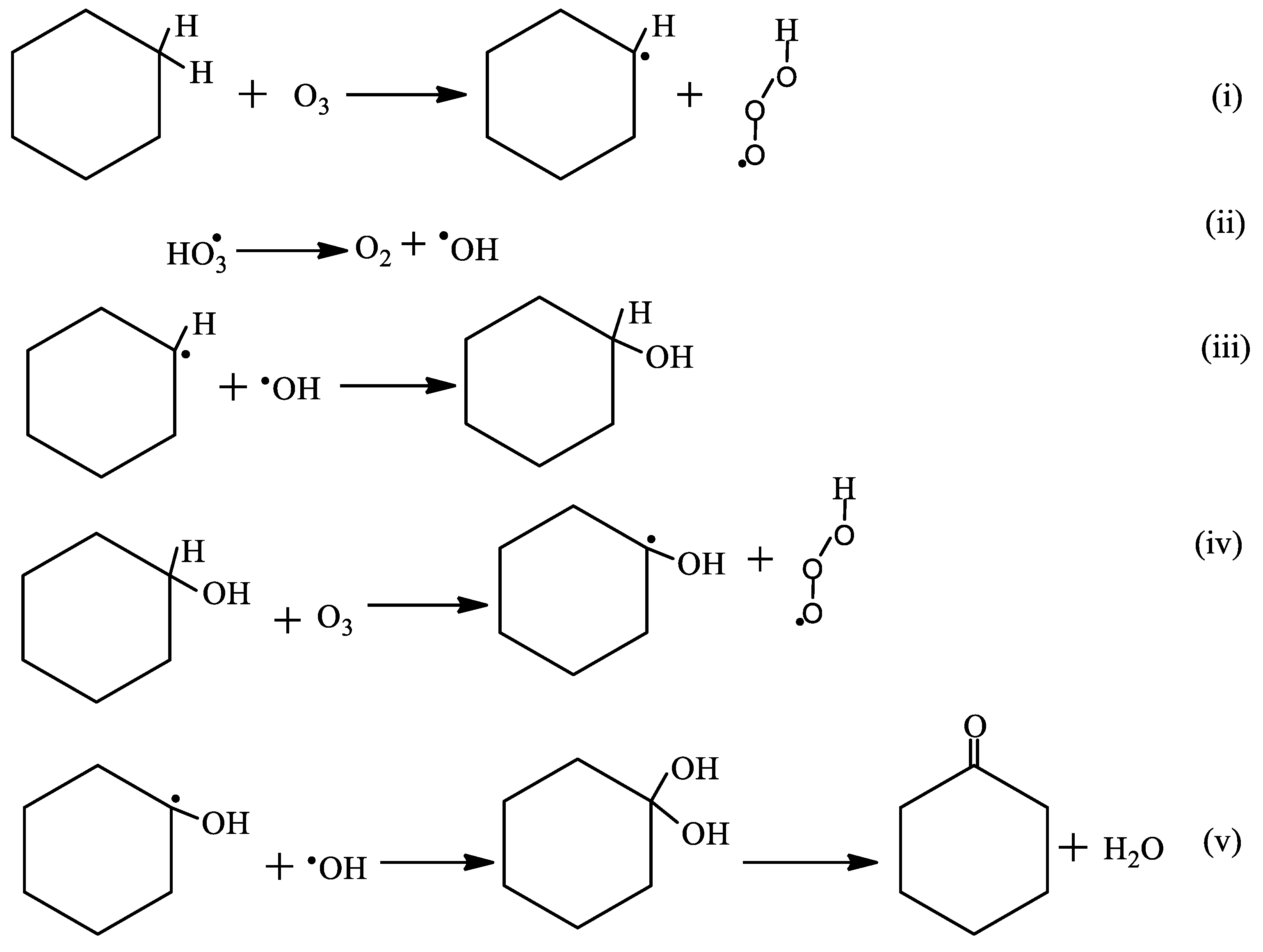
2.3. Oxidation of Cyclohexane
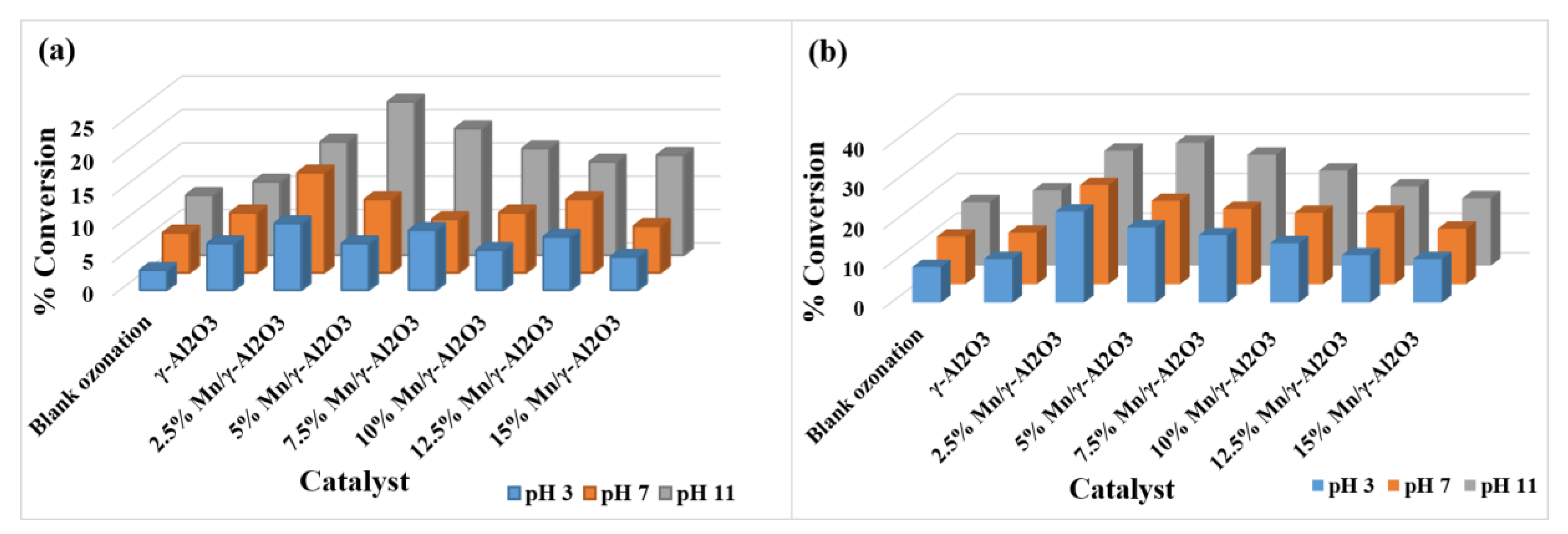
2.3.1. Percentage Conversion
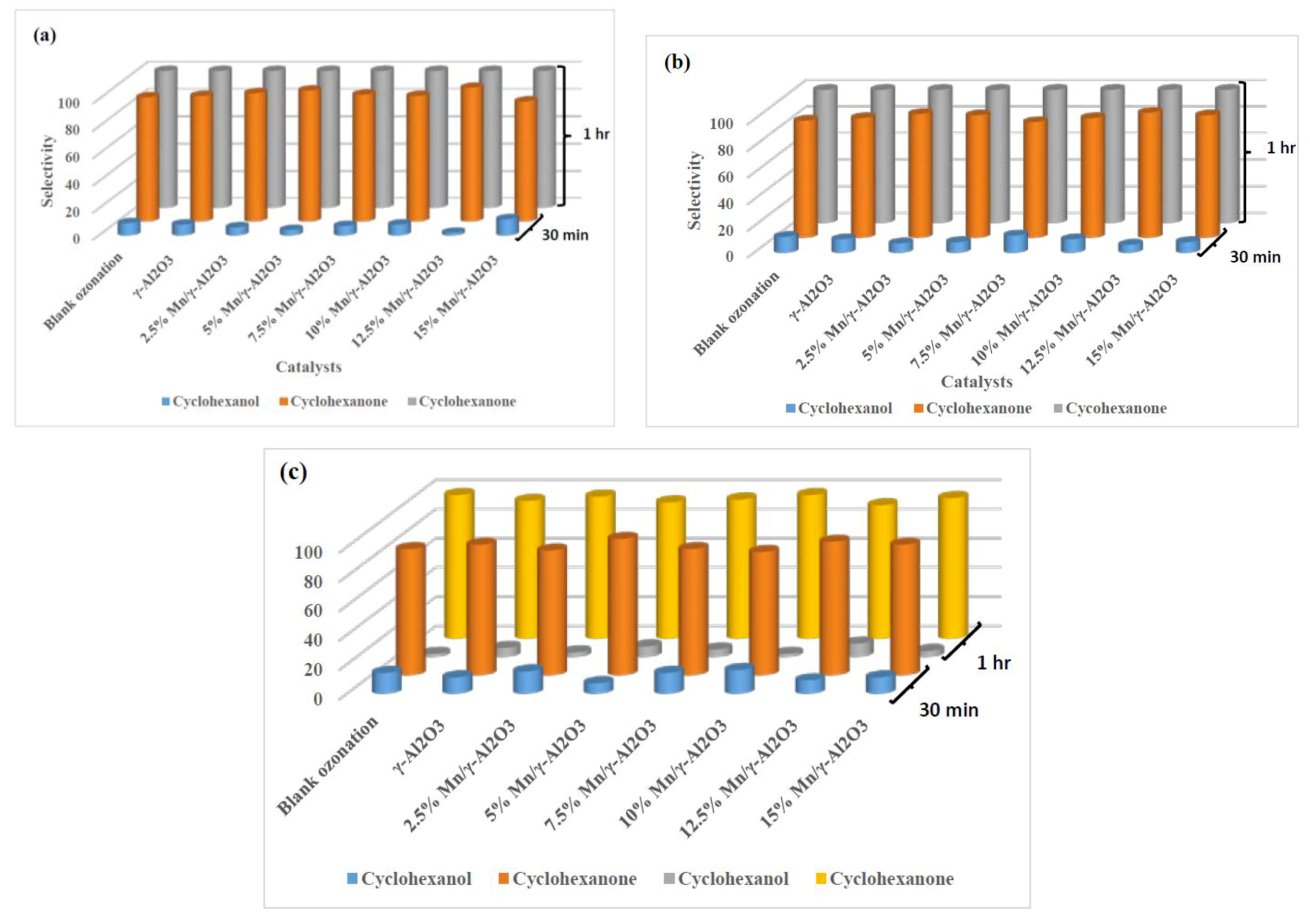
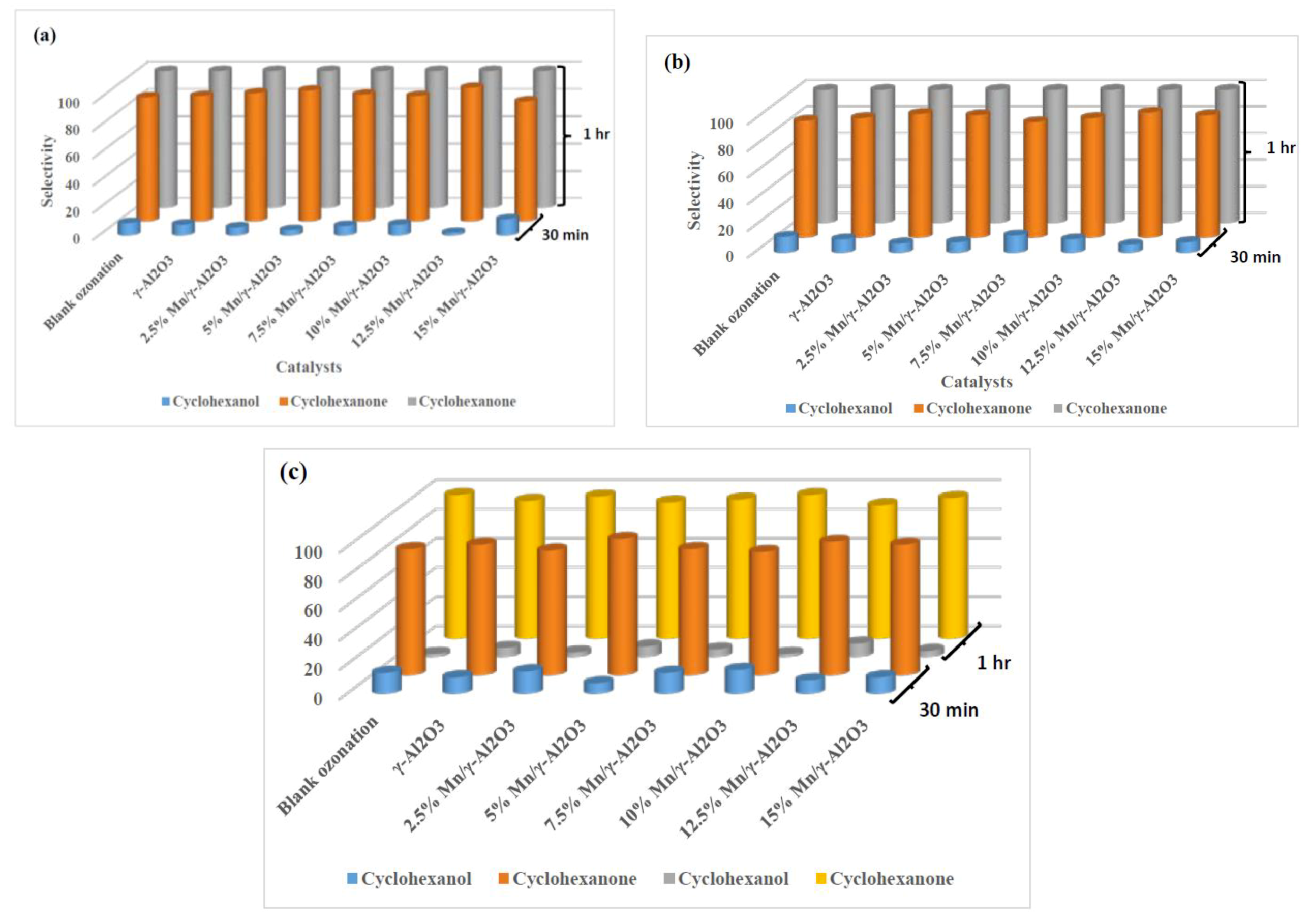
2.3.2. Percentage Selectivity
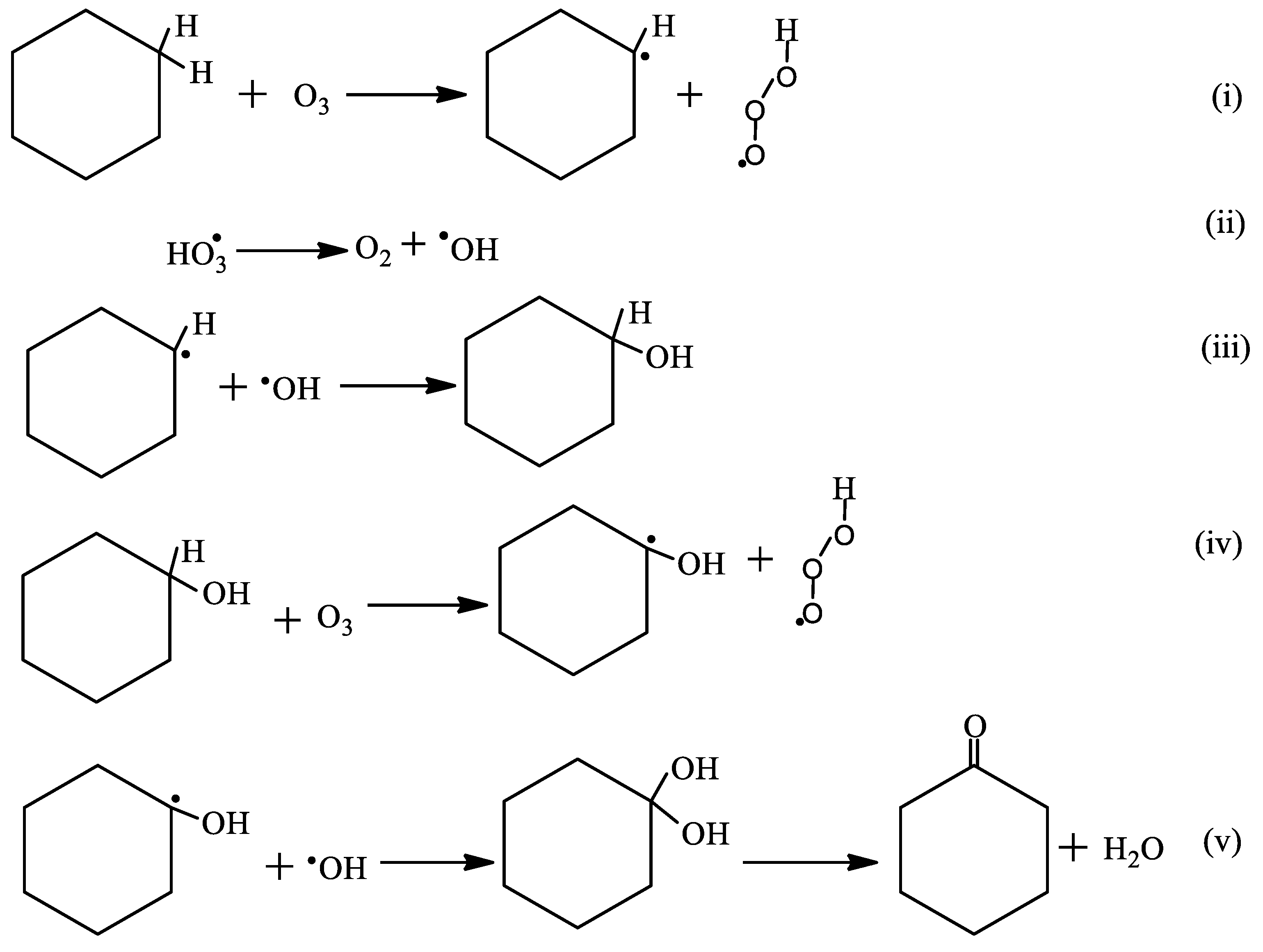
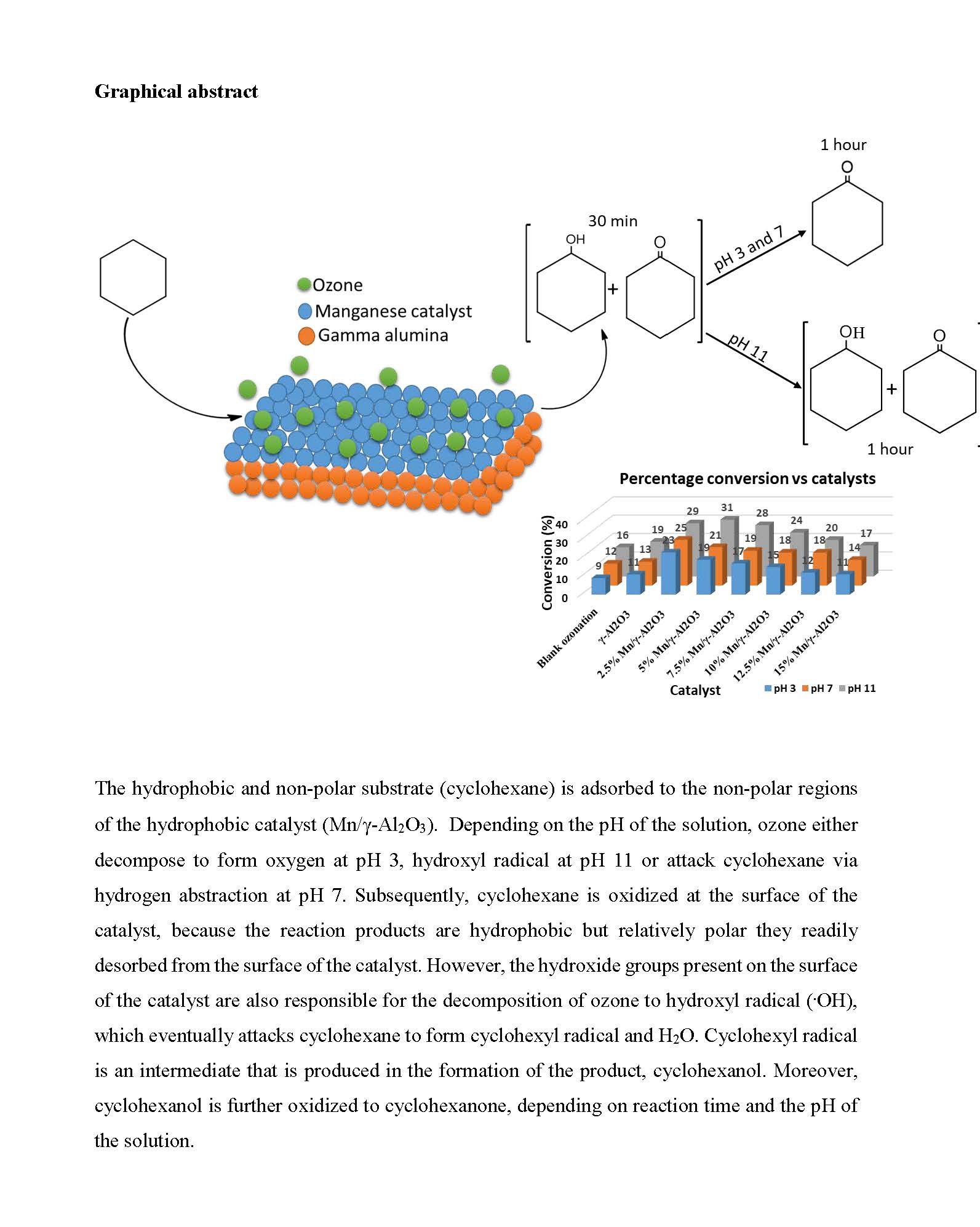
2.3.3. The Effects of the pH
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterisation
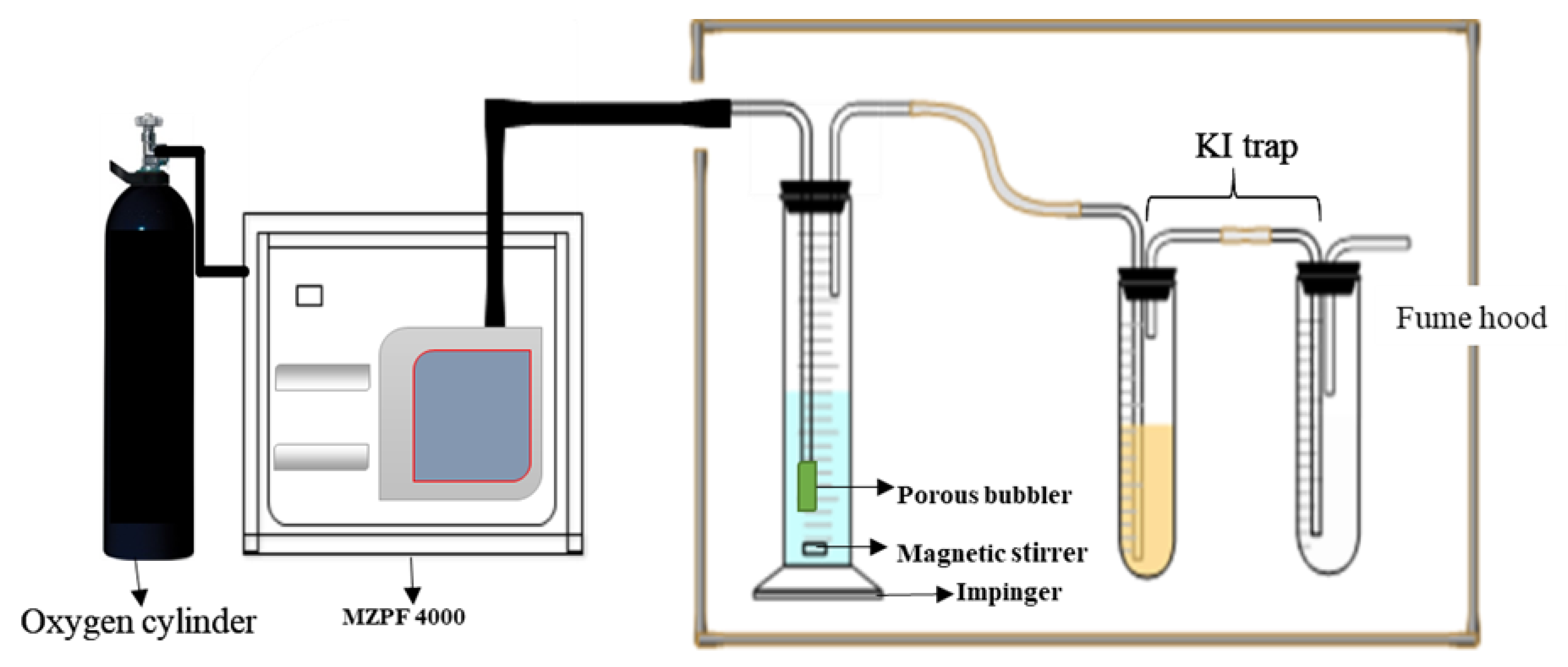
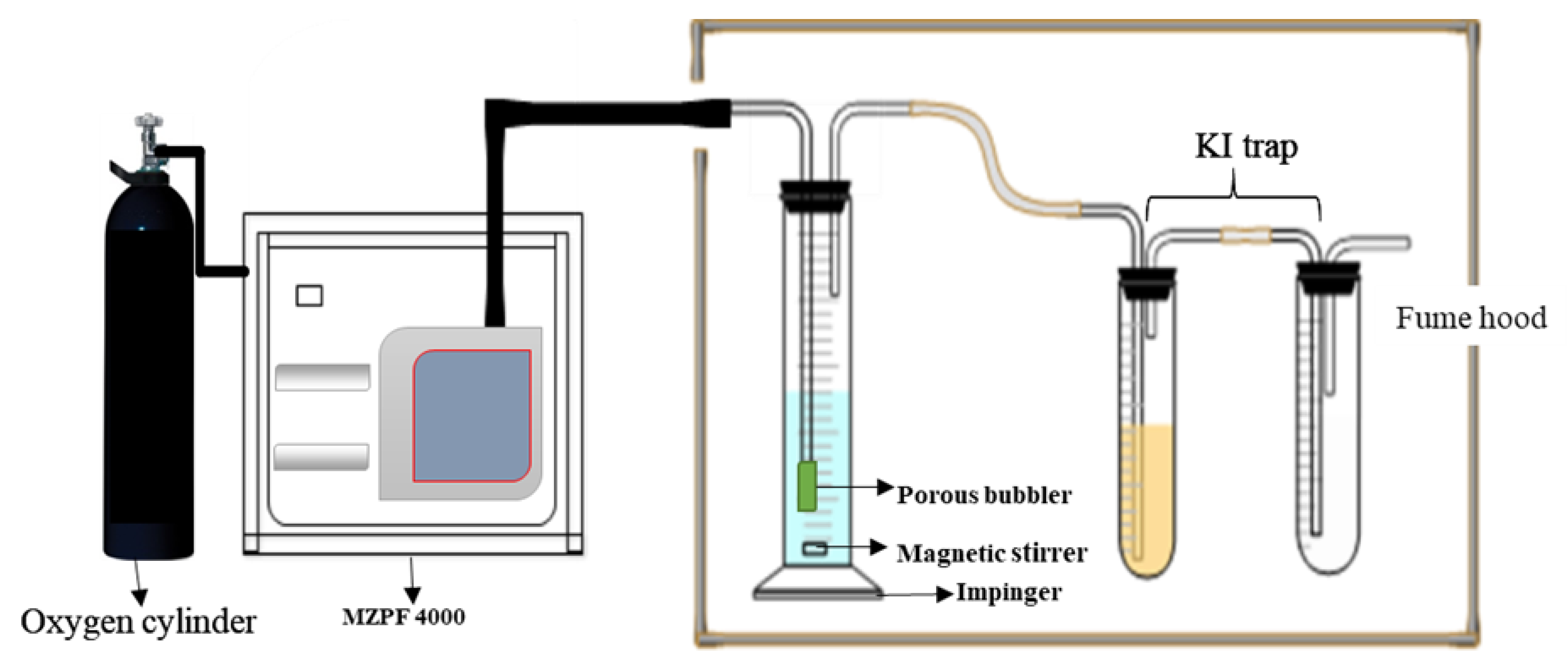
3.3. Oxidation of Cyclohexane
3.4. Reaction Products Characterisation
Gas Chromatography-Mass Spectroscopy (GC-MS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Son, L.; Liu, J.; Lou, W.; Yang, Y.; Wang, F.; Weerakkody, C.; Suib, S.L. Preparation of amorphous copper-chromium oxides catalyst for selective oxidation of cyclohexane. Mol. Catal. 2018, 460, 16–26. [Google Scholar] [CrossRef]
- Xu, L.X.; He, C.H.; Zhu, M.Q.; Fang, S. A highly active Au/Al2O3 catalyst for cyclohexane oxidation using molecular oxygen. Catal. Lett. 2007, 114, 202–205. [Google Scholar] [CrossRef]
- Savita, K.; Priti, S. Solvent-free oxidation of cyclohexane over covalently anchored transition-metal salicylaldimine complexes to α-zirconium phosphate using tert-butylhydroperoxide. J. Mol. Catal. A 2016, 411, 279–289. [Google Scholar]
- Zhou, L.; Xu, J.; Miao, H.; Wang, F.; Li, X. Catalytic oxidation of cyclohexane to cyclohexanol and cyclohexanone over Co3O4 nanocrystals with molecular oxygen. Appl. Catal. 2005, 292, 223–228. [Google Scholar] [CrossRef]
- Unnarkat, A.P.; Sridhar, T.; Wang, H.; Mahajani, S.; Suresh, A.K. Cobalt molebdenum oxide catalysts for selective oxidation of cyclohexane. React. Eng. Kinet. Catal. 2016, 62, 4384–4402. [Google Scholar]
- Raja, R.; Ratnasamy, P. Oxidation of cyclohexane over copper phthalocyanines encapsulated in zeolites. Catal. Lett. 1997, 48, 1–10. [Google Scholar] [CrossRef]
- Fu, Y.; Zhan, W.; Guo, Y.; Wang, Y.; Liu, X.; Guo, Y.; Wang, Y.; Lu, G. Effect of surface functionalization of cerium-doped MCM-48 on its catalytic performance for liquid-phase free solvent oxidation of cyclohexane with molecular oxygen. Microporous Mesoporous Mater. 2015, 214, 101–107. [Google Scholar] [CrossRef]
- Venart, J.E.S. The explosion and its aftermaths. Process Saf. Environ. Prot. 2004, 82, 105–127. [Google Scholar] [CrossRef]
- Sadie, C.; Samuels, D.E.; Obrien, T.P. The characteristics of the explosion of cyclohexane at the Nypro (UK) Flixborough plant on 1st June 1974. J. Occup. Accid. 1977, 1, 203–235. [Google Scholar] [CrossRef]
- Bose, P.; Glaze, W.H.; Maddox, D.S. Degradation of various advanced oxidation processes: 1. Reaction rates. Water Res. 1998, 23, 997–1004. [Google Scholar] [CrossRef]
- Wang, H.C.; Liang, H.S.; Chang, M.B. Oxidation of chlorobenzene using ozone over iron oxide and manganese oxide catalysts. J. Hazard. Mater. 2011, 186, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Walling, C. Fenton’s reagent revisited. Acc. Chem. Res. 1975, 8, 125–131. [Google Scholar] [CrossRef]
- Suh, J.H.; Mohsu, M. A study of relationship between biodegradability enhancement and oxidation of 1, 4-dioxane using ozone and hydrogen peroxide. Water Res. 2014, 126, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.F.; Pelizzetti, E.; Serpone, N. Photocatalyzed destruction of water contaminants. Environ. Sci. Technol. 1991, 25, 1523–1529. [Google Scholar] [CrossRef]
- Masten, S.J.; Davies, S.H. The use of ozonation to degrade organic contaminants in wastewaters. Environ. Sci. Technol. 1994, 28, 180–185. [Google Scholar] [CrossRef]
- Pullabhotla, V.S.R.R.; Jonnalagadda, S.B. Scope of Metal Loaded Microporous ZSM-5 Zeolites in the “Catazone” Process of n-Hexadecane at Moderate Conditions. Ind. Eng. Chem. Res. 2009, 48, 9097–9105. [Google Scholar] [CrossRef]
- Pegis, M.L.; Roberts, J.A.S.; Wasylenko, D.J.; Mader, E.A.; Appel, A.M.; Mayer, J.M. Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylformamide. Inorg. Chem. 2015, 54, 11883–11888. [Google Scholar] [CrossRef]
- Forni, L.; Behnemann, D.; Hart, E.J. Mechanism of the hydroxide ion initiated decomposition of ozone in aqueous solution. J. Phys. Chem. 1982, 86, 255–259. [Google Scholar] [CrossRef]
- Li, W.; Oyama, S.T. Mechanism of Ozone Decomposition on a Manganese Oxide Catalyst. 2. Steady-State and Transient Kinetic Studies. J. Am. Chem. Soc. 1998, 35, 9047–9052. [Google Scholar] [CrossRef]
- Nelieu, S.; Kerhoas, L.; Einhorn, J. Degradation of atrazine into ammeline by combined ozone/hydrogen peroxide treatment in water. Environ. Sci. Technol. 2000, 34, 430–437. [Google Scholar] [CrossRef]
- McGrane, W. Advanced oxidation processes for destruction and enhanced biodegradability of 1,4-dioxane. Chem. Oxid. 1997, 6, 231–245. [Google Scholar]
- Beltran, F.; Acedo, B.; Rivas, J. Use of ozone and hydrogen peroxide to remove alachlor from surface water. Bull. Environ. Contam. Toxicol. J. 1999, 63, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, H.; von Bismarck, R.; Hemmerling, L. Treatment of industrial wastewaters with ozone/hydrogen peroxide. Water Sci. Technol. 1995, 7, 127–134. [Google Scholar] [CrossRef]
- Wu, H.; Pantaleo, G.; Venezia, A.M.; Liotta, L.F. Mesoporous Silica Based Gold Catalysts: Novel Synthesis and Application in Catalytic Oxidation of CO and Volatile Organic Compounds (VOCs). Catalysts 2013, 3, 774–793. [Google Scholar] [CrossRef]
- Lan, S.; Guo, N.; Gan, S. Facile preparation of hierarchical hollow structure gamma alumina and study of its adsorption capacity. Appl. Catal. 2012, 97, 83–90. [Google Scholar] [CrossRef]
- Karim, M.R.; Rahman, M.A.; Ito, M. synthesis of gamma alumina catalysts and surface characterisation. Open Colloid Sci. J. 2011, 4, 32–36. [Google Scholar] [CrossRef]
- Ncanana, Z.S.; Pullabhotla, V.S.R.R. Ozone Initiated Oxidation of Cresol Isomers Using γ-Al2O3 and SiO2 as Adsorbents. Catal. Lett. 2018, 148, 1535–1546. [Google Scholar] [CrossRef]
- Maldonado, C.; Lucio-Ortiz, C.J.; De la Rosa, J.R.; Ramírez, A.H. Synthesis and characterization of Fe doped mesoporous Al2O3 by sol–Gel method and its use in trichloroethylene combustion. J. Sol Gel Sci. Technol. 2011, 58, 374–384. [Google Scholar]
- Syroezhko, A.M.; Begak, O.Y.; Proskuryakov, V.P. Catalytic Oxidation of Cyclohexane with Ozone-Oxygen Mixtures. Russ. J. Appl. Chem. 2004, 77, 51–56. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The Power of Single-Atom Catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Escaño, M.C.S.; Nakanishi, H.; Kasai, H.; Maekawa, H.; Osumi, K.; Sato, K. DFT+ U study on the oxygen adsorption and dissociation on CeO2-supported platinum cluster. Appl. Surf. Sci. 2014, 288, 244–250. [Google Scholar] [CrossRef]
- Védrine, J.C. Heterogeneous Catalysis on Metal Oxides. Catalysts 2017, 7, 341. [Google Scholar]
- Ross, J.R.H. Heterogeneous Catalysis; Elsevier: Amsterdam, The Netherlands, 2012; pp. 47–64. [Google Scholar]
- Conte, M.; Liu, X.; Murphy, D.M.; Whiston, K.; Hutchings, G.J. Cyclohexane oxidation using Au/MgO: An investigation of the reaction mechanism. Phys. Chem. Chem. Phys. 2012, 14, 16279–16285. [Google Scholar] [CrossRef] [PubMed]
- Kruanak, K.; Jarusutthirak, C. Degradation of 2,4,6-trichlorophenol in synthetic wastewater by catalytic 450 ozonation using alumina supported nickel oxides. J. Environ. Chem. Eng. 2019, 7, 102825. [Google Scholar] [CrossRef]
- Pullabhotla, V.S.R.R.; Jonnalagadda, S.B. Scope of Metal Loaded Microporous Zeolite-Y as Catalyst in Ozone Initiated Oxidation of n-hexadecane. J. Adv. Oxid. Technol. 2009, 12, 179–187. [Google Scholar] [CrossRef]
- Pullabhotla, V.S.R.R.; Rahman, A.; Jonnalagadda, S.B. Selective catalytic Knoevenagel condensation by Ni-SiO2 supported heterogeneous catalysts: An environmental benign approach. Catal. Commun. 2009, 10, 365–369. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Theoretical Mn wt % | ICP Mn wt % | EDX Mn wt% * |
|---|---|---|---|
| 2.5% Mn/γ-Al2O3 | 2.50 | 2.48 | 2.33 |
| 5% Mn/γ-Al2O3 | 5.00 | 5.39 | 5.9 |
| 7.5 % Mn/γ-Al2O3 | 7.50 | 7.46 | 7.57 |
| 10% Mn/γ-Al2O3 | 10.0 | 9.18 | 10.23 |
| 12.5% Mn/γ-Al2O3 | 12.5 | 12.48 | 12.42 |
| 15% Mn/γ-Al2O3 | 15.0 | 14.94 | 15.64 |
| Catalysts | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) |
|---|---|---|---|
| γ-Al2O3 | 295.0 | - | - |
| 2.5% Mn/γ-Al2O3 | 167.6 | 0.3827 | 9.13 |
| 7.5% Mn/γ-Al2O3 | 107.8 | 0.3022 | 11.21 |
| Catalyst | Reaction Time | Conversion (%) | Selectivity (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cyclohexanol | Cyclohexanone | |||||||||
| pH 3 | pH 7 | pH 11 | pH 3 | pH 7 | pH 11 | pH 3 | pH 7 | pH 11 | ||
| Blank ozonation | 30 min | 3 | 6 | 9 | 9 | 12 | 14 | 91 | 88 | 86 |
| 1 h | 9 | 12 | 16 | ----- | ----- | 2 | 100 | 100 | 98 | |
| γ-Al2O3 | 30 min | 7 | 9 | 11 | 8 | 10 | 11 | 92 | 90 | 89 |
| 1 h | 11 | 13 | 19 | ----- | ----- | 6 | 100 | 100 | 94 | |
| 2.5% Mn/γ-Al2O3 | 30 min | 9 | 15 | 17 | 6 | 7 | 15 | 94 | 93 | 95 |
| 1 h | 23 | 29 | 28 | ----- | ----- | 3 | 100 | 100 | 97 | |
| 5% Mn/γ-Al2O3 | 30 min | 7 | 11 | 23 | 4 | 8 | 7 | 86 | 92 | 93 |
| 1 h | 19 | 21 | 31 | ----- | ----- | 7 | 100 | 100 | 93 | |
| 7.5% Mn/γ-Al2O3 | 30 min | 9 | 8 | 19 | 7 | 13 | 13 | 93 | 87 | 87 |
| 1 h | 17 | 19 | 28 | ----- | ----- | 5 | 100 | 100 | 95 | |
| 10% Mn/γ-Al2O3 | 30 min | 6 | 9 | 16 | 8 | 10 | 6 | 92 | 90 | 94 |
| 1 h | 14 | 18 | 24 | ----- | ----- | 2 | 100 | 100 | 98 | |
| 12.5% Mn/γ-Al2O3 | 30 min | 8 | 11 | 14 | 2 | 6 | 9 | 98 | 94 | 91 |
| 1 h | 12 | 18 | 20 | ----- | ----- | 8 | 100 | 100 | 92 | |
| 15% Mn/γ-Al2O3 | 30 min | 5 | 7 | 15 | 12 | 8 | 11 | 88 | 92 | 89 |
| 1 h | 11 | 14 | 17 | ----- | ----- | 4 | 100 | 100 | 96 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mkhondwane, S.T.; Pullabhotla, V.S.R. Highly Selective pH-Dependent Ozonation of Cyclohexane over Mn/γ-Al2O3 Catalysts at Ambient Reaction Conditions. Catalysts 2019, 9, 958. https://doi.org/10.3390/catal9110958
Mkhondwane ST, Pullabhotla VSR. Highly Selective pH-Dependent Ozonation of Cyclohexane over Mn/γ-Al2O3 Catalysts at Ambient Reaction Conditions. Catalysts. 2019; 9(11):958. https://doi.org/10.3390/catal9110958
Chicago/Turabian StyleMkhondwane, Siphumelele Thandokwazi, and Viswanadha Srirama Rajasekhar Pullabhotla. 2019. "Highly Selective pH-Dependent Ozonation of Cyclohexane over Mn/γ-Al2O3 Catalysts at Ambient Reaction Conditions" Catalysts 9, no. 11: 958. https://doi.org/10.3390/catal9110958
APA StyleMkhondwane, S. T., & Pullabhotla, V. S. R. (2019). Highly Selective pH-Dependent Ozonation of Cyclohexane over Mn/γ-Al2O3 Catalysts at Ambient Reaction Conditions. Catalysts, 9(11), 958. https://doi.org/10.3390/catal9110958