2.1. Electronic Characteristics for the CeOx/Ag(111) Catalyst
A Bader analysis of the charge was performed firstly for CeO
x/Ag(111) catalyst, in order to reveal the electronic structure and interactions between ceria and Ag at the interface of this catalyst. As reported, the Bader charge of Ce in the particle is +2.00 e [
5]. While the calculated Bader charge for Ce in the CeO
2/Ag(111) are ~+2.1 e and ~+2.2 e, respectively (
Table 1). Thus it can be inferred that the formal oxidation states of Ce atoms in the ceria cluster are +4 and +3, respectively [
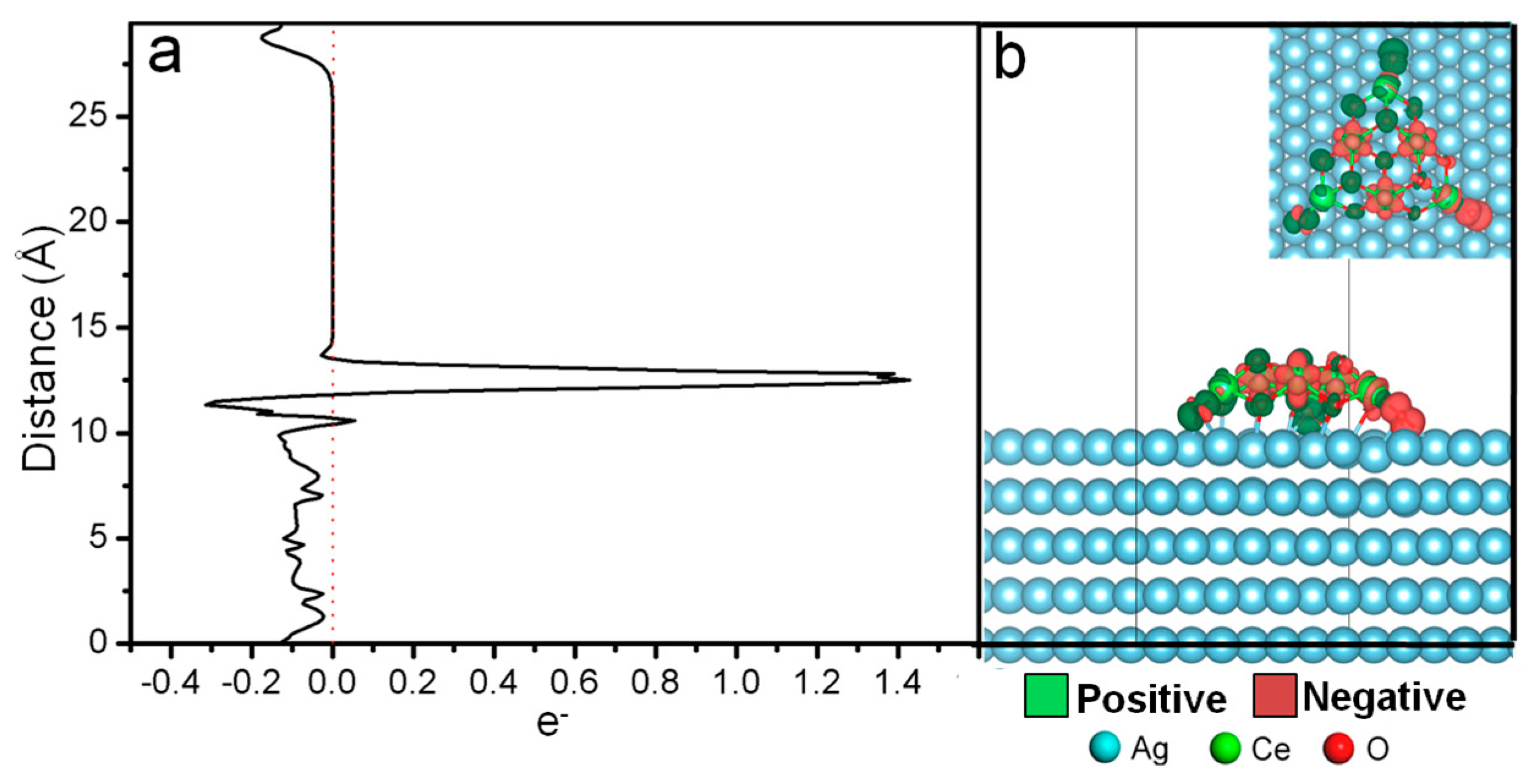
5]. Spin Density Difference (SDD) versus Z direction of the CeO
x/Ag(111) catalyst is plotted in Fig. 1a, the isosurface of SDD is plotted in
Figure 1b. From
Figure 1a,b, obvious electron accumulation is founded on Ce atoms at the edge of a cluster. Therefore it can be concluded that the electrons are mainly located on the edge of the CeO
x cluster. In other words, single electrons are concentrated in certain areas of ceria cluster, the computational results show that only parts of Ce atoms which located on the edges of ceria cluster have been reduced (
Figure 1b). All those Ce atoms can be considered as Ce
3+. In addition, the Electron Density Difference (EDD) versus Z direction of the CeO
x/Ag(111) catalyst is plotted in
Figure 2a, the isosurface of EDD is plotted in
Figure 2b. From
Figure 2, it was found that EDD indeed increased on CeO
x cluster and decreased on surface Ag atoms, which indicates that there is a charge transfer from the Ag slab to the ceria cluster. And the electrons are mainly coming from the surface Ag layer, which located under the CeO
x cluster, resulting in an oxidation of the Ag atoms and a reduction of the CeO
x cluster.
2.2. CO2 Adsorption and Activation
The adsorption and activation of CO
2 is considered as the first step for CO
2 reduction. In the initial CO
2 adsorption configurations, no matter where CO
2 was placed (on the interface of CeO
x/Ag(111), Ag and CeO
x, linear or bent structure). Only two stable structure of CO
2 adsorption (denoted as CO
2*, * = CeO
x/Ag(111)) were reached in the final (
Figure 3 and
Figure 4c). CO
2 is linearly adsorbed on the edge (Ce
3+) or corner (Ce
4+) of the CeO
x cluster using its O atom. The adsorption energy of CO
2 for those two structures is 0.42 eV and 0.40 eV, respectively. The shortest distance between O atom of CO
2 and Ce atom of CeO
x cluster is 3.03 Å and 3.04 Å. Gradient isosurfaces of reduced density gradient (RDG) versus sign(λ
2)/ρ (
Figure 3a) [
10] plot shows that there are Van Der Waals interactions between O-Ce and C-Ag for CO
2/*(Ce
3+) simultaneously. Whereas, in CO
2/*(Ce
4+), only O-Ce interaction has been found (
Figure 3c). Spikes near zero in the corresponding scatter plot (
Figure 3b,d) also reflect there exist weak interactions in CO
2/*(Ce
3+) than CO
2/*(Ce
4+). The synergism by Ag and CeO
x in CO
2/*(Ce
3+) configuration may be beneficial to the subsequent hydrogenating of CO
2. Therefore CO
2/*(Ce
3+) is chosen as the initial structure in this work.
2.3. Formation of Formic Acid and Methanol
The CO
2 adsorption configuration shown in
Figure 3a is set to be the starting point for further reduction. The first hydrogenation of CO
2* may occur at either the O or C atom, forming carboxyl (HOCO*) or formate (HCO
2*) species, respectively. Herein, the routes through both HOCO* and HCO
2* intermediates were investigated. The Gibbs free energy diagrams for all possible products involved in CO
2 reduction process on CeO
x/Ag(111) were also examined.
As mentioned above, hydrogenation of CO
2 could occur either on oxygen or carbon atom of CO
2. HOCO* or HCO
2* intermediates are eventually formed at the interface of CeO
x and Ag. The HCO
2* (
Figure 4d) intermediate is formed with a free energy release of −1.14 eV (
Figure 4a). While the HCO
2* intermediate can change to bidentate form using its O atoms with CeO
x cluster, which is endothermic with free energy barrier of 0.73 eV. When the hydrogenation of CO
2 occurs on the O atom (
Figure 4b), forming HOCO* (
Figure 4f) with a free energy barrier of 0.67 eV. This intermediate is bind to the catalyst using its C (C-Ag = 2.25 Å) and O (O-Ce = 2.56 Å) atom simultaneously. The Ag atom which bonds to C atom obviously migrates up from Ag surface. Obviously, the HCO
2* formation is thermodynamically preferable over HOCO*.
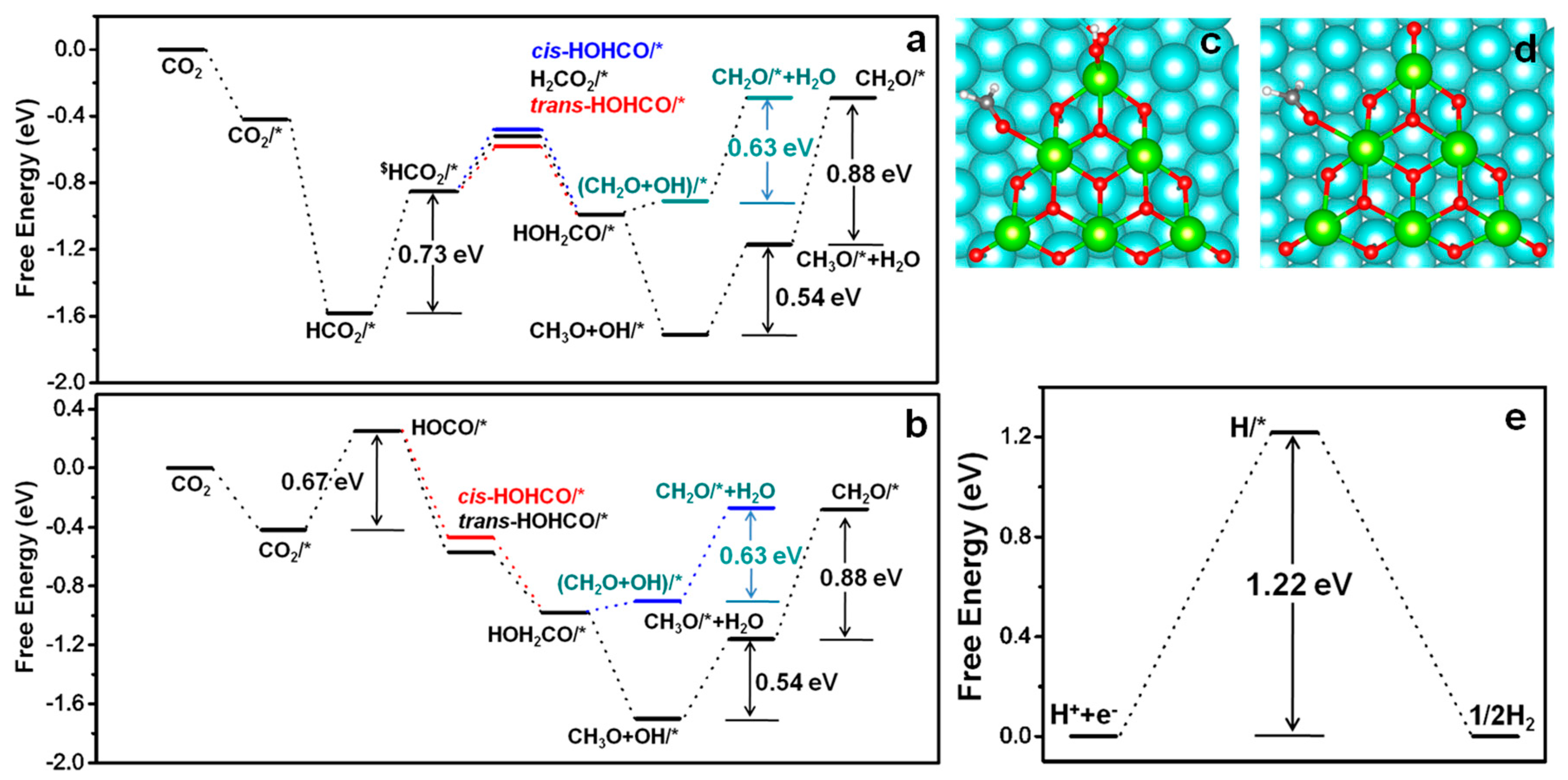
When HCO
2* or HOCO* intermediate is formed on the catalyst. Formic acid as an important product for CO
2 reduction can be generated firstly by hydrogenation of HCO
2* or HOCO* intermediate. For the hydrogenation of HOCO*, this is thermodynamically preferable. The formation of
cis-HOHCO* and trans-HOHCO* (
Figure 4h,i) is exothermic by 0.72 and 0.82 eV, respectively. For the hydrogenation of bidentate HCO
2*, subsequent reaction process can be achieved through two different ways. (a) The second hydrogen is bonded with carbon of HCO
2*, a H
2CO
2* intermediate is formed. The free energy change for this process is 0.42 eV endothermic. (b) The hydrogenation occurs on oxygen atom of HCO
2*, HOHCO* intermediate can be generated. There are also two possibilities here, (1) formation of
cis-HOHCO* (0.47 eV, endothermic), (2) formation of trans-HOHCO* (0.37 eV, endothermic). The free energy change for the rate-limiting step of formic acid formation from HOCO* and HCO
2* intermediate are 0.67 and 0.73 eV, respectively. Obviously, HOCO* reaction path is slightly more facile for the formation of formic acid.
As shown in
Figure 4, trans-HOHCO* can further hydrogenate by (H
+ + e
−) to generate HOH
2CO* intermediate. CH
3OH and O atom co-adsorbed on the catalyst can be generated through the further hydrogenation of HOH
2CO. Next, the formed CH
3OH can be desorbed from the surface of the catalyst, leaving only OH*. Thermodynamically, the formation of CH
3OH through HOH
2CO* is uphill in free energy by only 0.05 eV. In general, once HOHCO formed, it is very easy to convert into CH
3OH, although more hydrogenation steps are needed for this process.
2.4. Formation of Methane
With the aim of comparison, both HCO
2* and HOCH* reaction path have been plotted in
Figure 5. When the hydrogenation of HOH
2CO* firstly occurs on the carbon atom, methoxy (CH
3O*) and hydroxyl (OH*) intermediates are formed and co-adsorbed on the catalyst. This process is endothermic with free energy change of 0.71 eV. Further hydrogenation for the formation of H
2O and CH
4 are all uphill with free energy barrier of 0.54 eV and 0.20 eV, respectively. Obviously, the total free energy change from CO
2* to CH
3OH and CH
4 are almost the same. Thus it is speculated that both CH
3OH and CH
4 are easy to form on the interface of this catalyst, as both of process are free energy downhill from the CO
2*. However, it is noteworthy that the formation of CH
4 needs more steps than that of CH
3OH. Thus, CH
3OH formation is probably facile than the formation of CH
4. The most endothermic steps for the formation of HOCO* or trans-HOHCO* are observed, which will be the potential-limiting steps for the formation of CH
3OH and CH
4 on the CeO
x/Ag(111) catalyst.
2.5. Formation of Carbon Monoxide
Although CO
2 only weakly binds to CeO
x/Ag(111), less energy is required for the formation of HOCO* from CO
2* than that on pure Ag [
9], on which more than 1.2 eV is needed for the hydrogenating of CO
2 to form HOCO*. The formation of CO* (
Figure 6b) from HOCO* is endothermic on CeO
x/Ag(111) with a free energy change of 0.69 eV. However, further hydrogenating of HOCO* is exothermic with a free energy change of −0.82 eV. Clearly, further hydrogenating of HOCO* to form HOHCO* is more facile on CeO
x/Ag(111) than the formation of CO (0.18 eV, CO*). Simultaneously, CO adsorption is quite stable on CeO
x/Ag(111). As CO desorption is endothermic with a free energy change of 0.83 eV. Consequently, CO* may be further reduced on CeO
x/Ag(111) to other products rather than desorbed from the catalyst. The reduction pathway and the free energy profile of the corresponding intermediates from CO* have been mapped out (
Figure 6). As shown in
Figure 6, apart from the formation of HCO* (0.25 eV), all other processes after HCO* formation are downhill in free energy.
As shown in
Figure 6b, CO* is bonded to Ag only through C atom. Bond length of C-O and C-Ag are 1.15 and 2.15 Å, respectively. This indicates that CO is chemically adsorbed on the Ag surface and has been activated. With the formation of CHO*, O atom is weakly bonded to the Ce atom of the CeO
x cluster, distance of C-Ag and O-Ce are 2.20 and 2.60 Å. There is an obvious deformation of Ag surface. The Ag atom, which interacts with C atom, is moved upward from the Ag surface. Gradient isosurfaces of RDG and sign(λ
2)/ρ [
10] plot also shows clearly a strong attractive interaction between carbon and Ag (
Figure S1). With further hydrogenating of CHO*, the formed CH
2O* bonds to CeO
x/Ag(111) only through O-Ce interaction (C-Ag 4.28 Å, O-Ce 2.71 Å). The C-Ag in CHO* is completely broken upon formation of CH
2O*. The same situation occurs in the formation of CH
3O*, in which methoxyl bonds to the CeO
x/Ag(111) only through O-Ce (C-Ag 6.14 Å, O-Ce 2.13 Å). The high oxygen affinity of CeO
x/Ag(111) keeps the O atom of CH
3O* bound to Ce strongly. And this, in turn, will cause the breaking of the C-O bond of CH
3O*. Eventually, CH
4 can be formed in the subsequent hydrogenation step.
2.6. Formation of Acetaldehyde
The formation of formaldehyde can be achieved through (a) hydrogenation of CO* and HCO*, which is mentioned above in 2.5, or (b) dehydrogenation of methoxy in the (CH
3O_OH)* intermediate. As plotted in
Figure 7, Breaking C-O bond of HOH
2CO* intermediate can form CH
2O and OH co-adsorption intermediate (
Figure 7c). With the formation H
2O, CH
2O* is ultimately formed. Free energy change for this process is 0.63 eV, which is slightly lower than the formation of HOCO*, thus the rate-limiting step for this reaction path is also the HOCO* generation. Besides, the dehydrogenation of CH
3O* intermediate can also lead to the formation of CH
2O* (0.88 eV). Obviously, this reaction path is relatively difficult to achieve. Compared with the formation of CH
3OH and CH
4, more energy is needed for the generation of CH
2O. Thus CH
2O formation is unfavorable on this catalyst.
To sustain the catalytic cycle, no matter in what kind of reaction path, the O* species has to be cleared through consecutive hydrogenating to OH* and further to H
2O. The hydrogenating of OH* to H
2O is endothermic, with a free energy barrier of 0.70 eV, which indicate that OH is the most stable intermediate species at U = 0 V. This makes OH removal become the free energy limiting step. However, the free energy change is still lower than the desorption energy (1 eV) needed on TiO
2/Ag system [
9] or pure metals (such as Pt (0.75 eV)) [
11]. The free energy change needed for the formation and removal of H
2O is only slightly higher than the energy required for the generation of HOCO* intermediate (0.67 eV) on CeO
x/Ag(111) catalyst. The weak interaction between H
2O and CeO
x cluster is beneficial for the formation of H
2O (the distance of O-Ce has enlarged from 2.15 Å to 2.72 Å, for OH* and H
2O* intermediate), which makes the regeneration of the catalyst become easier, resulting in a great recyclability.
Finally, the hydrogen evolution reaction (HER), which is the main competitive reaction of CO
2RR, has been calculated. The computational result shows that the free energy change for the formation of H
2 is 1.22 eV, which is far higher than the rate-limiting step of CO
2RR on CeO
x/Ag(111) (
Figure 7e). Based on all those advantages mentioned above, it is reasonable to believe that the CeO
x/Ag(111)is a potential excellent catalyst for CO
2 electroreduction.
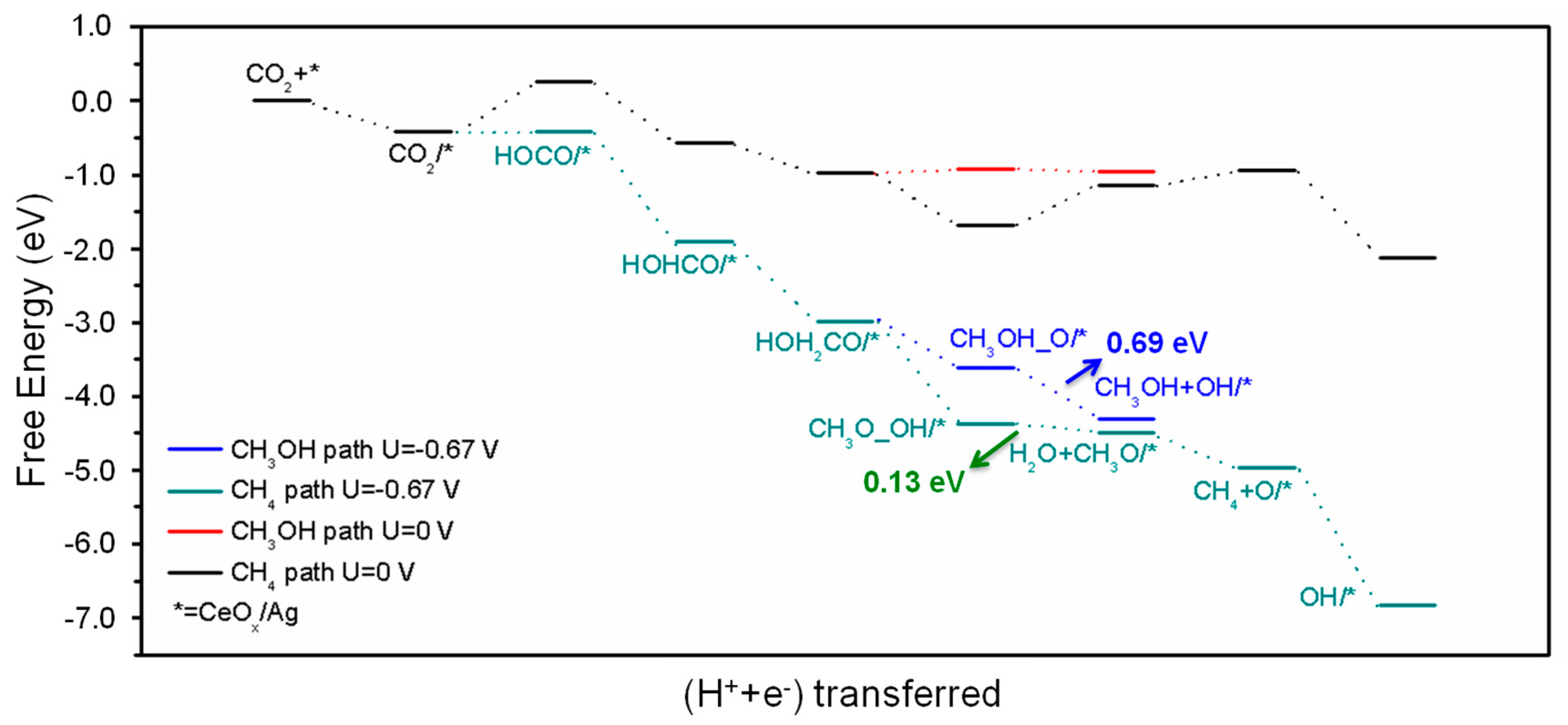
The above analysis shows that CH
3OH and CH
4 are the main products on the surface of CeO
x/Ag(111) catalyst. In order to change into exothermic and spontaneous process for all the reaction steps, the free energy diagram with an external potential of −0.67 V (RHE) was depicted and shown in
Figure 8. The potential is selected based on the potential-limiting step, that is the formation of HOCO*. Under this applied potential (U = −0.67 V vs RHE), the free energy change of all steps involving the proton-electron pairs will be corrected according to Equation (2). Under this potential, all steps following HOCO* formation become downhill on the free energy diagram.
When hydrogenation of CO
2 occurs on the oxygen atom, HOCO* is the key intermediate and plays a central role in the entire mechanism. Further hydrogenating HOCO* leads to the formation of HOHCO* (trans-formic acid) intermediate. When hydrogenation of CO
2 occurs on the carbon atom, trans-HOHCO becomes the critical intermediate. A HOH
2CO* intermediate can be formed through the hydrogenation of trans-HOHCO. Breaking different C-O bond of HOH
2CO* will lead to the formation of CH
3OH, CH
2O or an adsorbed methoxy (CH
3O*) species on the Ce site and an OH*. Subsequently, CH
3O* can be hydrogenated to produce CH
4 or CH
2O. And the catalyst will recover its original state with the desorption of H
2O by the hydrogenation of OH*. Based on the free energy profile for the HOCO* path shown in
Figure 8, CH
3OH desorption from the catalyst release more energy (−0.69 eV) than the formation of CH
3O* (−0.13 eV), whereas, the CH
3O* intermediate is more stable (0.19 eV lower) than CH
3OH + OH*. This result indicates that CH
3OH formation is less favorite than CH
4 thermodynamically. However, CH
3OH formation requires fewer proton reduction and hydrogenation steps, therefore, both CH
3OH and CH
4 are likely to be generated.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}