Active Site of O2 and Its Improvement Mechanism over Ce-Ti Catalyst for NH3-SCR Reaction


Abstract
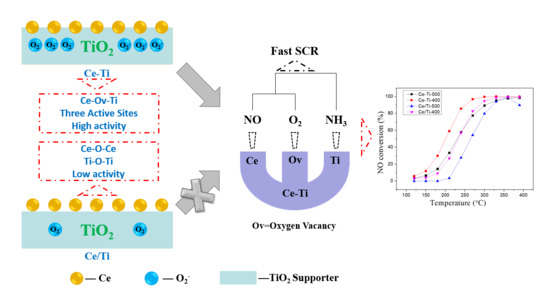
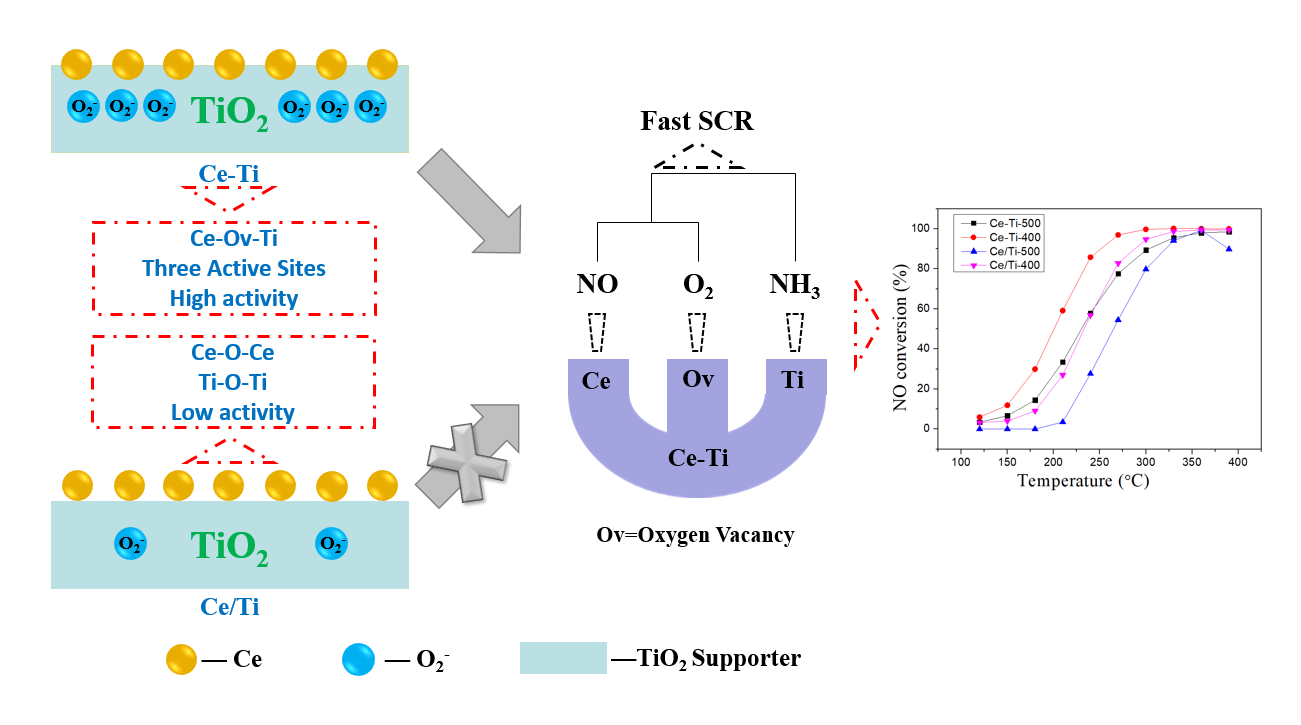
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Result and Discussion
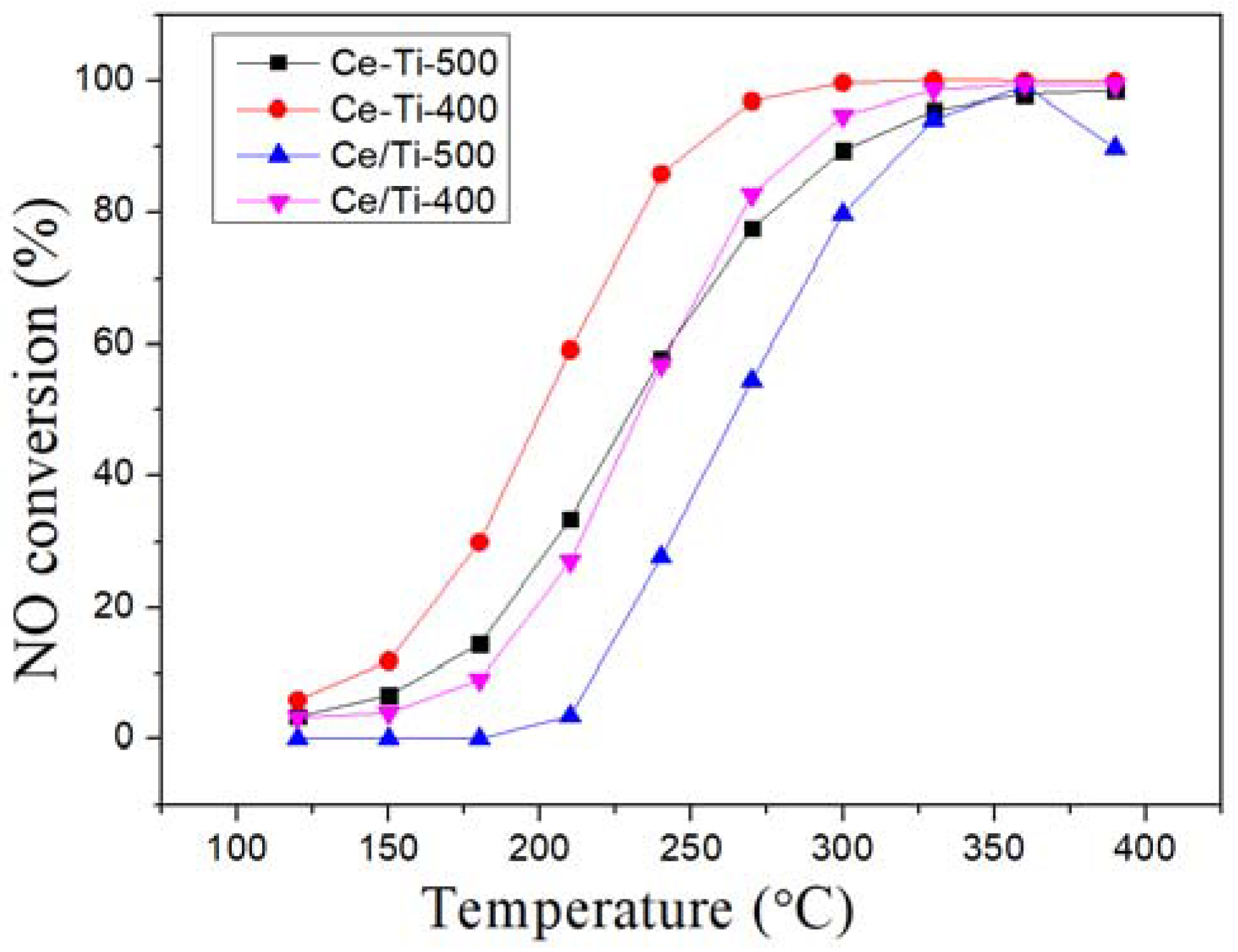
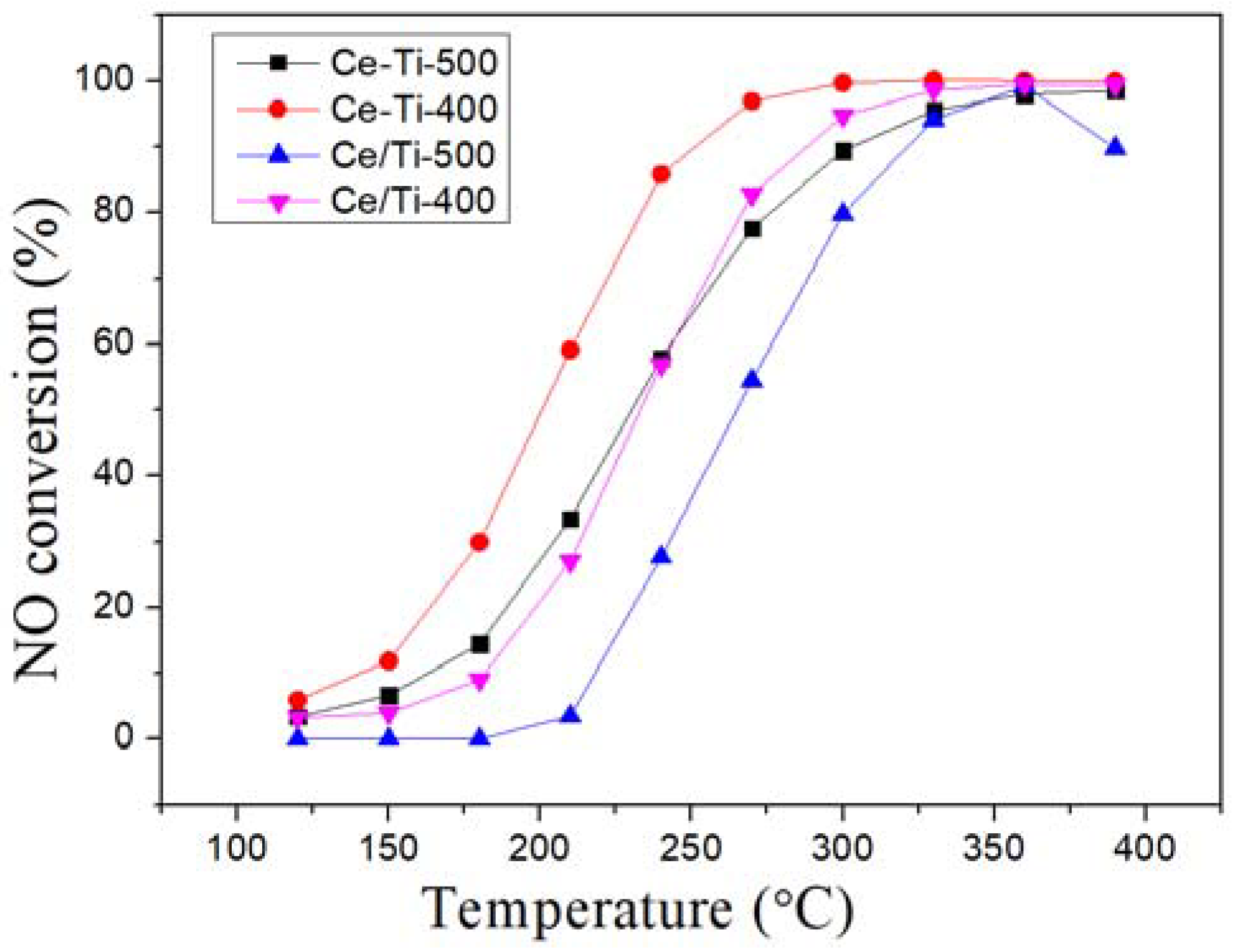
2.1. Catalytic Activity Tests
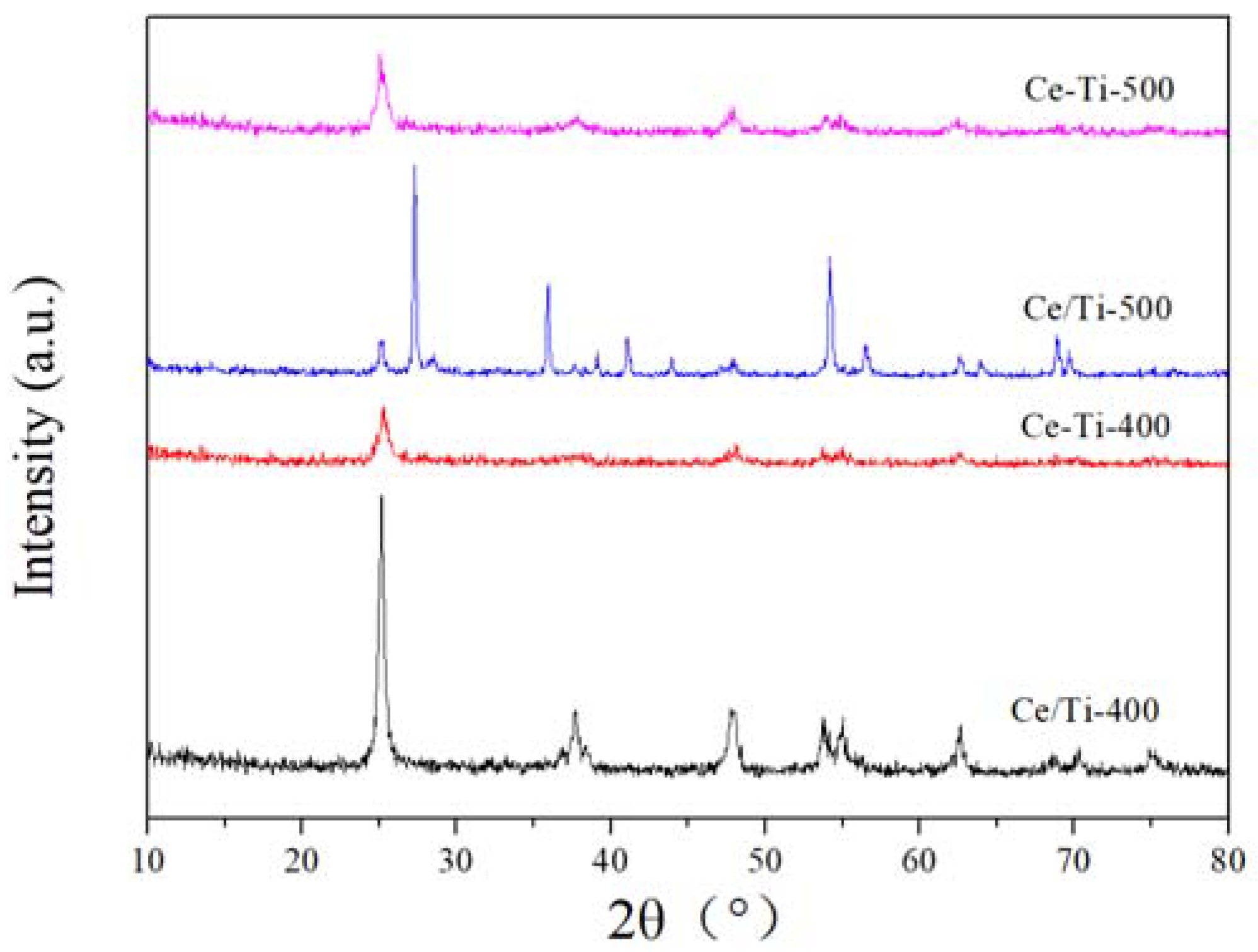
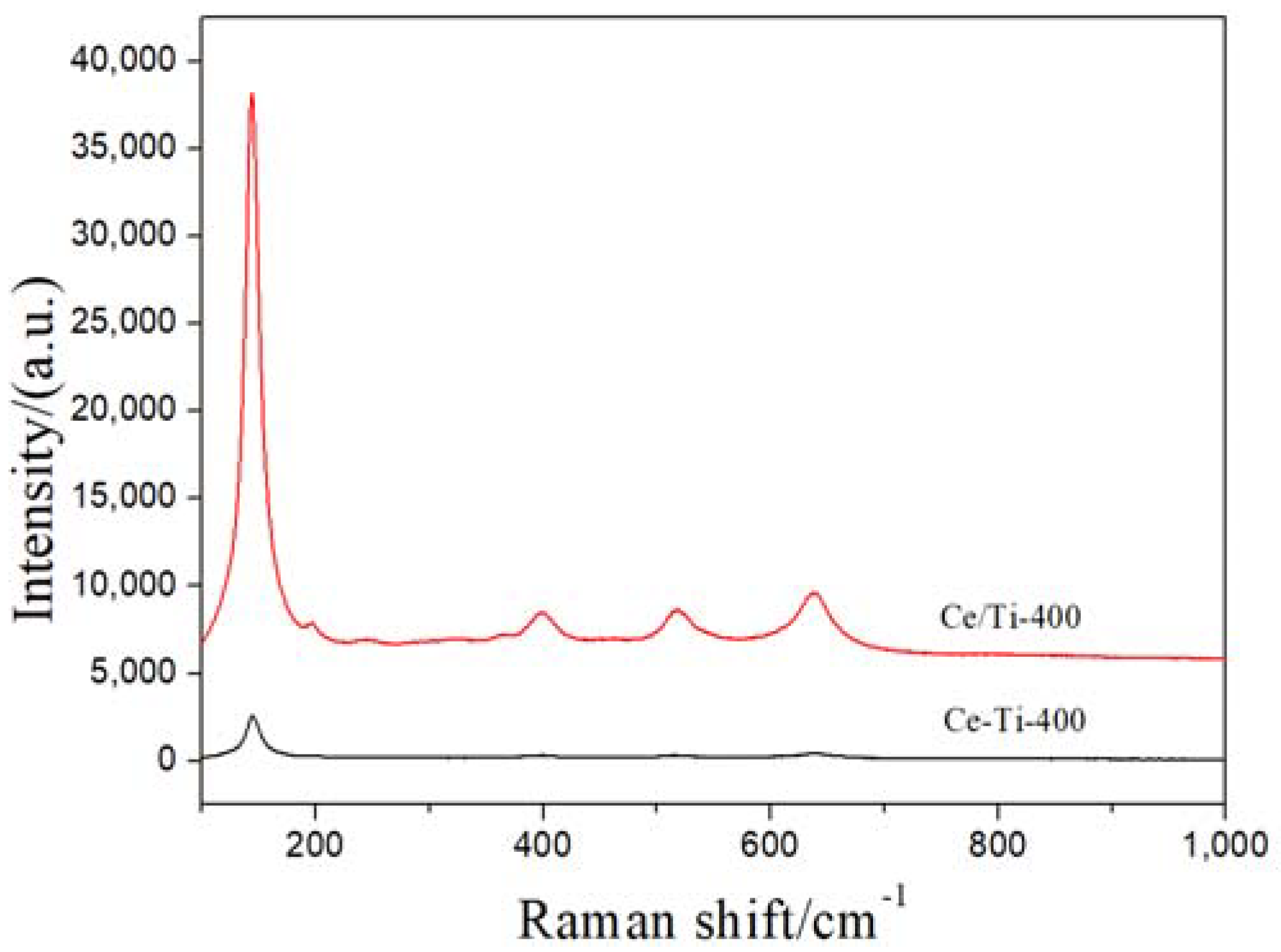
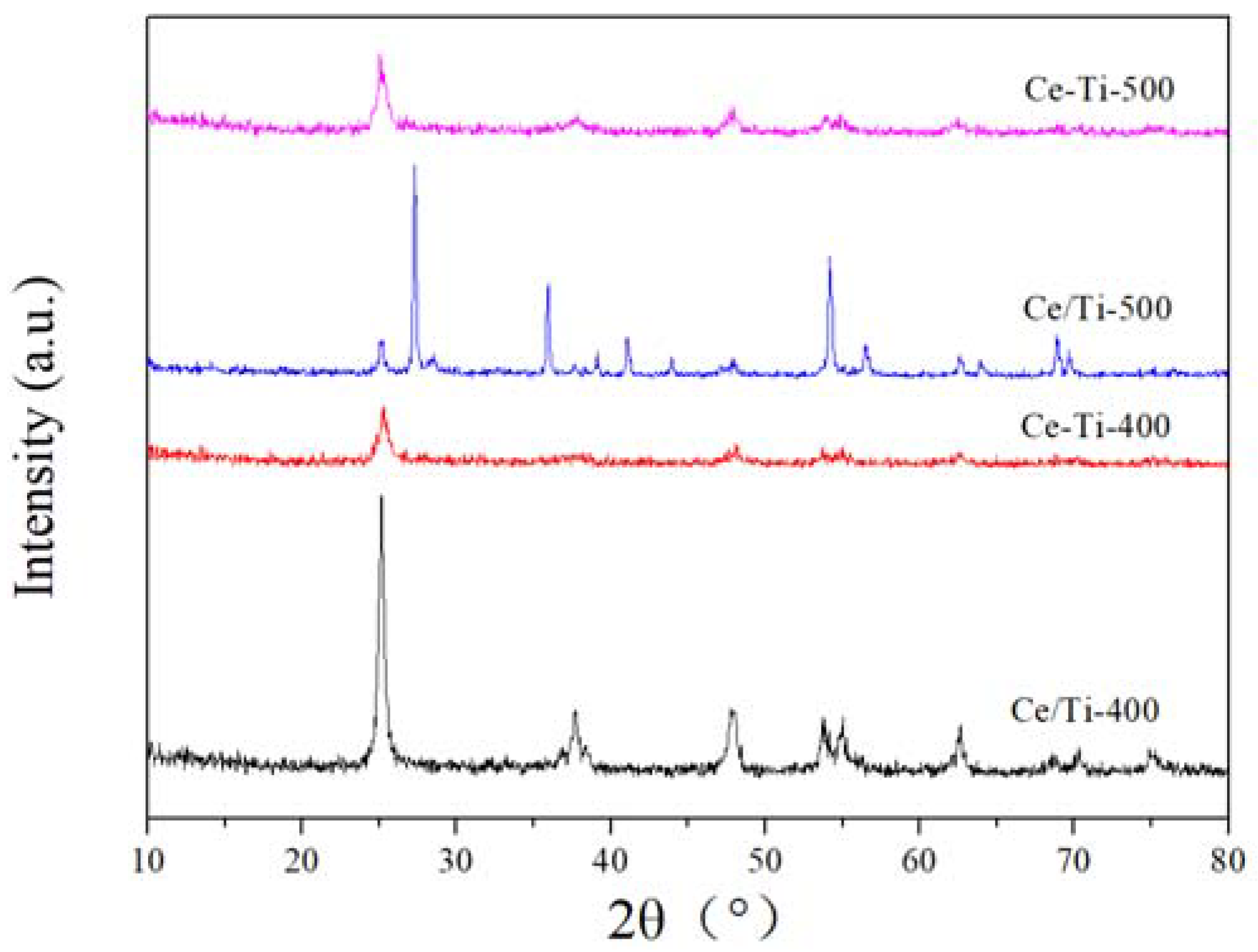
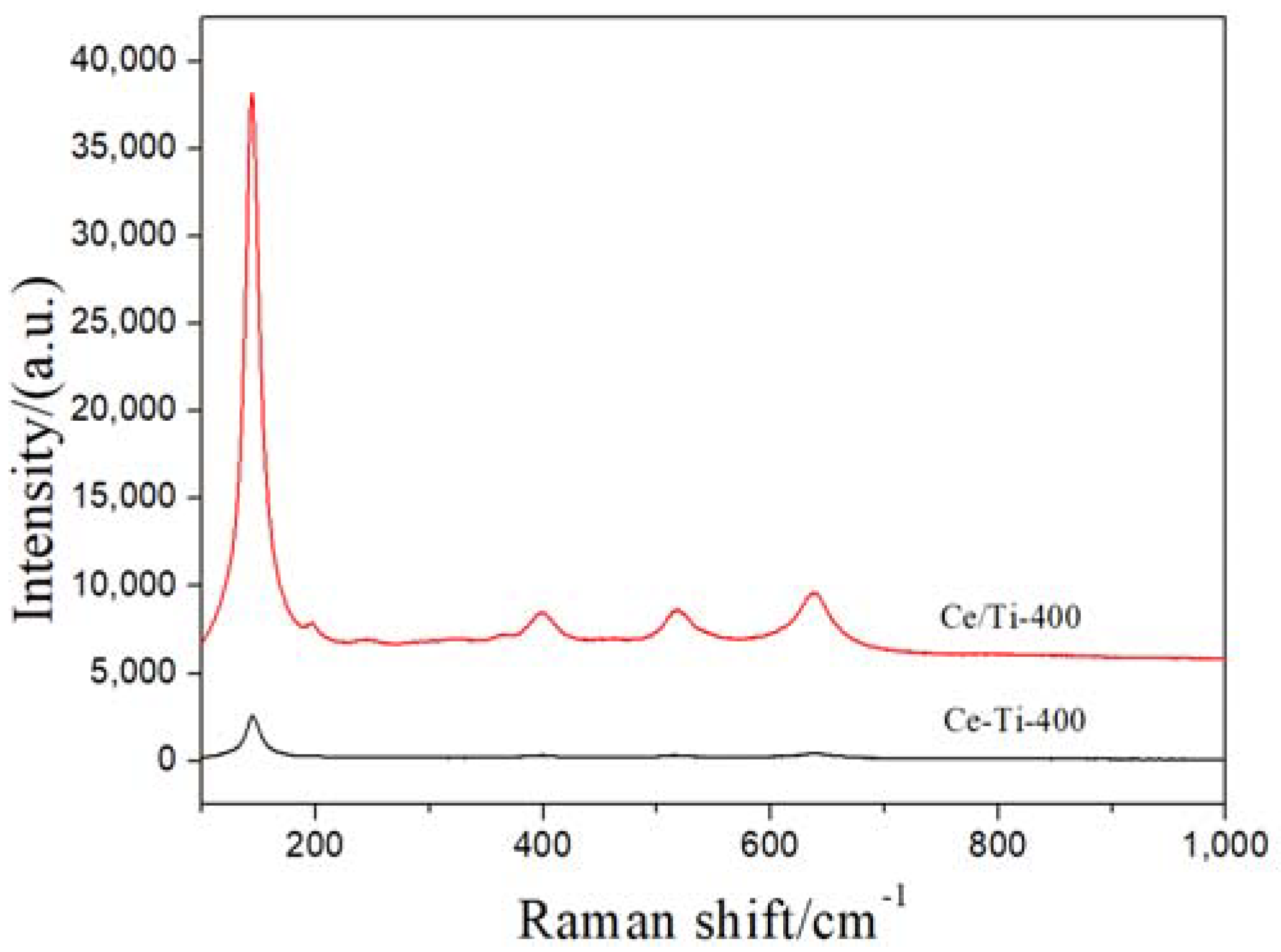
2.2. XRD and Raman Analysis
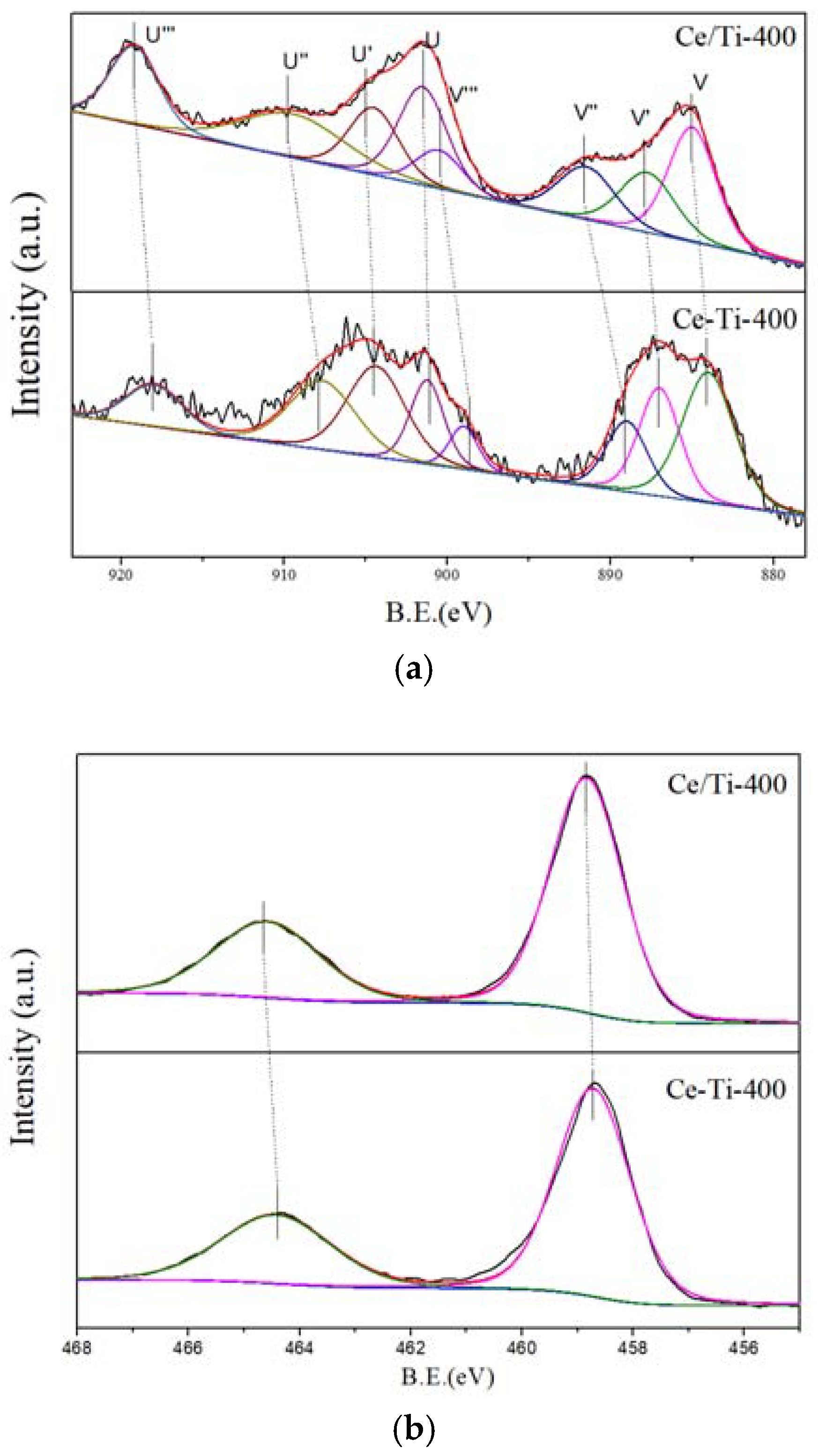
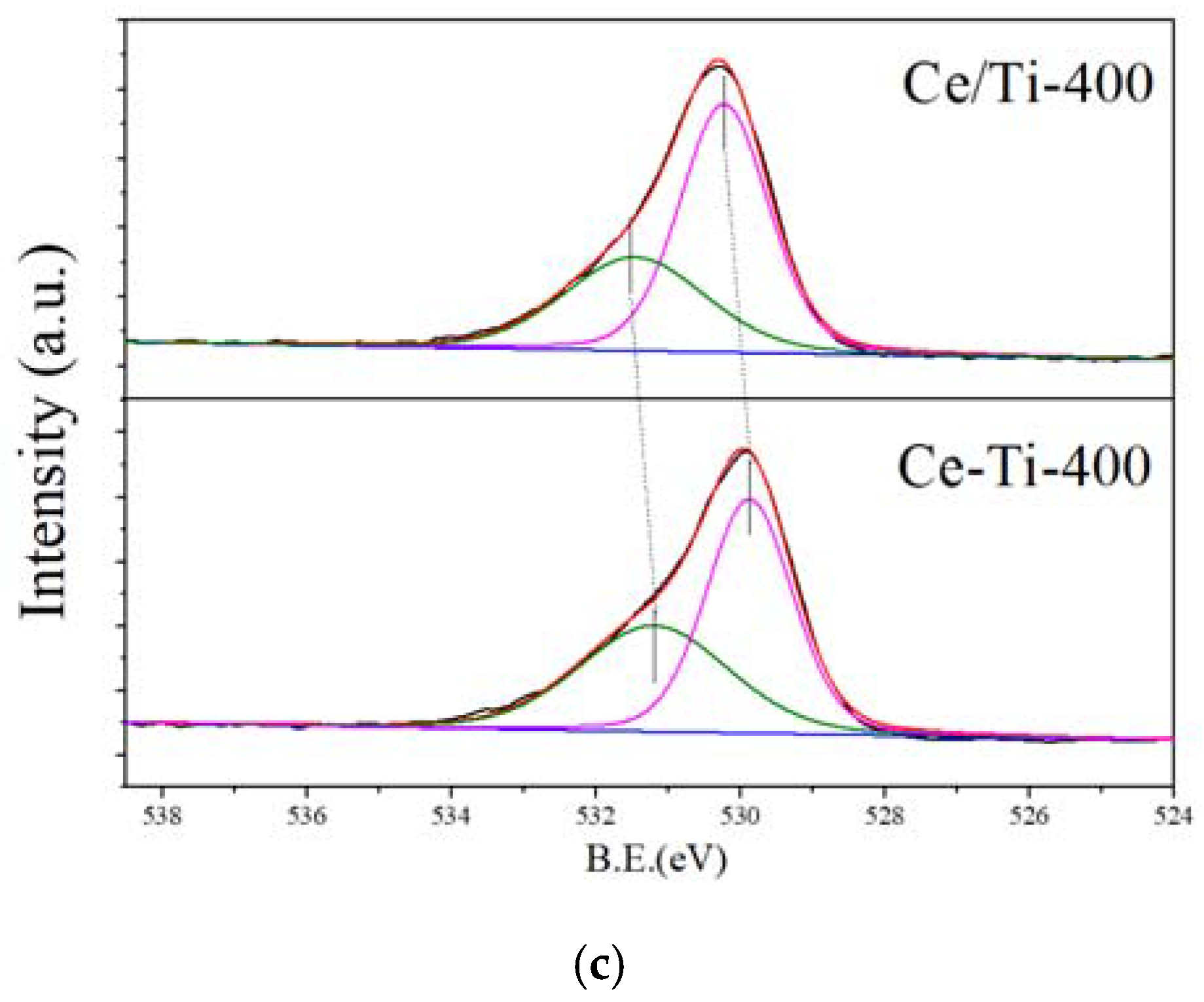
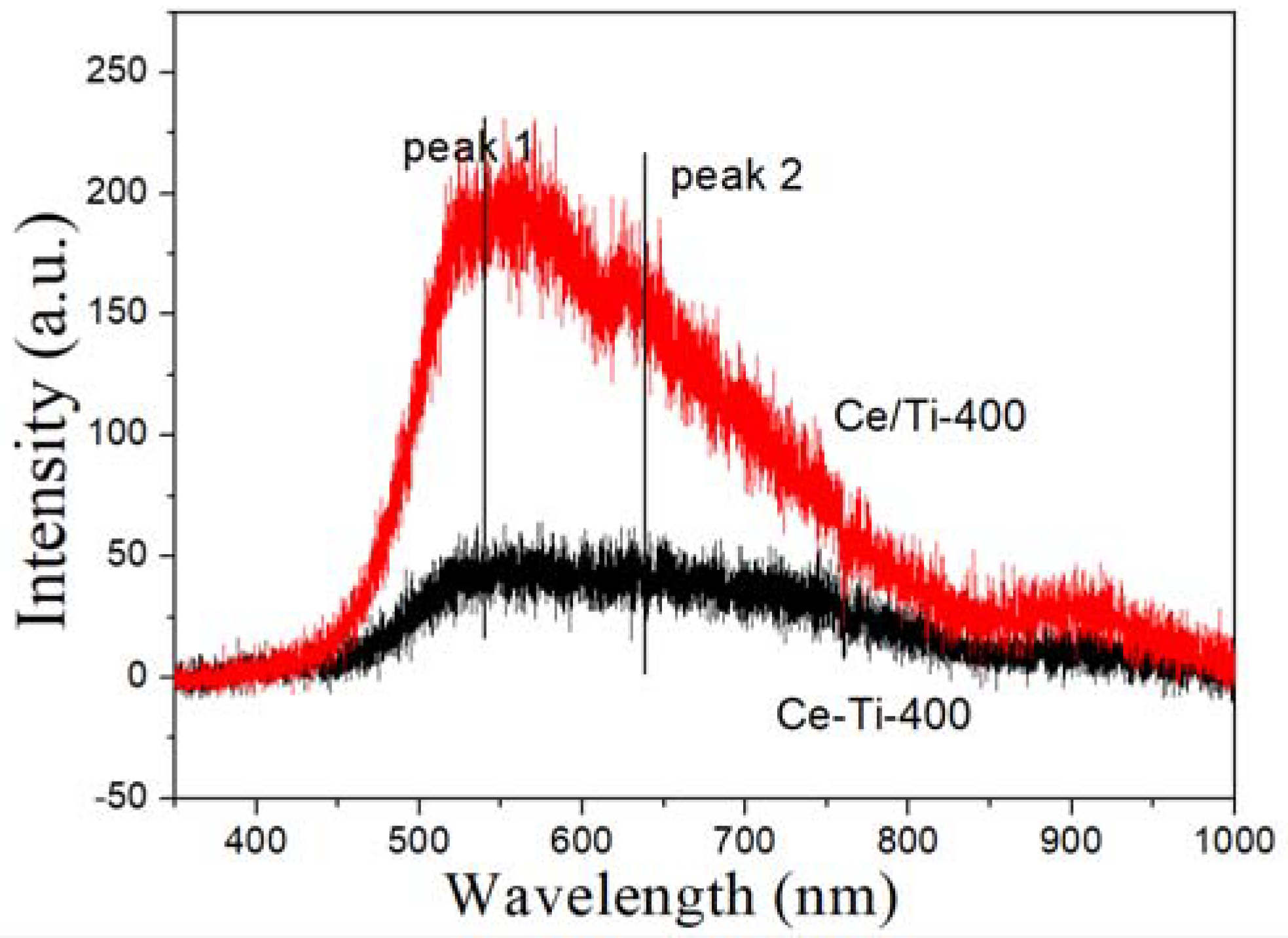
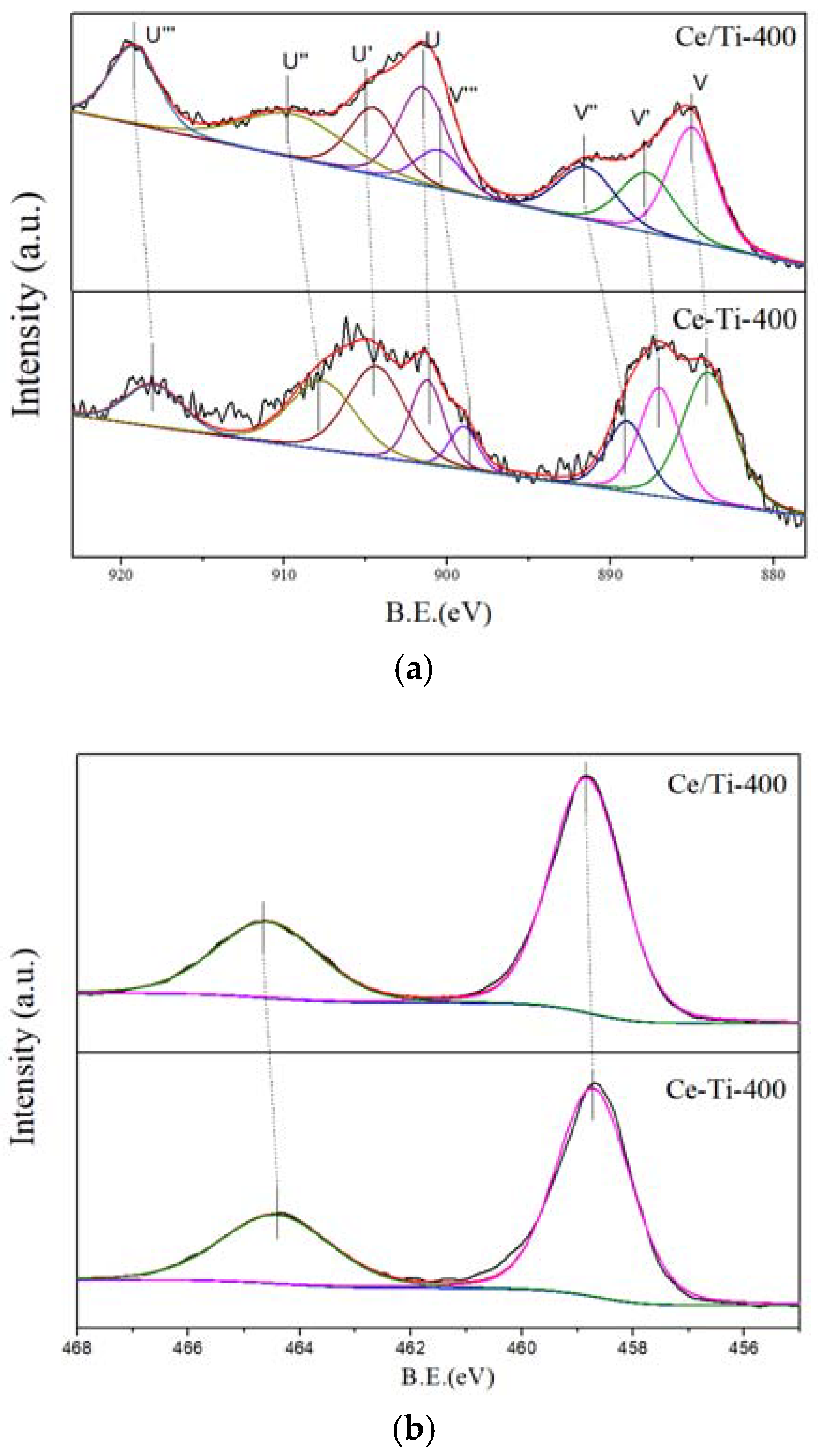
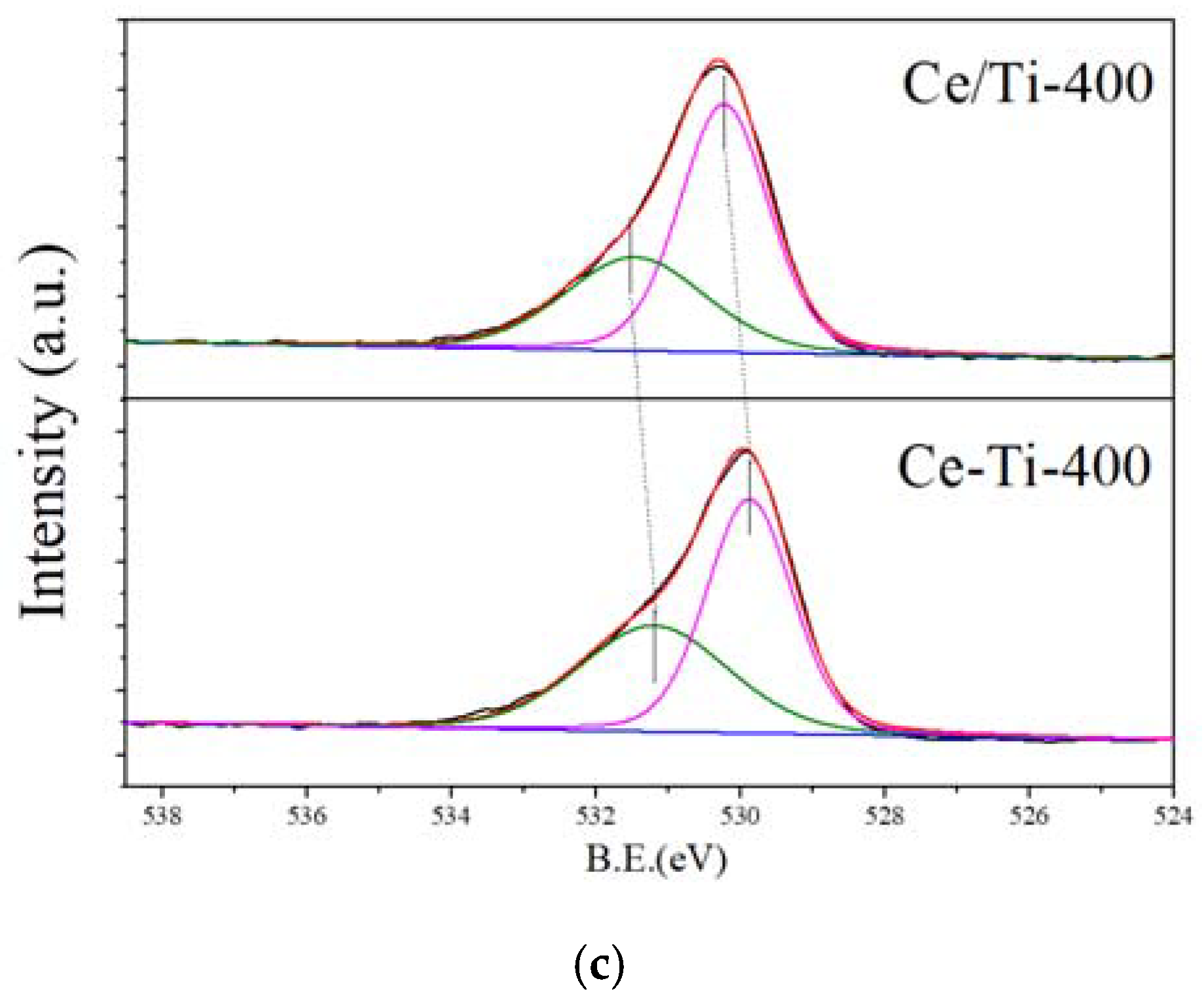
2.3. XPS and PL Analysis
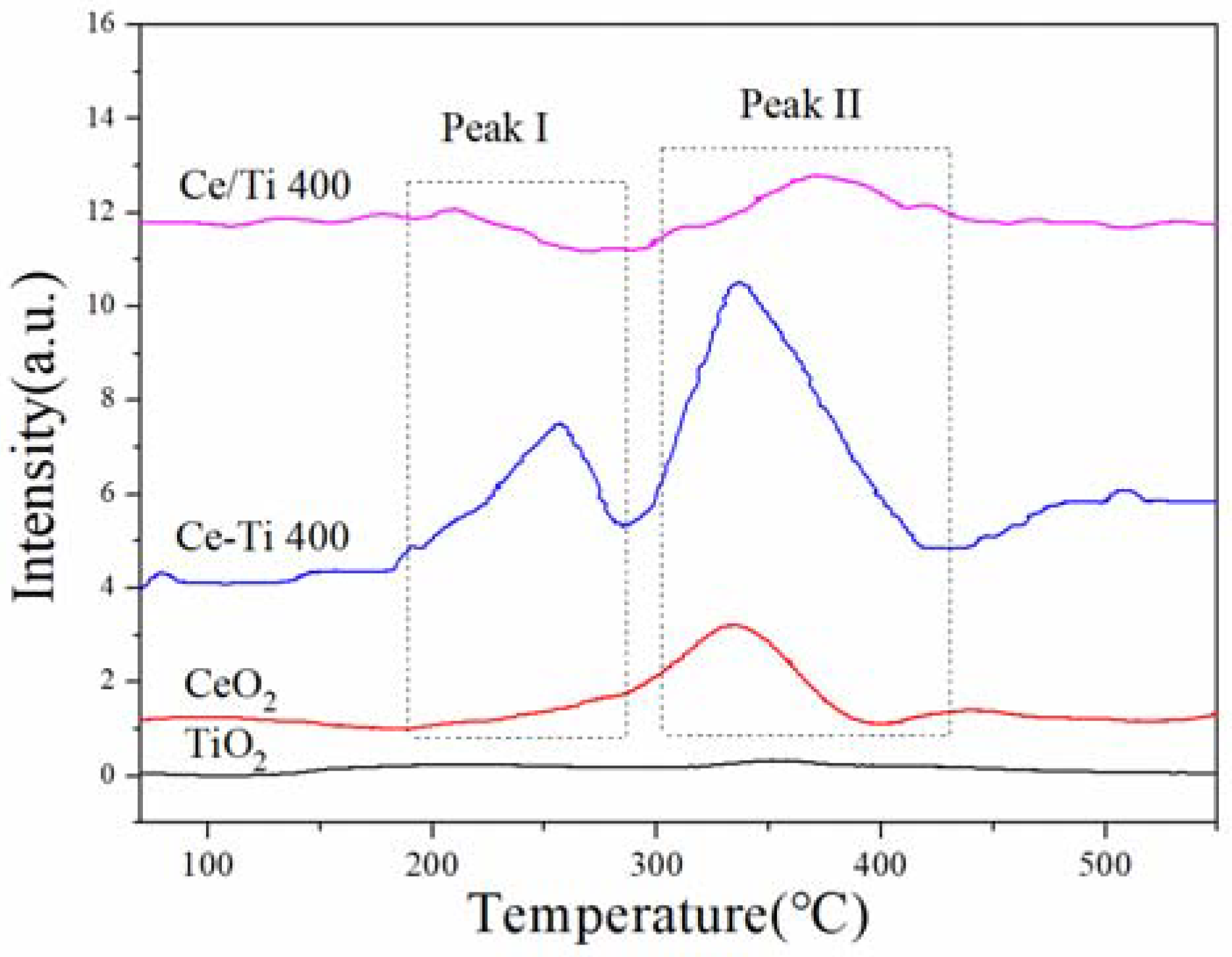
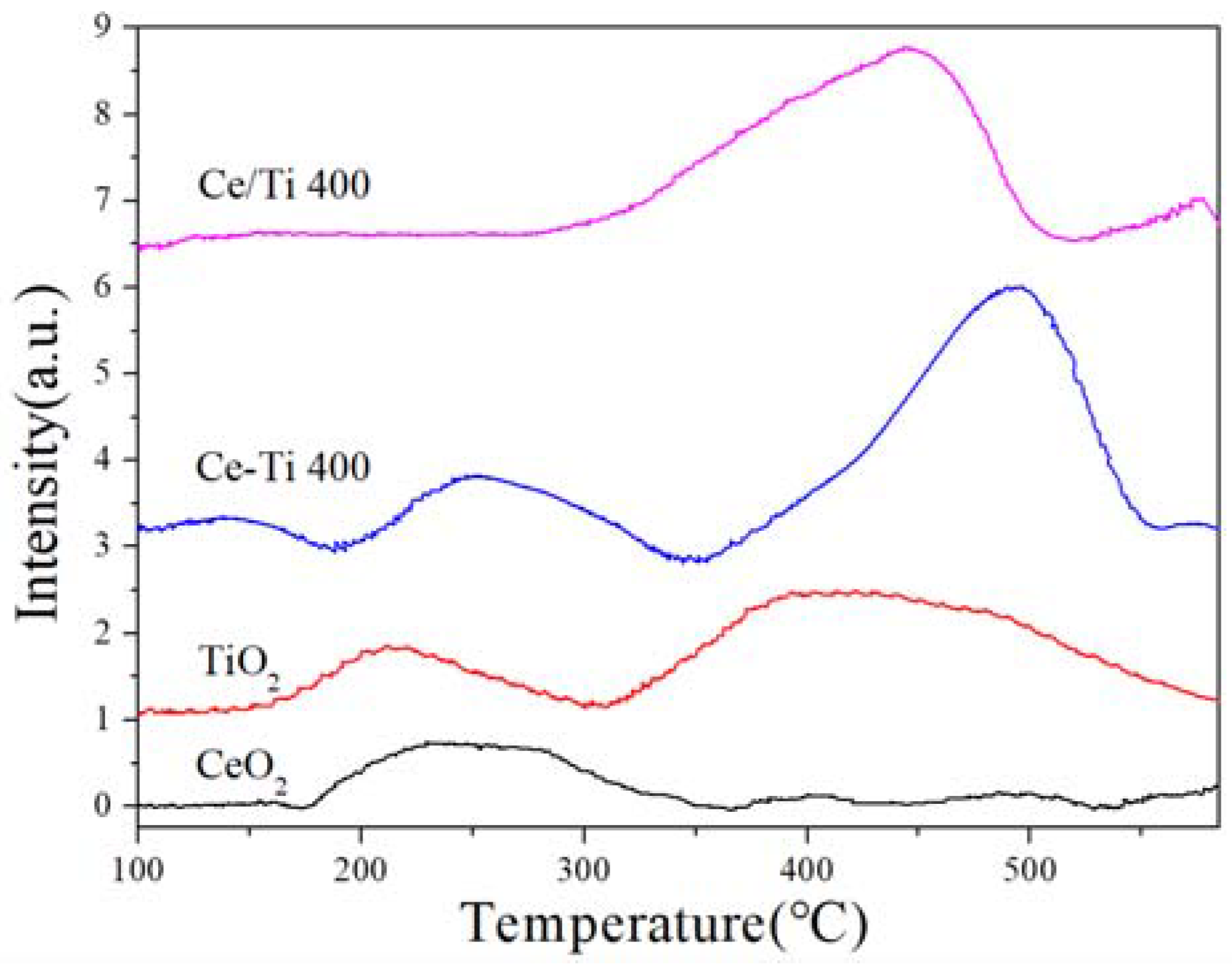
2.4. NO-TPD and NH3-TPD
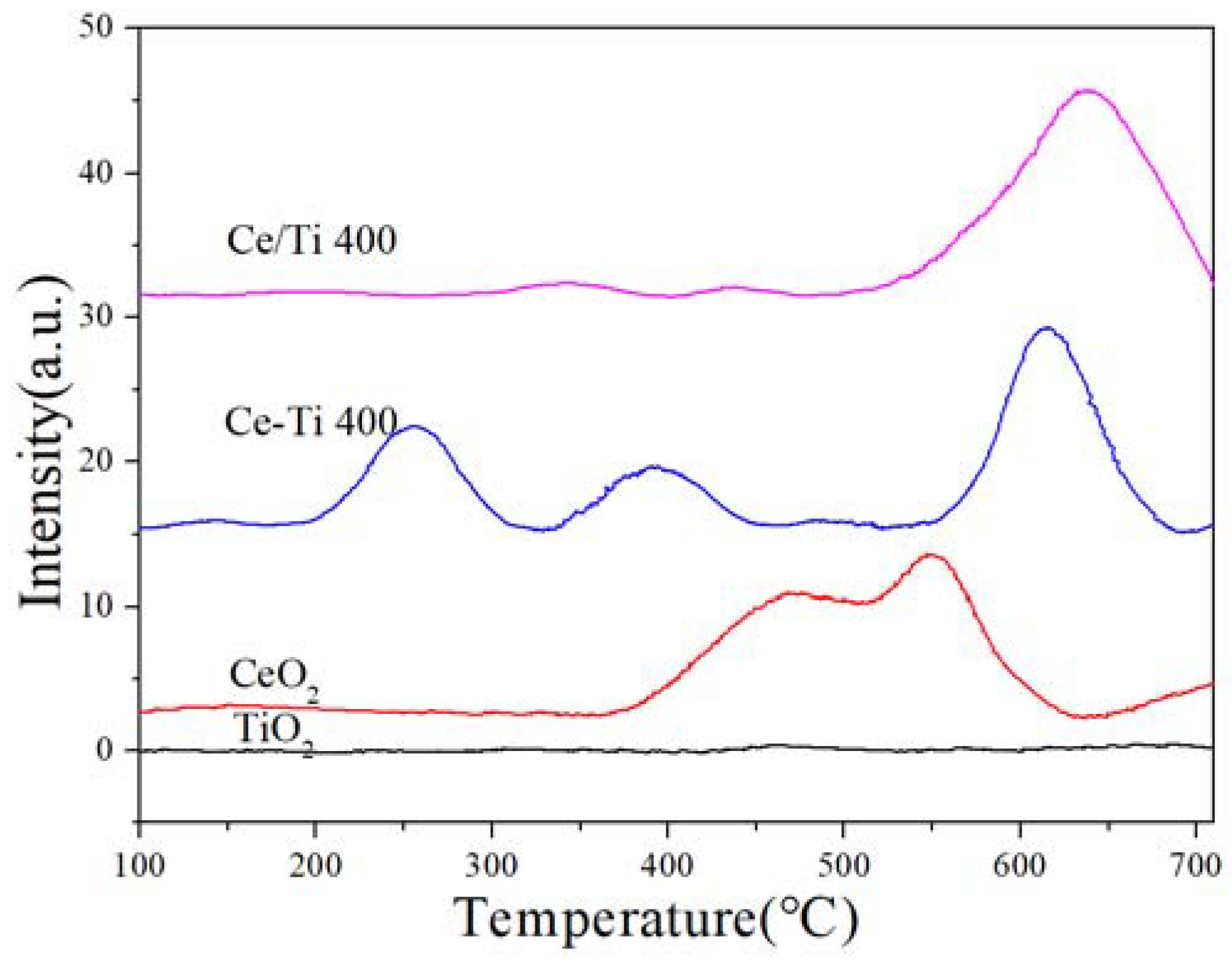
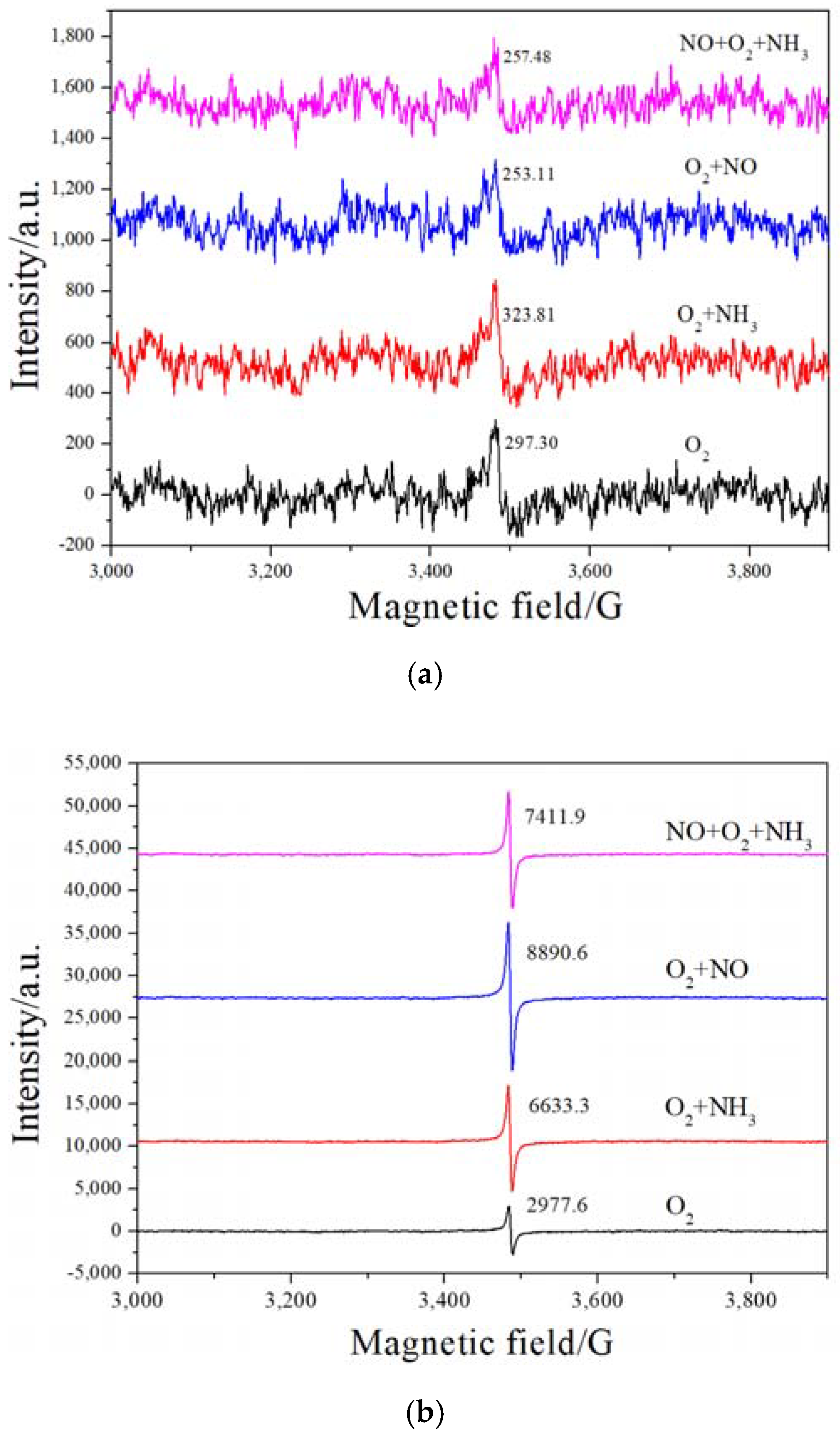
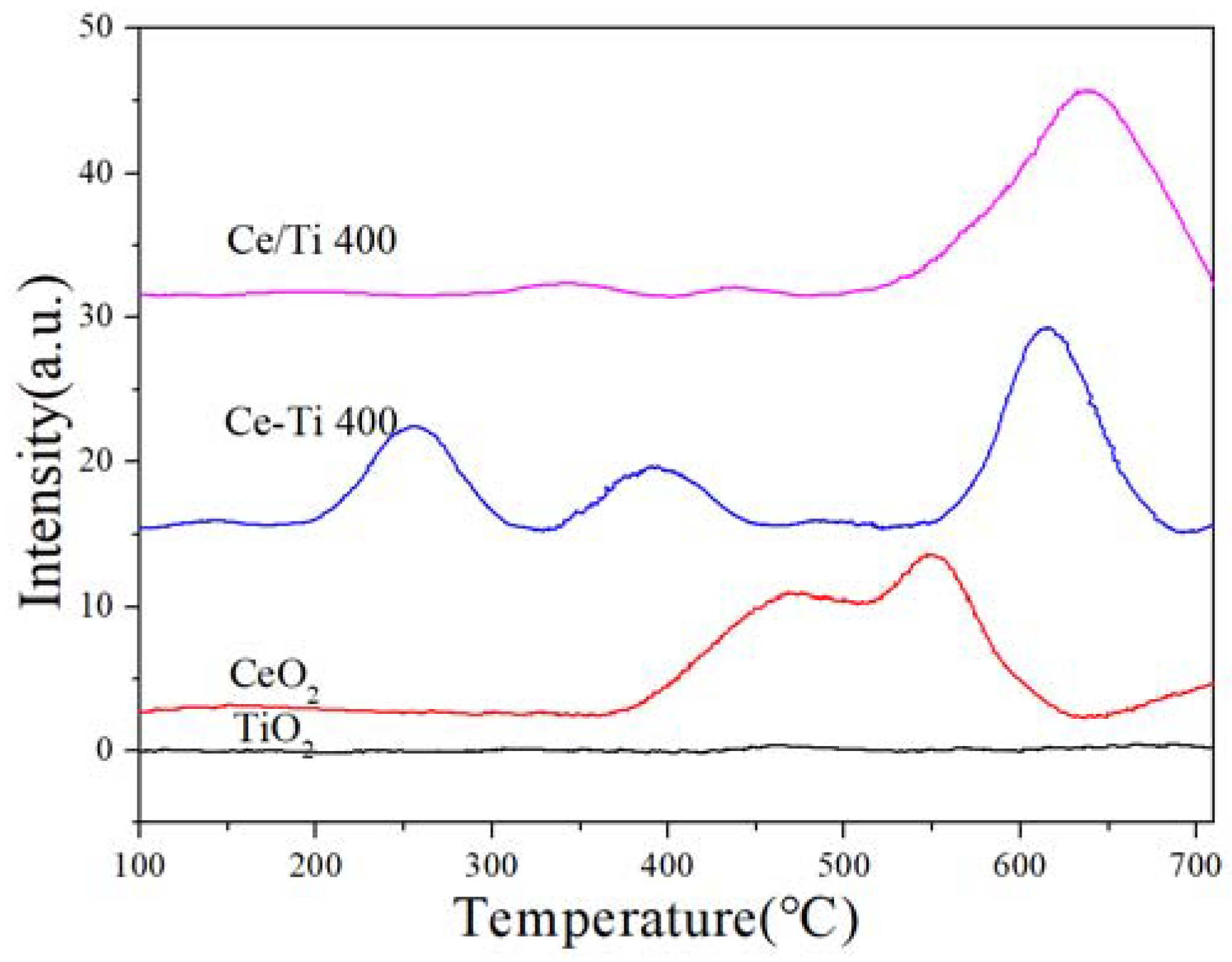
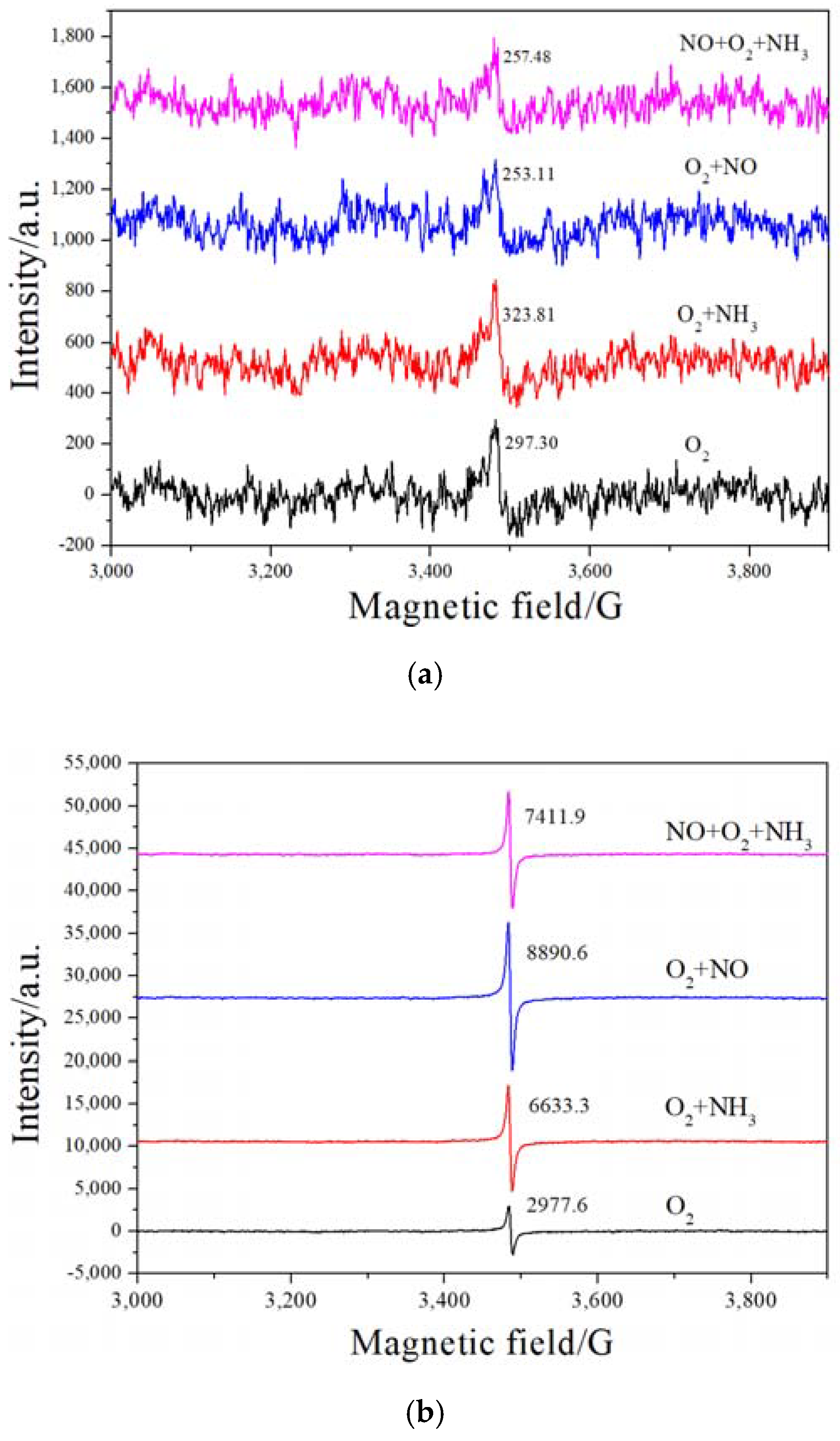
2.5. H2-TPR and EPR Analysis
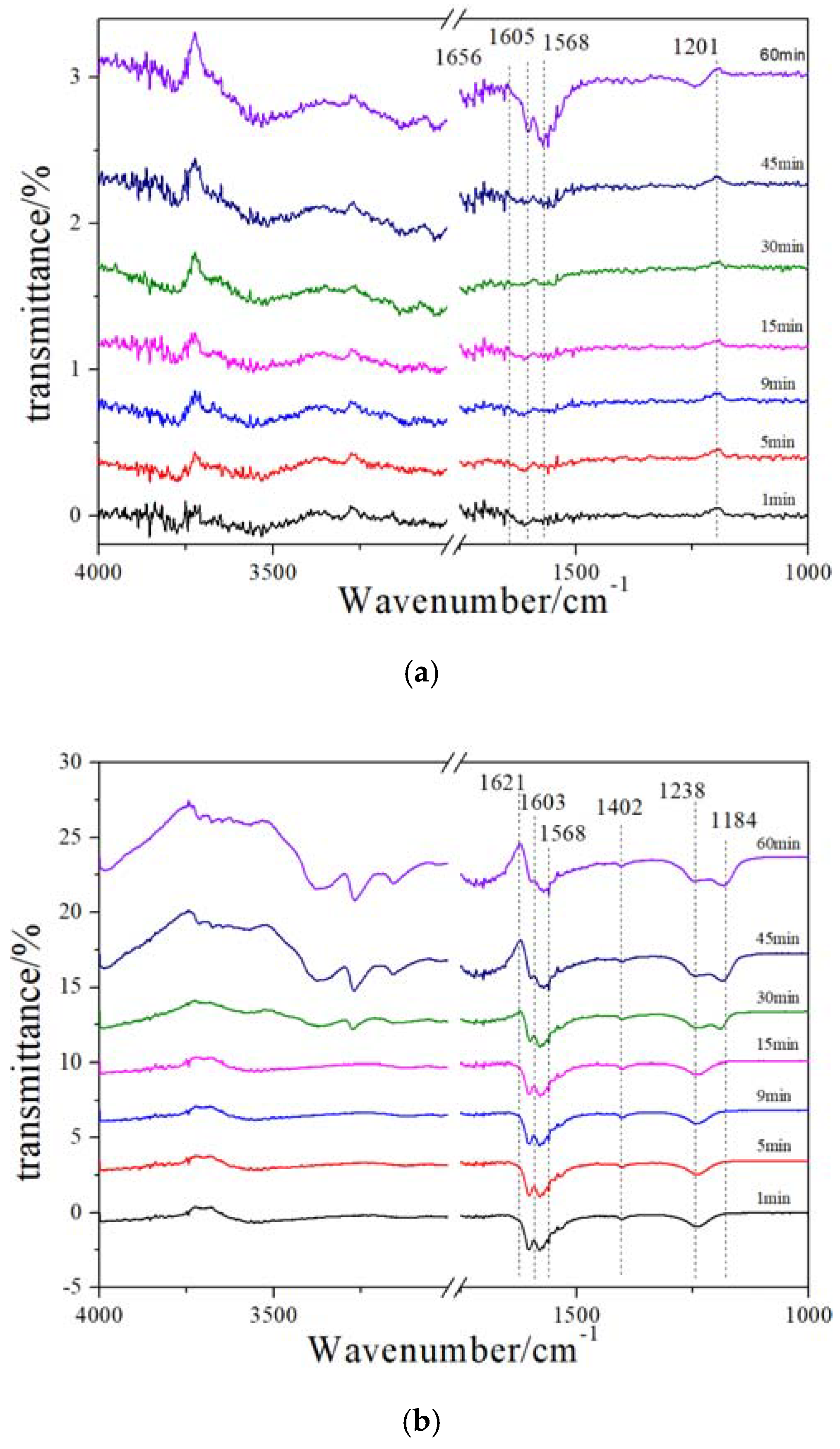
2.6. In Situ DRIFT (NH3 Adsorption after NO + O2 Pre-Adsorption)
3. Experimental
3.1. Synthesis of Catalysts
3.1.1. Synthesis of Doped Catalyst
3.1.2. Synthesis of Supported Catalyst
3.2. Catalyst Activity Tests
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Liu, G.; Gao, P.-X. A review of NOx storage/reduction catalysts: Mechanism, materials and degradation studies. Catal. Sci. Technol. 2011, 1, 552–568. [Google Scholar] [CrossRef]
- Xie, X.; Lu, J.; Hums, E.; Huang, Q.; Lu, Z. Study on the Deactivation of V2O5-WO3/TiO2 Selective Catalytic Reduction Catalysts through Transient Kinetics. Energy Fuels 2015, 29, 3890–3896. [Google Scholar] [CrossRef]
- Ding, S.; Liu, F.; Shi, X.; Liu, K.; Lian, Z.; Xie, L.; He, H. Significant Promotion Effect of Mo Additive on a Novel Ce-Zr Mixed Oxide Catalyst for the Selective Catalytic Reduction of NO(x) with NH3. ACS Appl. Mater. Interfaces 2015, 7, 9497–9506. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Yang, R.T.; Chang, R. MnOx-CeO2 mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2004, 51, 93–106. [Google Scholar] [CrossRef]
- Contreras-García, M.E.; García-Benjume, M.L.; Macías-Andrés, V.I.; Barajas-Ledesma, E.; Medina-Flores, A.; Espitia-Cabrera, M.I. Synergic effect of the TiO2-CeO2 nanoconjugate system on the band-gap for visible light photocatalysis. Mater. Sci. Eng. B 2014, 183, 78–85. [Google Scholar] [CrossRef]
- Muñoz-Batista, M.J.; Gómez-Cerezo, M.N.; Kubacka, A.; Tudela, D.; Fernández-García, M. Role of Interface Contact in CeO2–TiO2 Photocatalytic Composite Materials. ACS Catal. 2013, 4, 63–72. [Google Scholar] [CrossRef]
- Zhang, R.; Zhong, Q.; Zhao, W.; Yu, L.; Qu, H. Promotional effect of fluorine on the selective catalytic reduction of NO with NH3 over CeO2-TiO2 catalyst at low temperature. Appl. Surface Sci. 2014, 289, 237–244. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, Y.; Zhong, Y.; Luo, Z.; Cen, K. The activity and characterization of CeO2-TiO2 catalysts prepared by the sol-gel method for selective catalytic reduction of NO with NH3. J. Hazard. Mater. 2010, 174, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xin, Y.; Li, Q.; Wang, Z.; Zhang, Z.; Zheng, L. Ce-Ti amorphous oxides for selective catalytic reduction of NO with NH3: Confirmation of Ce-O-Ti active sites. Environ. Sci. Technol. 2012, 46, 9600–9605. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gu, H.; Li, P.; Zhou, Y.; Liu, Y.; Qi, Z.; Xin, Y.; Zhang, Z. In situ IR studies of selective catalytic reduction of NO with NH3 on Ce-Ti amorphous oxides. Chin. J. Catal. 2014, 35, 1289–1298. [Google Scholar] [CrossRef]
- Mosrati, J.; Atia, H.; Eckelt, R.; Lund, H.; Agostini, G.; Bentrup, U.; Rockstroh, N.; Keller, S.; Armbruster, U.; Mhamdi, M. Nb-Modified Ce/Ti Oxide Catalyst for the Selective Catalytic Reduction of NO with NH3 at Low Temperature. Catalysts 2018, 8, 175. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Feng, X.; Ma, L.; Cao, X.; Wang, B. Ni-Ce-Ti as a superior catalyst for the selective catalytic reduction of NOx with NH3. J. Mol. Catal. 2018, 445, 179–186. [Google Scholar] [CrossRef]
- Zhao, W.; Tang, Y.; Wan, Y.; Li, L.; Yao, S.; Li, X. Promotion effects of SiO2 or/and Al2O3 doped CeO2/TiO2 catalysts for selective catalytic reduction of NO by NH3. J. Hazard. Mater. 2014, 278, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, H.; Zeng, H.; Xu, Q. A novel Ce–Sb binary oxide catalyst for the selective catalytic reduction of NOx with NH3. Catal. Sci. Technol. 2016, 6, 8063–8071. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuwono, A.H.; Wang, J.; Li, J. Enhanced Photocatalysis by Doping Cerium into Mesoporous Titania Thin Films. J. Phys. Chem. C 2009, 113, 21406–21412. [Google Scholar] [CrossRef]
- Kityakarn, S.; Worayingyong, A.; Suramitr, A.; Smith, M.F. Ce-doped nanoparticles of TiO2: Rutile-to-brookite phase transition and evolution of Ce local-structure studied with XRD and XANES. Mater. Chem. Phys. 2013, 139, 543–549. [Google Scholar] [CrossRef]
- Mateos-Pedrero, C.; González-Carrazán, S.R.; Soria, M.A.; Ruíz, P. Effect of the nature of TiO2 support over the performances of Rh/TiO2 catalysts in the partial oxidation of methane. Catal. Today 2013, 203, 158–162. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, X.; Zhang, N.; Zhao, Q.; He, X.; Feng, J. Photocatalytic mechanism of TiO2–CeO2 films prepared by magnetron sputtering under UV and visible light. Surf. Sci. 2005, 595, 203–211. [Google Scholar] [CrossRef]
- Liu, Z.; Yi, Y.; Li, J.; Woo, S.I.; Wang, B.; Cao, X.; Li, Z. A superior catalyst with dual redox cycles for the selective reduction of NO(x) by ammonia. Chem. Commun. 2013, 49, 7726–7728. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Zhong, Q.; Cai, W. High performance of NO oxidation over Ce–Co–Ti catalyst: The interaction between Ce and Co. Appl. Surf. Sci. 2015, 325, 211–216. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G. Co3O4/CeO2 composite oxides for methane emissions abatement: Relationship between Co3O4–CeO2 interaction and catalytic activity. Appl. Catal. B Environ. 2006, 66, 217–227. [Google Scholar] [CrossRef]
- Nasir, M.; Xi, Z.; Xing, M.; Zhang, J.; Chen, F.; Tian, B.; Bagwasi, S. Study of Synergistic Effect of Ce- and S-Codoping on the Enhancement of Visible-Light Photocatalytic Activity of TiO2. J. Phys. Chem. C 2013, 117, 9520–9528. [Google Scholar] [CrossRef]
- Liu, J.; Han, R.; Zhao, Y.; Wang, H.; Lu, W.; Yu, T.; Zhang, Y. Enhanced Photoactivity of V−N Codoped TiO2 Derived from a Two-Step Hydrothermal Procedure for the Degradation of PCP−Na under Visible Light Irradiation. J. Phys. Chem. C 2011, 115, 4507–4515. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Meng, Y.; Ding, H.; Shan, Y.; Zhao, X.; Tang, X. A Highly Efficient Visible-Light-Activated Photocatalyst Based on Bismuth- and Sulfur-Codoped TiO2. J. Phys. Chem. C 2008, 112, 6620–6626. [Google Scholar] [CrossRef]
- Zhao, L.; Park, S.G.; Magyari-Köpe, B.; Nishi, Y. First principles modeling of charged oxygen vacancy filaments in reduced TiO2 –implications to the operation of non-volatile memory devices. Math. Comput. Model. 2013, 58, 275–281. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H.; Labhsetwar, N.K.; Hishita, S.; Ohashi, N. Visible-light-driven photocatalysis on fluorine-doped TiO2 powders by the creation of surface oxygen vacancies. Chem. Phys. Lett. 2005, 401, 579–584. [Google Scholar] [CrossRef]
- Li, D.; Ohashi, N.; Hishita, S.; Kolodiazhnyi, T.; Haneda, H. Origin of visible-light-driven photocatalysis: A comparative study on N/F-doped and N–F-codoped TiO2 powders by means of experimental characterizations and theoretical calculations. J. Solid State Chem. 2005, 178, 3293–3302. [Google Scholar] [CrossRef]
- Yu, L.; Zhong, Q.; Deng, Z.; Zhang, S. Enhanced NOx removal performance of amorphous Ce-Ti catalyst by hydrogen pretreatment. J. Mol. Catal. A Chem. 2016, 423, 371–378. [Google Scholar] [CrossRef]
- Li, H.; Zhang, S.; Zhong, Q. Effect of nitrogen doping on oxygen vacancies of titanium dioxide supported vanadium pentoxide for ammonia-SCR reaction at low temperature. J. Colloid Interface Sci. 2013, 402, 190. [Google Scholar] [CrossRef] [PubMed]
- Funk, S.; Hokkanen, B.; Burghaus, U.; Ghicov, A.; Schmuki, P. Unexpected adsorption of oxygen on TiO2 nanotube arrays: Influence of crystal structure. Nano Lett. 2007, 7, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, L.; Lu, G. A highly effective catalyst of Sm-MnOx for the NH3-SCR of NOx at low temperature: The promotional role of Sm and its catalytic performance. ACS Catal. 2015, 5, 5973–5983. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, Q.; Zhao, W.; Li, Y. Surface characterization studies on F-doped V2O5/TiO2 catalyst for NO reduction with NH 3 at low-temperature. Chem. Eng. J. 2014, 253, 207–216. [Google Scholar] [CrossRef]
- Ballinger, T.H.; Yates, J.T. IR spectroscopic detection of Lewis acid sites on alumina using adsorbed carbon monoxide. Correlation with aluminum-hydroxyl group removal. Langmuir 1991, 7, 3041–3045. [Google Scholar] [CrossRef]
- Jiang, Y.; Xing, Z.; Wang, X.; Huang, S.; Wang, X.; Liu, Q. Activity and characterization of a Ce–W–Ti oxide catalyst prepared by a single step sol-gel method for selective catalytic reduction of NO with NH3. Fuel 2015, 151, 124–129. [Google Scholar] [CrossRef]
- Chen, H.; Sayari, A.; Adnot, A.; Larachi, F.C. Composition–activity effects of Mn-Ce-O composites on phenol catalytic wet oxidation. Appl. Catal. B 2001, 32, 195–204. [Google Scholar] [CrossRef]
- Inturi, S.N.R.; Boningari, T.; Suidan, M.; Smirniotis, P.G. Visible-light-induced photodegradation of gas phase acetonitrile using aerosol-made transition metal (V, Cr, Fe, Co, Mn, Mo, Ni, Cu, Y, Ce, and Zr) doped TiO2. Appl. Catal. B 2014, 144, 333–342. [Google Scholar] [CrossRef]
- Dall’acqua, L.; Nova, I.; Lietti, L.; Ramis, G.; Busca, G.; Giamello, E. Spectroscopic characterisation of MoO3/TiO2 deNOx-SCR catalysts: Redox and coordination properties. Phys. Chem. Chem. Phys. 2000, 2, 4991–4998. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, X.; Lin, B.; Gao, B. Origin of the visible-light photoactivity of NH3-treated TiO2: Effect of nitrogen doping and oxygen vacancies. Appl Surf. Sci. 2013, 264, 845–852. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, C.Z.; Zhan, L.; Li, C.; Qiao, W.; Ling, L. MnOx-CeO2/Activated Carbon Honeycomb Catalyst for Selective Catalytic Reduction of NO with NH3 at Low Temperatures. Ind. Eng. Chem. Res. 2012, 51, 11667–11673. [Google Scholar] [CrossRef]
- Yao, X.; Zhao, R.; Chen, L.; Du, J.; Tao, C.; Yang, F. Selective catalytic reduction of NOx by NH3 over CeO2 supported on TiO2: Comparison of anatase, brookite, and rutile. Appl. Catal. B Environ. 2017, 208, 82–93. [Google Scholar] [CrossRef]
- Wang, C.; Yang, S.; Chang, H.; Peng, Y.; Li, J. Dispersion of tungsten oxide on SCR performance of V2O5WO3/TiO2: Acidity, surface species and catalytic activity. Chem. Eng. J. 2013, 225, 520–527. [Google Scholar] [CrossRef]
- Mu, W.; Zhu, J.; Zhang, S.; Guo, Y.; Su, L.; Li, X.; Li, Z. Novel proposition on mechanism aspects over Fe-Mn/ZSM-5 catalyst for NH3-SCR of NOx at low temperature: Rate and direction of multifunctional electron-transfer-bridge and in-situ DRIFTs analysis. Catal. Sci. Technol. 2016. [Google Scholar] [CrossRef]
- Volodin, A.M.; Cherkashin, A.E.; Zakharenko, V.S. Influence of physically adsorbed oxygen on the separation of electron-hole pairs on anatase irradiated by visible light. React. Kinet. Catal. Lett. 1979, 11, 103–106. [Google Scholar] [CrossRef]
- Volodin, A.M.; Cherkashin, A.E.; Zakharenko, V.S. Formation of O2− ion-radicals on reduced anatase. Influence of adsorbed CO on the stabilization of O2−. React. Kinet. Catal. Lett. 1979, 11, 107–111. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. DRIFT study on cerium-tungsten/titania catalyst for selective catalytic reduction of NOx with NH3. Environ. Sci. Technol. 2010, 44, 9590. [Google Scholar] [CrossRef] [PubMed]
- Ellmers, I.; Vélez, R.P.; Bentrup, U.; Schwieger, W.; Brückner, A.; Grünert, W. SCR and NO oxidation over Fe-ZSM-5—The influence of the Fe content. Catal. Today 2015, 258, 337–346. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, D.; Zhang, S.; Zeng, Y.; Wang, P.; Zhong, Q. Active Site of O2 and Its Improvement Mechanism over Ce-Ti Catalyst for NH3-SCR Reaction. Catalysts 2018, 8, 336. https://doi.org/10.3390/catal8080336
Jiang D, Zhang S, Zeng Y, Wang P, Zhong Q. Active Site of O2 and Its Improvement Mechanism over Ce-Ti Catalyst for NH3-SCR Reaction. Catalysts. 2018; 8(8):336. https://doi.org/10.3390/catal8080336
Chicago/Turabian StyleJiang, Dong, Shule Zhang, Yiqing Zeng, Pengfei Wang, and Qin Zhong. 2018. "Active Site of O2 and Its Improvement Mechanism over Ce-Ti Catalyst for NH3-SCR Reaction" Catalysts 8, no. 8: 336. https://doi.org/10.3390/catal8080336
APA StyleJiang, D., Zhang, S., Zeng, Y., Wang, P., & Zhong, Q. (2018). Active Site of O2 and Its Improvement Mechanism over Ce-Ti Catalyst for NH3-SCR Reaction. Catalysts, 8(8), 336. https://doi.org/10.3390/catal8080336