Fischer-Tropsch Synthesis: Cd, In and Sn Effects on a 15%Co/Al2O3 Catalyst



 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. BET and Porosity Measurements
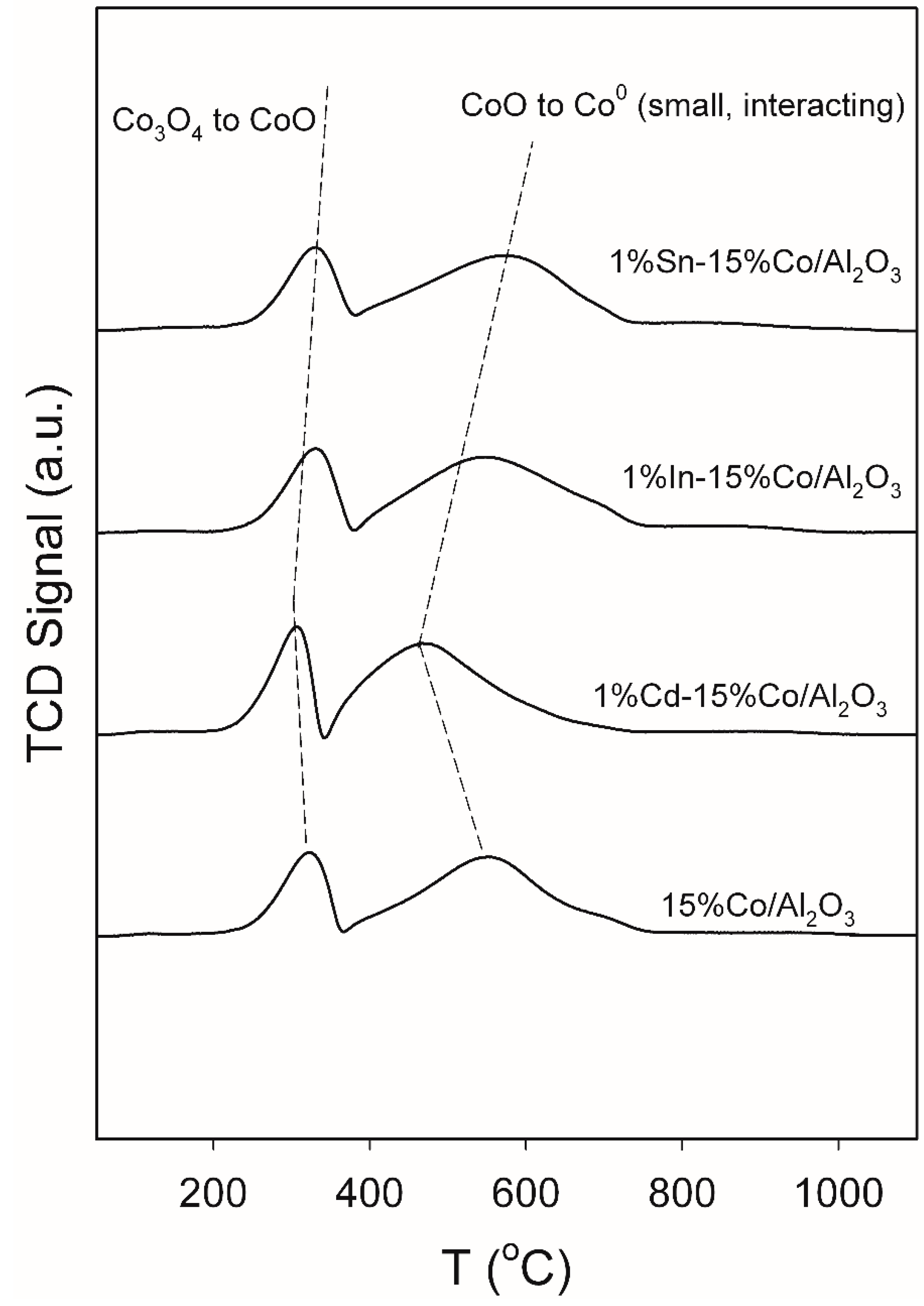
2.2. TPR and Hydrogen Chemisorption/Pulse Reoxidation
2.3. NH3 TPD Study of Cd, In and Sn Promoted Catalysts
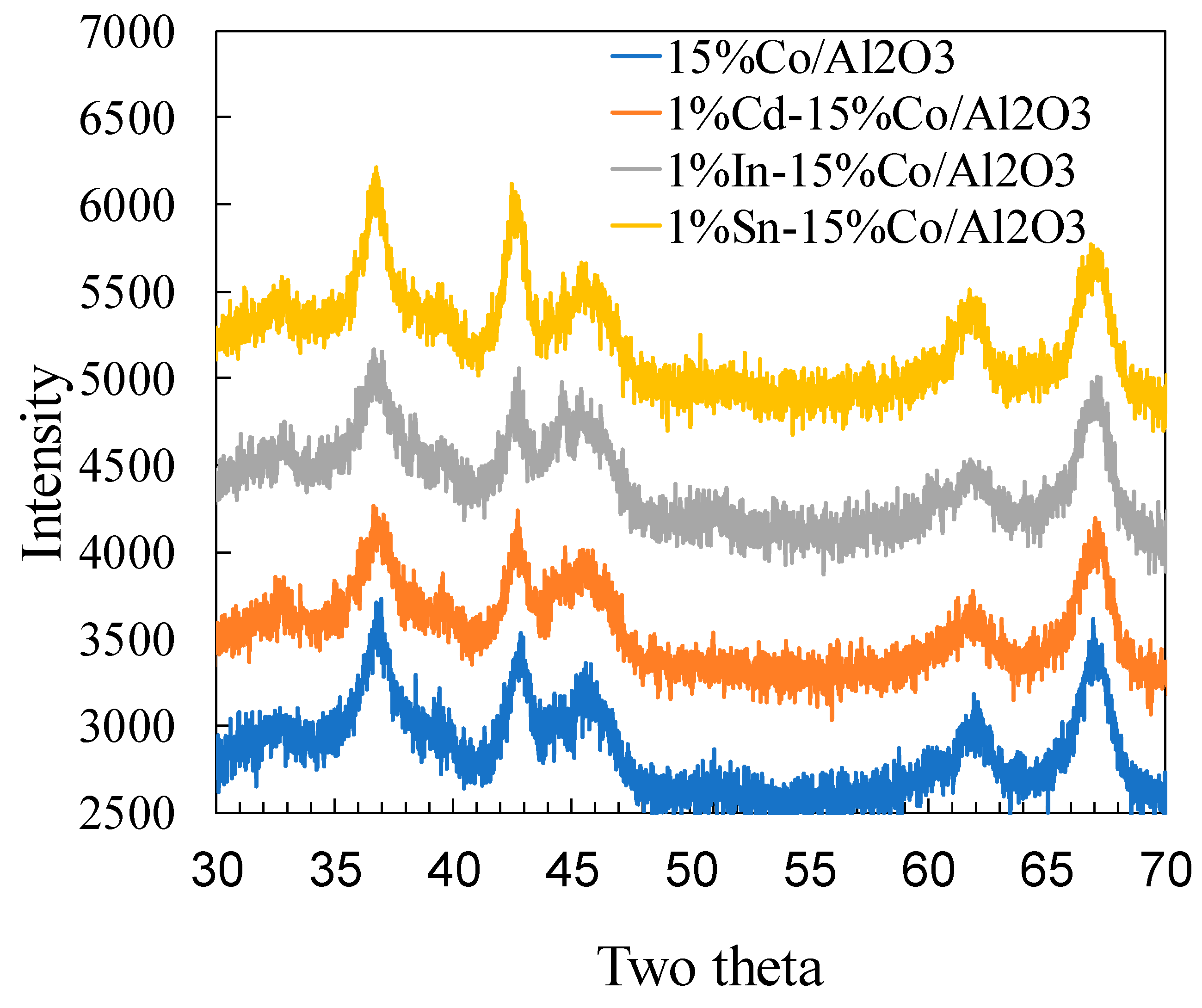
2.4. XRD Study of Unpromoted and Cd, In and Sn Promoted Cobalt Catalysts
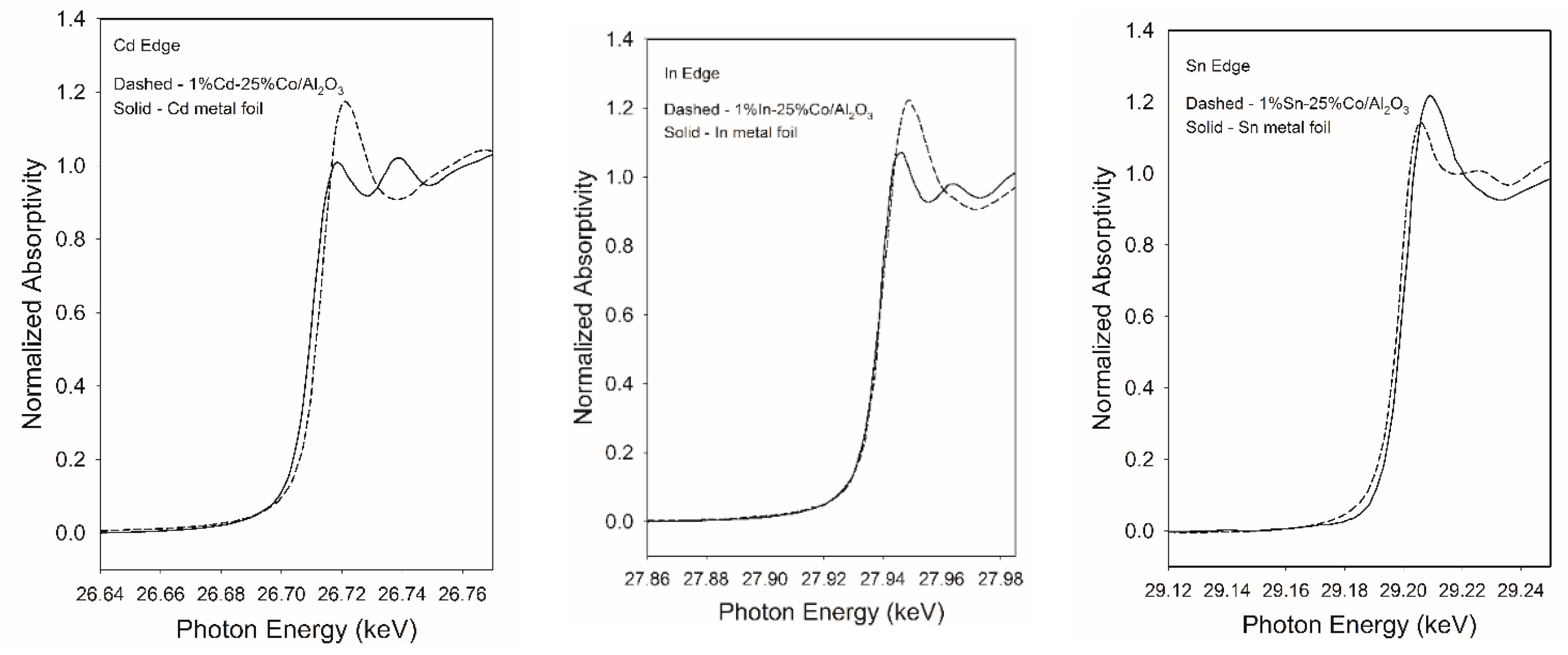
2.5. XANES Study of Cd, In and Sn Promoted Catalysts
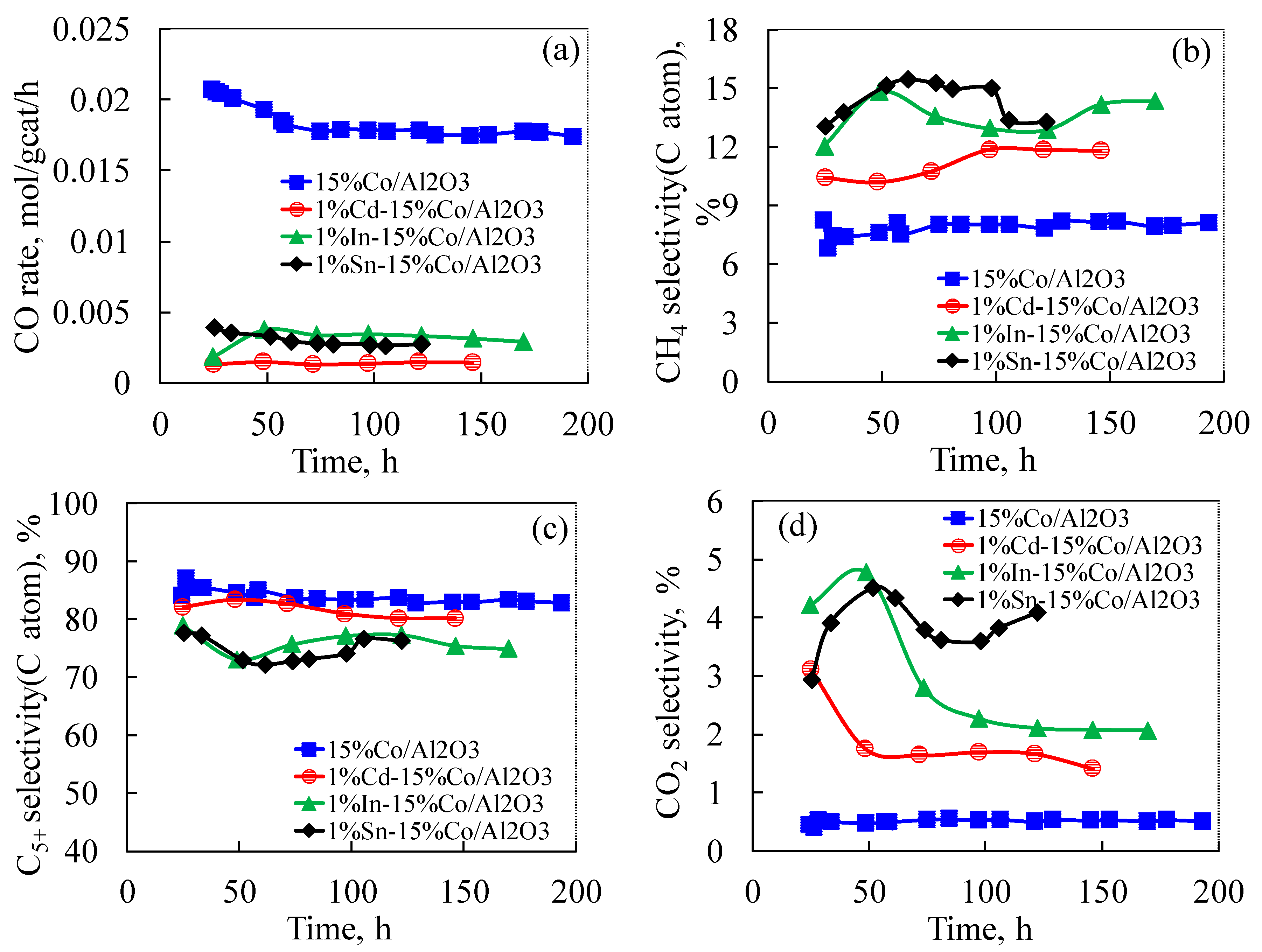
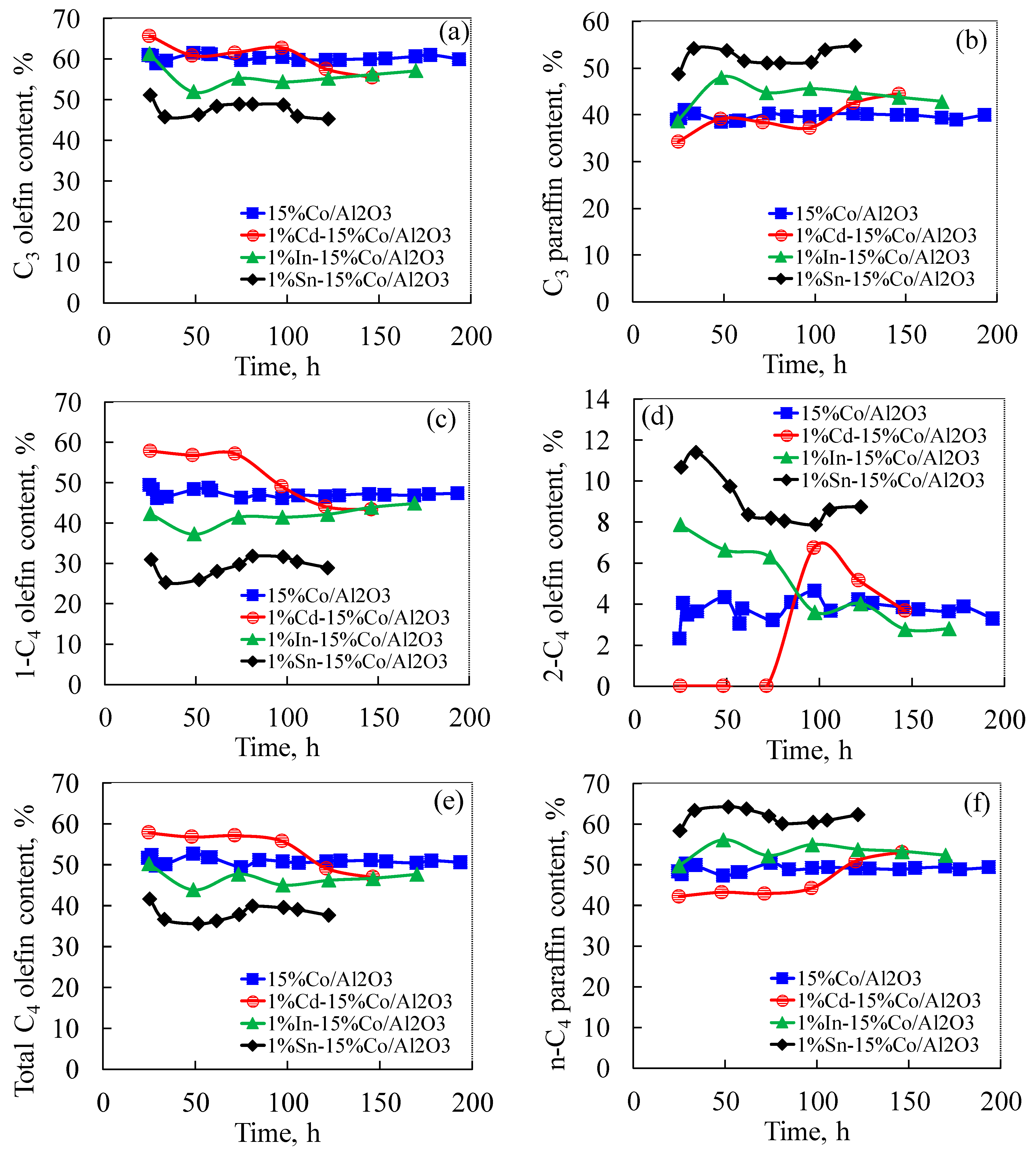
2.6. Effect of Cd, In and Sn Additives on Fischer-Tropsch Synthesis
3. Experimental
3.1. Catalyst Preparation
3.2. BET Surface Area and Porosity Measurements
3.3. Temperature Programmed Reduction
3.4. Hydrogen Chemisorption/Pulse Reoxidation
3.5. NH3 Temperature Programmed Desorption
3.6. X-ray Absorption Near Edge Spectroscopy (XANES)
3.7. X-ray Diffraction (XRD)
3.8. Catalytic Activity Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Espinoza, R.L.; van Berge, P.J.; Bolder, F.H. Catalysts. U.S Patent 5,733,839, 31 March 1998. [Google Scholar]
- van Berge, P.J.; Barradas, S.; van de Loosdrecht, J.; Visagie, J.L. Advances in the cobalt catalyzed Fischer-Tropsch synthesis. Erdoel Erdgas Kohle 2001, 117, 138–142. [Google Scholar]
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer-Tropsch synthesis: Support, loading and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Jacobs, G.; Ji, Y.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer-Tropsch synthesis: Temperature programmed EXAFS/XANES investigation of the influence of support type, cobalt loading, and noble metal promoter addition to the reduction behavior of cobalt oxide particles. Appl. Catal. A Gen. 2007, 333, 177–191. [Google Scholar] [CrossRef]
- Jacobs, G.; Ribeiro, M.C.; Ma, W.; Ji, Y.; Khalid, S.; Sumodjo, P.T.A.; Davis, B.H. Group 11 (Cu, Ag, Au) promotion of 15%Co/Al2O3 Fischer-Tropsch catalysts. Appl. Catal. A: Gen. 2009, 361, 137–151. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.; Davis, B.H. Influence of reduction promoters on stability of cobalt/γ-alumina Fischer-Tropsch synthesis catalysts. Catalysts 2014, 4, 49–76. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Keogh, R.A.; Bukur, D.B.; Davis, B.H. Fischer-Tropsch synthesis: Effect of Pd, Pt, Re, and Ru noble metal promoters on the activity and selectivity of a 25%Co/Al2O3 catalyst. Appl. Catal. A Gen. 2012, 437–438, 1–9. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Gao, P.; Jermwongratanachai, T.; Shafer, W.D.; Pendyala, V.R.R.; Chia, H.Y.; Klettlinger, J.L.S.; Davis, B.H. Fischer-Tropsch synthesis: Pore size and Zr promotional effects on the activity and selectivity of 25%Co/Al2O3 catalysts. Appl. Catal. A Gen. 2014, 475, 314–324. [Google Scholar] [CrossRef]
- Chonco, Z.H.; Nabaho, D.; Claeys, M.; van Steen, E. The role of reduction promoters in Fischer-Tropsch catalysts for the production of liquid fuels. In Proceedings of the 23rd Meeting of the North American Catalysis Society, Louisville, KY, USA, 2–7 June 2013. [Google Scholar]
- Hilmen, A.M.; Schanke, D.; Holmen, A. TPR study of the mechanism of rhenium promotion of alumina-supported cobalt Fischer-Tropsch catalysts. Catal. Lett. 1996, 38, 143–147. [Google Scholar] [CrossRef]
- Cook, K.M.; Perez, H.D.; Bartholomew, C.H.; Hecker, W.C. Effect of promoter deposition order on platinum-, ruthenium-, or rhenium-promoted cobalt Fischer-Tropsch catalysts. Appl. Catal. A Gen. 2014, 482, 275–286. [Google Scholar] [CrossRef]
- Vada, S.; Hoff, A.; Ådnanes, E.; Schanke, D.; Holmen, A. Fischer-Tropsch synthesis on supported cobalt catalysts promoted by platinum and rhenium. Top. Catal. 1995, 2, 155–162. [Google Scholar] [CrossRef]
- Cook, K.M.; Hecker, W.C. Reducibility of alumina-supported cobalt Fischer-Tropsch catalysts: Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size. Appl. Catal. A Gen. 2012, 449, 69–80. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Maillot, J.C.; Davis, B.H. Fischer-Tropsch synthesis: Study of the promotion of Pt on the reduction property of Co/Al2O3 catalysts by in situ EXAFS of Co K and Pt LIII edges and XPS. J. Synchrotron Radiat. 2004, 11, 414–422. [Google Scholar] [CrossRef]
- Ma, W.; Ding, Y.J.; Lin, L.W. Fischer-Tropsch Synthesis over Activated-Carbon-Supported Cobalt Catalysts: Effect of Co Loading and Promoters on Catalyst Performance. Ind. Eng. Chem. Res. 2004, 43, 2391–2398. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.; Gao, P.; Todic, B.; Bhatelia, T.; Bukur, D.B.; Khalid, S.; Davis, B.H. Fischer-Tropsch Synthesis: Differences Observed in Local Atomic Structure and Selectivity with Pd Compared to Typical Promoters (Pt, Re, Ru) of Co/Al2O3 Catalysts. Top. Catal. 2012, 55, 811–817. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Ma, W.; Shafer, W.D.; Gnanamani, M.K.; Gao, P.; Kitiyanan, B.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H.; et al. Studies on the regeneration of sulfur-poisoned NOx storage and reduction catalysts, including a Ba composite oxide. Appl. Catal. A Gen. 2013, 464–465, 165–180. [Google Scholar] [CrossRef]
- Guerrero-Ruiz, A.; Sepulveda-Escribano, A.; Rodriguez-Ramos, I. Carbon monoxide hydrogenation over carbon supported cobalt or ruthenium catalysts. Promoting effects of magnesium, vanadium and cerium oxides. Appl. Catal. A Gen. 1994, 120, 71–83. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, H.; Liew, K.Y.; Li, J. Effect of magnesia on alumina-supported cobalt Fischer-Tropsch synthesis catalysts. J. Mol. Catal. 2005, 237, 172–181. [Google Scholar] [CrossRef]
- Kikuchi, E.; Sorita, R.; Takahashi, H.; Matsuda, T. Catalytic performances of cobalt-based ultrafine particles prepared by chemical reduction in slurry-phase Fischer-Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 121–128. [Google Scholar] [CrossRef]
- Van der Riet, M.; Hutchings, G.J.; Copperthwaite, R.G. Selective formation of C3 hydrocarbons from carbon monoxide and hydrogen using cobalt-manganese oxide catalysts. J. Chem. Soc. Chem. Commun. 1986, 10, 798–799. [Google Scholar] [CrossRef]
- Mendes, F.M.T.; Perez, C.A.C.; Noronha, F.B.; Schmal, M. TPSR of CO hydrogenation on Co/Nb2O5/Al2O3 catalysts. Catal. Today 2005, 101, 45–50. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Graham, U.M.; Pendyala, V.R.R.; Martinelli, M.; MacLennan, A.; Hu, Y.; Davis, B.H. Effect of sequence of P and Co addition over silica for Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2017, 538, 190–198. [Google Scholar] [CrossRef]
- Huffman, G.P.; Shah, N.; Zhao, J.M.; Huggins, F.E.; Hoost, T.E.; Halvorsen, S.; Goodwin, J.G. In situ XAFS investigation of K-promoted Co catalysts. J. Catal. 1995, 151, 17–25. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Keogh, R.A.; Yen, C.H.; Klettlinger, J.L.S.; Davis, B.H. Fischer-Tropsch synthesis: Effect of Pt promoter on activity, selectivities to hydrocarbons and oxygenates, and kinetic parameters over 15%Co/Al2O3. In Synthetic Liquids Production and Refining; Klerk, A.D., King, D.L., Eds.; American Chemical Society: Washington, DC, USA, 2011; pp. 127–153. [Google Scholar]
- Cho, H.J.; Dornath, P.; Fan, W. Synthesis of Hierarchical Sn-MFI as Lewis Acid Catalysts for Isomerization of Cellulosic Sugars. ACS Catal. 2014, 4, 2029–2037. [Google Scholar] [CrossRef]
- Srinivasan, R.; Rice, L.A.; Davis, B.H. Electron microdiffraction study of platinum-tin-alumina reforming catalysts. J. Catal. 1991, 129, 257–268. [Google Scholar] [CrossRef]
- Passos, F.B.; Aranda, D.A.G.; Schmal, M. Characterization and catalytic activity of bimetallic Pt-In/Al2O3 and Pt-Sn/Al2O3 catalysts. J. Catal. 1998, 178, 478–488. [Google Scholar] [CrossRef]
- Coleto, I.; Roldan, R.; Jimenez-Sanchidrian, C.; Gomez, J.P.; Romero-Salguero, F.J. Transformation of α-olefins over Pt-M (M = Re, Sn, Ge) supported chlorinated alumina catalysts. Fuel 2007, 86, 1000–1007. [Google Scholar] [CrossRef]
- Mazzieri, V.A.; Grau, J.M.; Vera, C.R.; Yori, J.C.; Parera, J.M.; Pieck, C.L. Role of Sn in Pt-Re-Sn/Al2O3-Cl catalysts for naphtha reforming. Catal. Today 2005, 107–108, 643–650. [Google Scholar] [CrossRef]
- Ressler, T. WinXAS: A program for X-ray absorption spectroscopy data analysis under MS-Windows. J. Synchrotron Radiat. 1998, 5, 118–122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support/Catalyst | BET SA | Pore Volume (BJH Adsorp) | Average Pore Radius (BJH Adsorp) | H2 Desorbed | Co Reduction Degree | Apparent Corrected Co Disperion | Apparent Corrected Average Co Diamter |
|---|---|---|---|---|---|---|---|
| (m2/g) | (cm3/g) | (nm) | (µmol/gcat) | (%) | (%) | (nm) | |
| 15% Co/Al2O3-150 | 116.2 | 0.3096 | 4.72 | 57.3 | 48.5 | 9.28 | 11.1 |
| 1%Cd 15% Co/Al2O3 | 110.4 | 0.3024 | 4.75 | 52.6 | 58.8 | 7.19 | 14.3 |
| 1%In 15% Co/Al2O3 | 111.6 | 0.3053 | 4.73 | 45.6 | 56.6 | 6.34 | 16.3 |
| 1%Sn 15% Co/Al2O3 | 115.0 | 0.3051 | 4.68 | 36.6 | 46.2 | 6.23 | 13.8 |
| TOS | CO Conversion | GHSV | H2/CO | Selectivity (C atom %) | CO2 Selectivity | ||
|---|---|---|---|---|---|---|---|
| (hrs) | (%) | (NL/gcat/h) | CH4 | C2-C4 | C5+ | (%) | |
| 15%Co/Al2O3-150 | |||||||
| 11–57 | 48–51 | 2.78 | 2.10 | 7.44 | 7.02 | 85.54 | 0.47 |
| 59–193 | 50–52 | 2.44 | 2.10 | 8.00 | 8.63 | 83.37 | 0.52 |
| 1%Cd-15%Co/Al2O3-150 | |||||||
| 25–72 | 18–20 | 0.50 | 2.10 | 10.45 | 6.88 | 82.67 | 2.17 |
| 97–146 | 38–40 | 0.26 | 2.10 | 11.50 | 7.39 | 81.11 | 1.59 |
| 1%In-15%Co/Al2O3-150 | |||||||
| 49–97 | 47–52 | 0.50 | 2.10 | 13.77 | 10.95 | 75.28 | 3.28 |
| 146–170 | 40–44 | 0.50 | 2.10 | 14.25 | 10.59 | 75.16 | 2.07 |
| 1%Sn-15%Co/Al2O3-150 | |||||||
| 25.3 | 27.07 | 1.00 | 2.10 | 13.05 | 9.31 | 77.64 | 2.93 |
| 33–52 | 47–51 | 0.51 | 2.27 | 13.46 | 9.98 | 76.56 | 4.21 |
| 74–98 | 37 | 0.51 | 2.17 | 14.56 | 11.34 | 74.09 | 3.83 |
| 106–122 | 47 | 0.47 | 2.11 | 14.06 | 10.92 | 75.01 | 3.59 |
| TOS | CO | Olefin Content (%) | Total C4 Olefin | Paraffin Content (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| (hrs) | conv. (%) | 1-C2 | 2-C3 | 1-C4 | 2-C4 | Content (%) | C2 | C3 | C4 |
| 15%Co/Al2O3-150 | |||||||||
| 11–57 | 48–51 | 7.43 | 60.67 | 48.09 | 3.47 | 51.56 | 92.57 | 39.33 | 48.44 |
| 59–193 | 50–52 | 7.38 | 60.24 | 46.96 | 3.83 | 50.79 | 92.62 | 39.76 | 49.21 |
| 1%Cd-15%Co/Al2O3-150 | |||||||||
| 25–72 | 18–20 | 27.89 | 62.72 | 57.25 | 0.00 | 57.25 | 72.11 | 37.28 | 42.75 |
| 97–146 | 38–40 | 13.18 | 58.59 | 45.43 | 5.19 | 50.62 | 86.82 | 41.41 | 49.38 |
| 1%In-15%Co/Al2O3-150 | |||||||||
| 49–97 | 47–52 | 5.52 | 53.87 | 40.06 | 5.51 | 45.57 | 94.48 | 46.13 | 54.43 |
| 146–170 | 40–44 | 6.79 | 56.68 | 44.42 | 2.78 | 47.20 | 93.21 | 43.32 | 52.80 |
| 1%Sn-15%Co/Al2O3-150 | |||||||||
| 25.3 | 27.07 | 10.61 | 51.23 | 30.90 | 10.69 | 41.59 | 89.39 | 48.77 | 58.41 |
| 33–52 | 47–51 | 9.30 | 46.07 | 25.57 | 10.58 | 36.15 | 90.70 | 53.93 | 63.85 |
| 74–98 | 37 | 8.37 | 47.78 | 28.78 | 9.03 | 37.81 | 91.63 | 52.22 | 62.19 |
| 106–122 | 47 | 7.30 | 47.59 | 30.10 | 8.53 | 38.63 | 92.70 | 52.41 | 61.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Jacobs, G.; Shafer, W.D.; Ji, Y.; Klettlinger, J.L.S.; Khalid, S.; Hopps, S.D.; Davis, B.H. Fischer-Tropsch Synthesis: Cd, In and Sn Effects on a 15%Co/Al2O3 Catalyst. Catalysts 2019, 9, 862. https://doi.org/10.3390/catal9100862
Ma W, Jacobs G, Shafer WD, Ji Y, Klettlinger JLS, Khalid S, Hopps SD, Davis BH. Fischer-Tropsch Synthesis: Cd, In and Sn Effects on a 15%Co/Al2O3 Catalyst. Catalysts. 2019; 9(10):862. https://doi.org/10.3390/catal9100862
Chicago/Turabian StyleMa, Wenping, Gary Jacobs, Wilson D. Shafer, Yaying Ji, Jennifer L. S. Klettlinger, Syed Khalid, Shelley D. Hopps, and Burtron H. Davis. 2019. "Fischer-Tropsch Synthesis: Cd, In and Sn Effects on a 15%Co/Al2O3 Catalyst" Catalysts 9, no. 10: 862. https://doi.org/10.3390/catal9100862
APA StyleMa, W., Jacobs, G., Shafer, W. D., Ji, Y., Klettlinger, J. L. S., Khalid, S., Hopps, S. D., & Davis, B. H. (2019). Fischer-Tropsch Synthesis: Cd, In and Sn Effects on a 15%Co/Al2O3 Catalyst. Catalysts, 9(10), 862. https://doi.org/10.3390/catal9100862