Enantioselective Mannich Reaction Promoted by Chiral Phosphinoyl-Aziridines


 and
and 
Abstract

1. Introduction
2. Results and Discussion
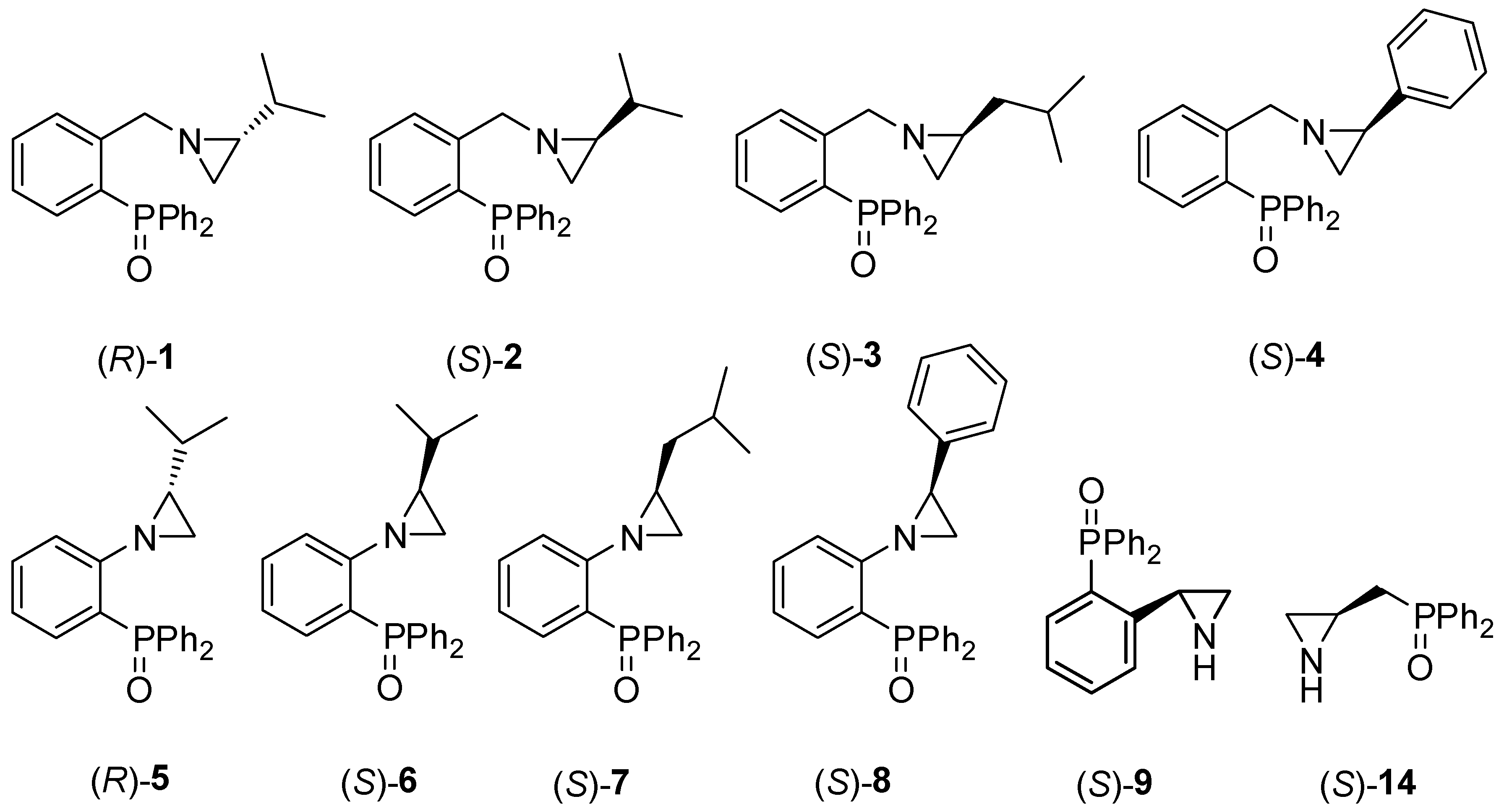
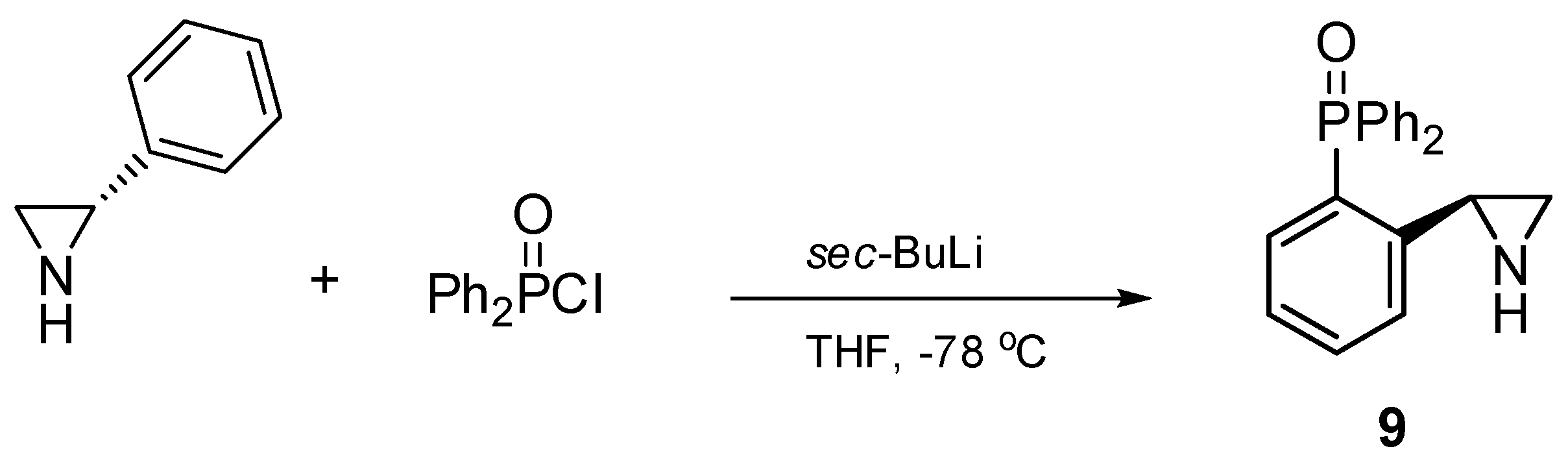
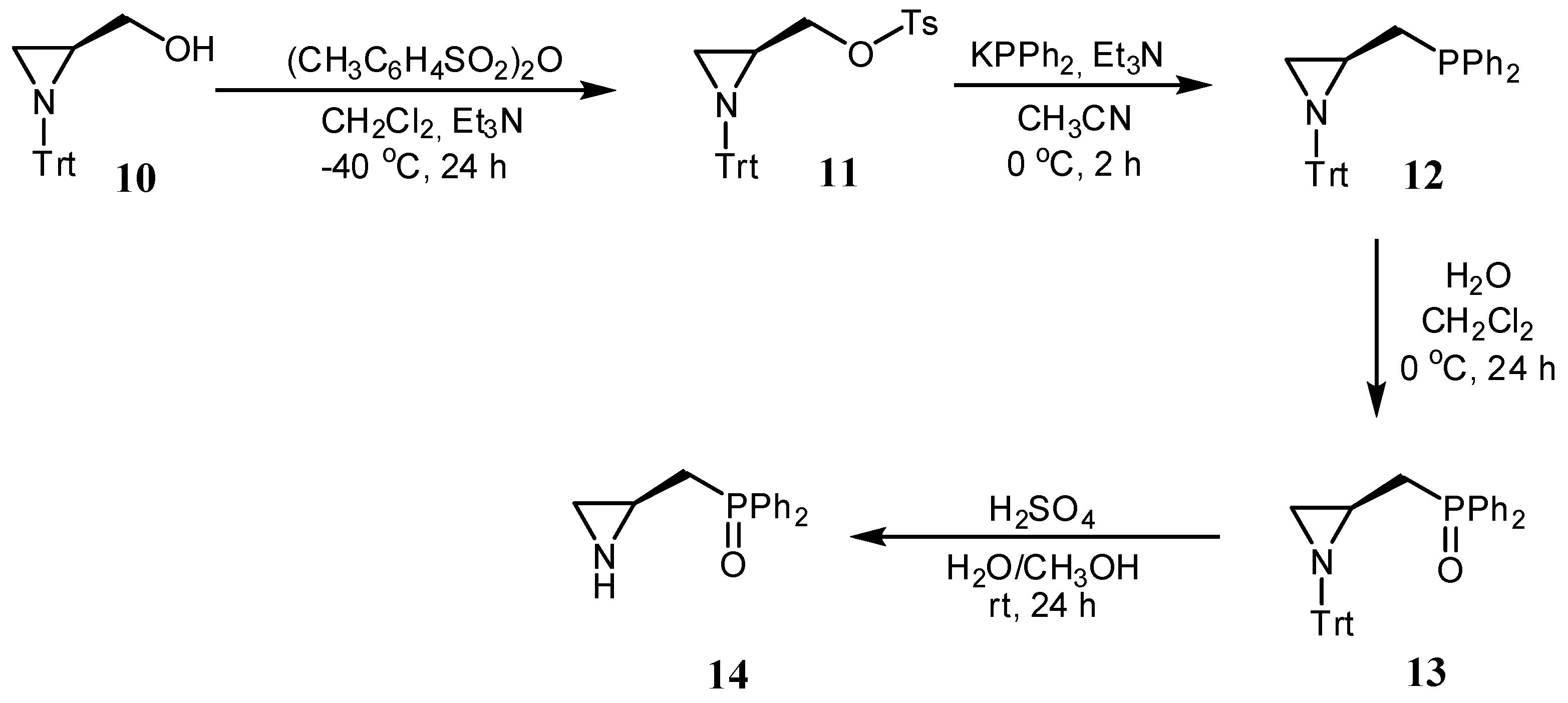
2.1. Synthesis of the Chiral Aziridines 1–9 and 14
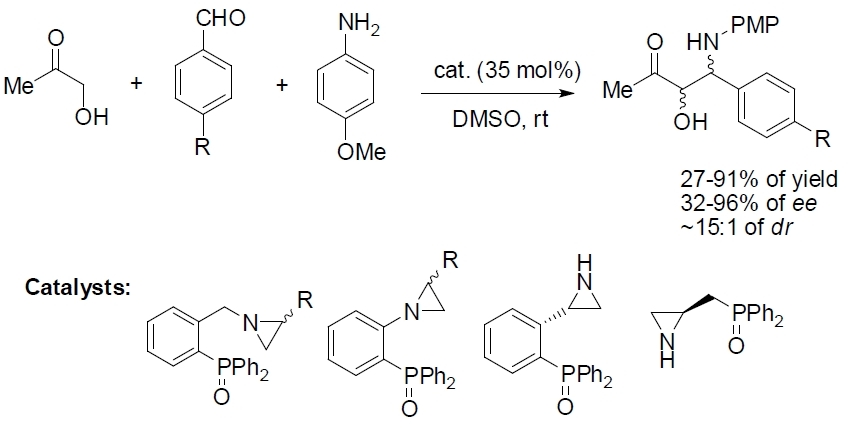
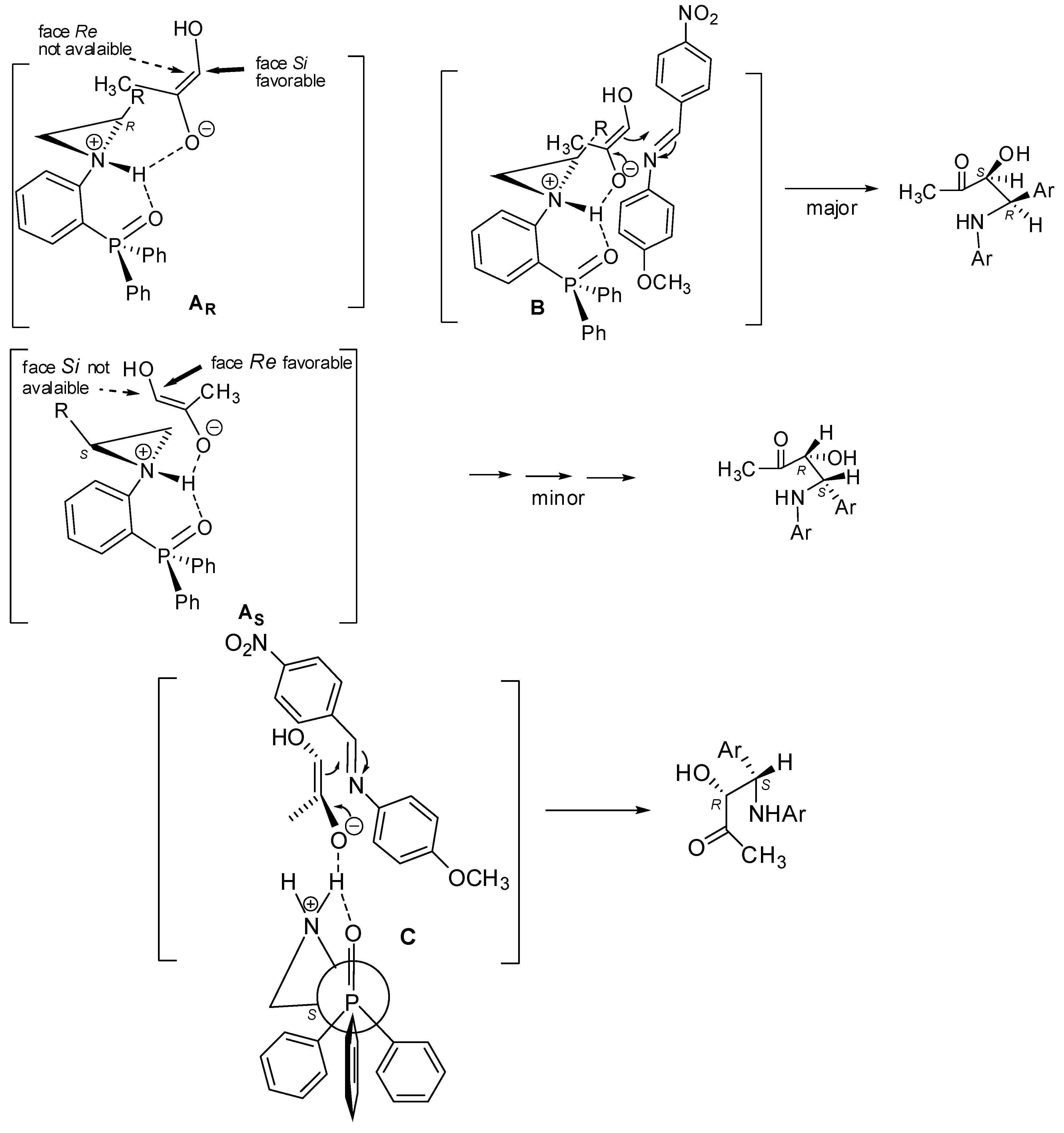
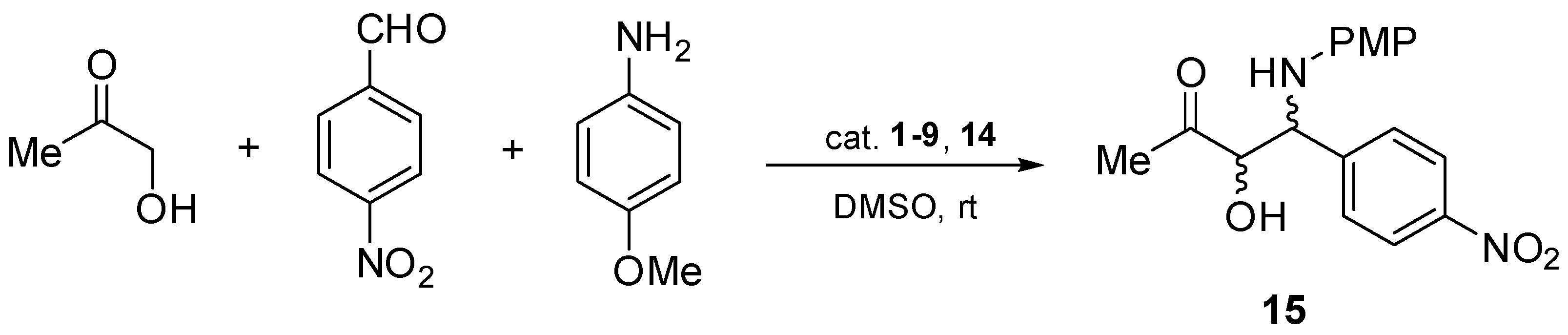
2.2. Asymmetric Three-Component Mannich Reaction Promoted by Aziridines 1–9, 14
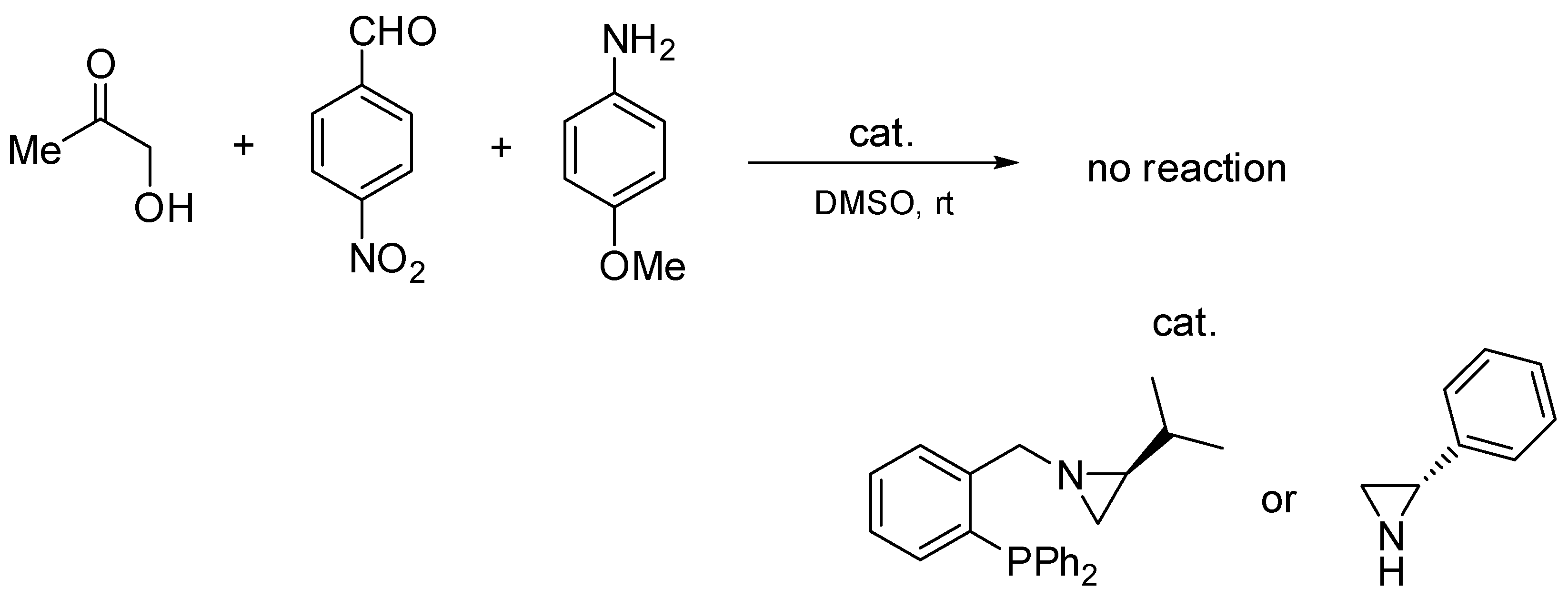
2.3. Asymmetric Mannich Reaction in the Presence of Catalyst 9
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis of (S)-2-diphenylphosphinoyl-2-phenylaziridine 9
3.2.2. Synthesis of [(S)-1-Tritylaziridin-2-yl]methyl-4-methylbenzenesulfonate 11
3.2.3. Synthesis of Diphenyl-[[(S)-1-tritylaziridin-2-yl]methyl]phosphine 12
3.2.4. Synthesis of (S)-2-(Diphenylphosphinoylmethyl)-1-trityl-aziridine 13
3.2.5. Synthesis of (S)-2-(Diphenylphosphinoylmethyl)aziridine 14
3.2.6. Organocatalytic Asymmetric Mannich Reaction–General Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Seayad, J.; List, B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Da Gama Oliveira, V.; do Carmo Cardoso, M.F.; da Silva Magalhães Forezi, L. Organocatalysis: A brief overview on its evolution and application. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leenders, T.; Kaczmarczyk, S.; Kiełbasiński, P.; Leśniak, S.; Rutjes, F.P.J.T. Efficient catalysts for asymmetric Mannich reactions. Org. Biomol. Chem. 2013, 11, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Chimni, S.S. Mechanochemistry assisted asymmetric organocatalysis: A sustainable approach. Beilstein J. Org. Chem. 2012, 8, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Chen, D.-F.; Gong, L.-Z. Recent progress in organocatalytic asymmetric total syntheses of complex indole alkaloids. Natl. Sci. Rev. 2017, 4, 381–396. [Google Scholar] [CrossRef]
- Hui, C.; Pu, F.; Xu, J. Metal-catalyzed asymmetric Michael addition in natural product synthesis. Chem. Eur. J. 2017, 23, 4023–4036. [Google Scholar] [CrossRef]
- Chanda, T.; Zhao, J.C.-G. Recent progress in organocatalytic asymmetric domino transformations. Adv. Synth. Catal. 2018, 360, 2–79. [Google Scholar] [CrossRef]
- Szöllösi, G. Asymmetric one-pot reactions using heterogeneous chemical catalysis: Recent steps towards sustainable processes. Catal. Sci. Technol. 2018, 8, 389–422. [Google Scholar] [CrossRef]
- Verkade, J.M.M.; van Hemert, L.J.C.; Quaedflieg, P.J.L.M.; Rutjes, F.P.J.T. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41. [Google Scholar] [CrossRef]
- Torii, M.; Kato, K.; Uraguchi, D.; Ooi, T. Chiral ammonium betaine-catalyzed asymmetric Mannich-type reaction of oxindoles. Beilstein J. Org. Chem. 2016, 12, 2099–2103. [Google Scholar] [CrossRef]
- Sharma, A.; Peddinti, R.K. Direct asymmetric Mannich reaction catalyzed by a D-glucosamine-derived organocatalyst. Synlett 2018, 29, 630–634. [Google Scholar] [CrossRef]
- Karimi, B.; Enders, D.; Jafari, E. Recent advances in metal-catalyzed asymmetric Mannich reactions. Synthesis 2013, 45, 2769–2812. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Xiang, Y.; He, Y.-H.; Guan, Z. Anti-selective direct asymmetric Mannich reaction catalyzed by protease. Tetrahedron Lett. 2019, 60, 1066–1071. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Q.; Gao, S. Recent advances in the intramolecular Mannich reaction in natural products total synthesis. Org. Chem. Front. 2018, 5, 1049–1066. [Google Scholar] [CrossRef]
- Bai, S.; Zhu, Y.; Wu, Q. Asymmetric Mannich reaction: Synthesis of novel chiral 5-(substituted aryl)-1,3,4-thiadiazole derivatives with anti-plant-virus potency. Heterocycl. Commun. 2019, 25, 47–51. [Google Scholar] [CrossRef]
- Trost, B.M.; Saget, T.; Hung, C.-I. Direct catalytic asymmetric Mannich reactions for the construction of quaternary carbon stereocenters. J. Am. Chem. Soc. 2016, 138, 3659–3662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, L.; Hu, Y.; Zha, Z.; Wang, Z.; Loh, T.-P. Bifunctional amino sulfonohydrazide catalyzed direct asymmetric Mannich reaction of cyclic ketimines with ketones: Highly diastereo- and enantioselective construction of quaternary carbon stereocenters. Org. Lett. 2015, 17, 1050–1053. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Nie, J.; Cai, H.; Ma, J.-A. Cyclic aldimines as superior electrophiles for Cu-catalyzed decarboxylative Mannich reaction of β-ketoacids with a broad scope and high enantioselectivity. Org. Lett. 2014, 16, 2542–2545. [Google Scholar] [CrossRef]
- Doğan, Ö.; Çağli, E. PFAM catalyzed enantioselective diethylzinc addition to imines. Turk. J. Chem. 2015, 39, 290–296. [Google Scholar] [CrossRef]
- Dogan, Ö.; Tan, D. Enantioselective direct aldol reactions promoted by phosphine oxide aziridinyl phosphonate organocatalysts. Tetrahedron Asymmetry 2015, 26, 1348–1353. [Google Scholar] [CrossRef]
- Eröksüz, S.; Dogan, Ö.; Garner, P.P. A new chiral phosphine oxide ligand for enantioselective 1,3-dipolar cycloaddition reactions of azomethine ylides. Tetrahedron Asymmetry 2010, 21, 2535–2541. [Google Scholar] [CrossRef]
- Dogan, Ö.; Isci, M.; Aygun, M. New phosphine oxide aziridinyl phosphonates as chiral Lewis bases for the Abramov-type phosphonylation of aldehydes. Tetrahedron Asymmetry 2013, 24, 562–567. [Google Scholar] [CrossRef]
- Dogan, Ö.; Bulut, A.; Ali Tecimer, M. Chiral phosphine oxide aziridinyl phosphonate as a Lewis base catalyst for enantioselective allylsilane addition to aldehydes. Tetrahedron Asymmetry 2015, 26, 966–969. [Google Scholar] [CrossRef]
- Wujkowska, Z.; Zawisza, A.; Leśniak, S.; Rachwalski, M. Phosphinoyl-aziridines as a new class of chiral catalysts for enantioselective Michael addition. Tetrahedron 2019, 75, 230–235. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leśniak, S.; Kiełbasiński, P. Highly enantioselective aza-Henry reaction promoted by amine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymmetry 2011, 22, 1087–1089. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leśniak, S.; Sznajder, E.; Kiełbasiński, P. Highly enantioselective Henry reaction catalyzed by chiral tridentate heteroorganic ligands. Tetrahedron Asymmetry 2009, 20, 1547–1549. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leśniak, S.; Kiełbasiński, P. Highly enantioselective asymmetric direct aldol reaction catalyzed by amine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymmetry 2011, 22, 1325–1327. [Google Scholar] [CrossRef]
- Jarzyński, S.; Leśniak, S.; Pieczonka, A.M.; Rachwalski, M. N-Trityl-aziridinyl alcohols as highly efficient chiral catalysts in asymmetric additions of organozinc species to aldehydes. Tetrahedron Asymmetry 2015, 26, 35–40. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Yield [%] | ee [%] a | drb | Abs. Conf. c |
|---|---|---|---|---|---|
| 1 | 1 | 37 | 34 | 8:1 | (3S,4R) |
| 2 | 2 | 43 | 40 | 10:1 | (3R,4S) |
| 3 | 3 | 30 | 37 | 8:1 | (3R,4S) |
| 4 | 4 | 27 | 32 | 8:1 | (3R,4S) |
| 5 | 5 | 60 | 48 | 10:1 | (3S,4R) |
| 6 | 6 | 69 | 60 | 15:1 | (3R,4S) |
| 7 | 7 | 65 | 56 | 12:1 | (3R,4S) |
| 8 | 8 | 62 | 52 | 10:1 | (3R,4S) |
| 9 | 9 | 91 | 96 | 20:1 | (3R,4S) |
| 10 | 14 | 90 | 96 | 20:1 | (3R,4S) |
| Entry | Ar | Product | Yield [%] | ee [%] a | drb |
|---|---|---|---|---|---|
| 1 | Ph | 16 | 88 | 89 | 20:1 |
| 2 | 2-MeOC6H4 | 17 | 85 | 82 | 20:1 |
| 3 | 4-MeOC6H4 | 18 | 90 | 90 | 20:1 |
| 4 | 4-BrC6H4 | 19 | 89 | 91 | 20:1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchcic, A.; Zawisza, A.; Leśniak, S.; Adamczyk, J.; Pieczonka, A.M.; Rachwalski, M. Enantioselective Mannich Reaction Promoted by Chiral Phosphinoyl-Aziridines. Catalysts 2019, 9, 837. https://doi.org/10.3390/catal9100837
Buchcic A, Zawisza A, Leśniak S, Adamczyk J, Pieczonka AM, Rachwalski M. Enantioselective Mannich Reaction Promoted by Chiral Phosphinoyl-Aziridines. Catalysts. 2019; 9(10):837. https://doi.org/10.3390/catal9100837
Chicago/Turabian StyleBuchcic, Aleksandra, Anna Zawisza, Stanisław Leśniak, Justyna Adamczyk, Adam Marek Pieczonka, and Michał Rachwalski. 2019. "Enantioselective Mannich Reaction Promoted by Chiral Phosphinoyl-Aziridines" Catalysts 9, no. 10: 837. https://doi.org/10.3390/catal9100837
APA StyleBuchcic, A., Zawisza, A., Leśniak, S., Adamczyk, J., Pieczonka, A. M., & Rachwalski, M. (2019). Enantioselective Mannich Reaction Promoted by Chiral Phosphinoyl-Aziridines. Catalysts, 9(10), 837. https://doi.org/10.3390/catal9100837