1. Introduction
Bifunctional oxygen evolution (OE) and oxygen reduction (OR) reactions are the anodic and cathodic half-cell reactions taking place in the air electrode during charging and discharging of the Metal-Air Batteries (MAB). These reactions are also the backbone of regenerative fuel cells, electrolysers, and photo-electrochemical cells converting electrical energy into chemical energy or vice versa, which could be drawn back when there is a need [
1]. The OE and OR reactions involve the two-electron or four-electron transfer reactions that are hindered by significant overpotentials and slow kinetics. In order to overcome these limitations, the OE and OR reactions should employ an active catalyst to accelerate the reactions. Platinum group metal (PGM) elements Pt, Ir, and Ru are known as the best catalysts for these reactions [
2,
3,
4]. However, these PGM elements’ availability, stability, and cost are prohibitive to commercialize on a broader scale [
3,
5].
In this scenario, transition metal oxides based on perovskites, pyrochlores, layered double hydroxide (LDH), and spinels are promising candidates for oxygen reduction and evolution reactions. Because of their structural robustness, cost, accessibility, ease and mode of synthesis, desirable activity, low electrical resistance, and thermodynamic stability in the aqueous alkaline electrolyte [
5,
6], they are preferred as alternatives for both reactions. Among these different structures, spinel-based oxides pose superior OE and OR performances, due to their mixed valence states [
7]. Another important class of materials is LDH, which are ionically conducting-mixed valence compounds with layered structure. These LDH phases are more active than the spinel or rock salt type structures due to the fact they are readily oxidizable to metal hydroxide/oxyhydroxide interphase that is highly active for the OE reaction [
8]. There have been different LDH phases such as Ni-Fe, Ni-Co, Co-Fe, Ni-Ni, Ni-Mn, Mn-Co, Co-Mn, and Co-Ni, which were reported as efficient OER catalysts in alkaline media [
8,
9,
10,
11]. These LDH phases are easily convertible to different oxide or spinel phases upon the removal of intercalated anions. These oxides and LDH have the mixed valency state that provides them donor-acceptor chemisorption sites for reversible oxygen adsorption [
12]. Besides the electron, conduction between mixed valence of spinels enables high conductivity and dramatically changes the bond strength of surface–intermediate species [
12,
13]. Spinels (AB
2O
4) with earth abundant transition metals (Fe, Co, Ni, Cu, and Mn) are considered to be good options compared to the noble metals [
14]. The substitution of the B site ion or using different A and B combinations are tuned to surface redox sites and metal-oxygen bonds to give a desirable state, subsequently leading to superior performance. Recently, several transition metal oxide (A
2+A
3+2O
4) spinels and mixed transition metal oxide (A
2+B
3+2O
4) spinels have been reported as either bifunctional or single OE or OR electrocatalysts in an alkaline environment, such as cobaltites, ferrites, and manganates [
15,
16,
17]. Among them, inverse spinel type-NiFe
2O
4 gained tremendous interest owing to its low cost, environmental friendliness, widespread availability, activity, high conductivity, and stability. However, the electrocatalyst activity varies by structure, valence, composition, and morphology, which in its turn depends on synthesis methods and conditions [
18]. The crystallinity, phase, and crystallite size and oxidation state of the metal center could be varied during the preparation, for example by calcination at different temperatures [
19].
The most commonly used synthesis methods are conventional solid-state, vapor phase deposition, and solution-based. The solid-state method consumes a lot of energy and time to produce the micro-sized particles. Even though the vapor phase deposition could produce an ultra-pure atomic thick-porous structure, its mass production will be expensive and time-consuming [
17]. The solution-based methods are easily scalable and low temperature-driven, and morphological control is also possible. These methods include sol-gel, co-precipitation, hydrothermal, micro-emulsion, microwave, and electrochemical synthesis. Among them, hydrothermal synthesis has several desirable features: low cost, non-toxic chemicals, high purity homogeneous phase, uniform sized nanoparticle, and preferentially oriented crystal facets [
20,
21,
22]. This method could be used to synthesize Ni ferrite nanoparticles with the increased surface area and surface properties.
Herein, we prepared Ni and Fe oxide phases through the urea-assisted hydrothermal method, and the samples were sintered within a temperature range of 100 to 600 °C. The samples were characterized using XRD (X-ray Diffraction), TEM (Transmission Electron Microscopy), N2 adsorption isotherm, and Raman and X-ray photoelectron spectroscopy (XPS). Electrochemical performance such as bifunctional activity, as well as the electrode kinetics, were studied using Cyclic Voltammetry (CV) and linear sweep voltammetry (LSV) coupled with RDE (Rotating Disk electrode) in alkaline electrolyte.
2. Results and Discussions
Ni-Fe carbonate hydroxide was synthesized using the urea assisted-hydrothermal method. After the hydrothermal synthesis, the centrifuged products were further subjected to calcination to result in the final product (Ni-ferrite). The reaction sequence is given in the following Equations (1)–(3)
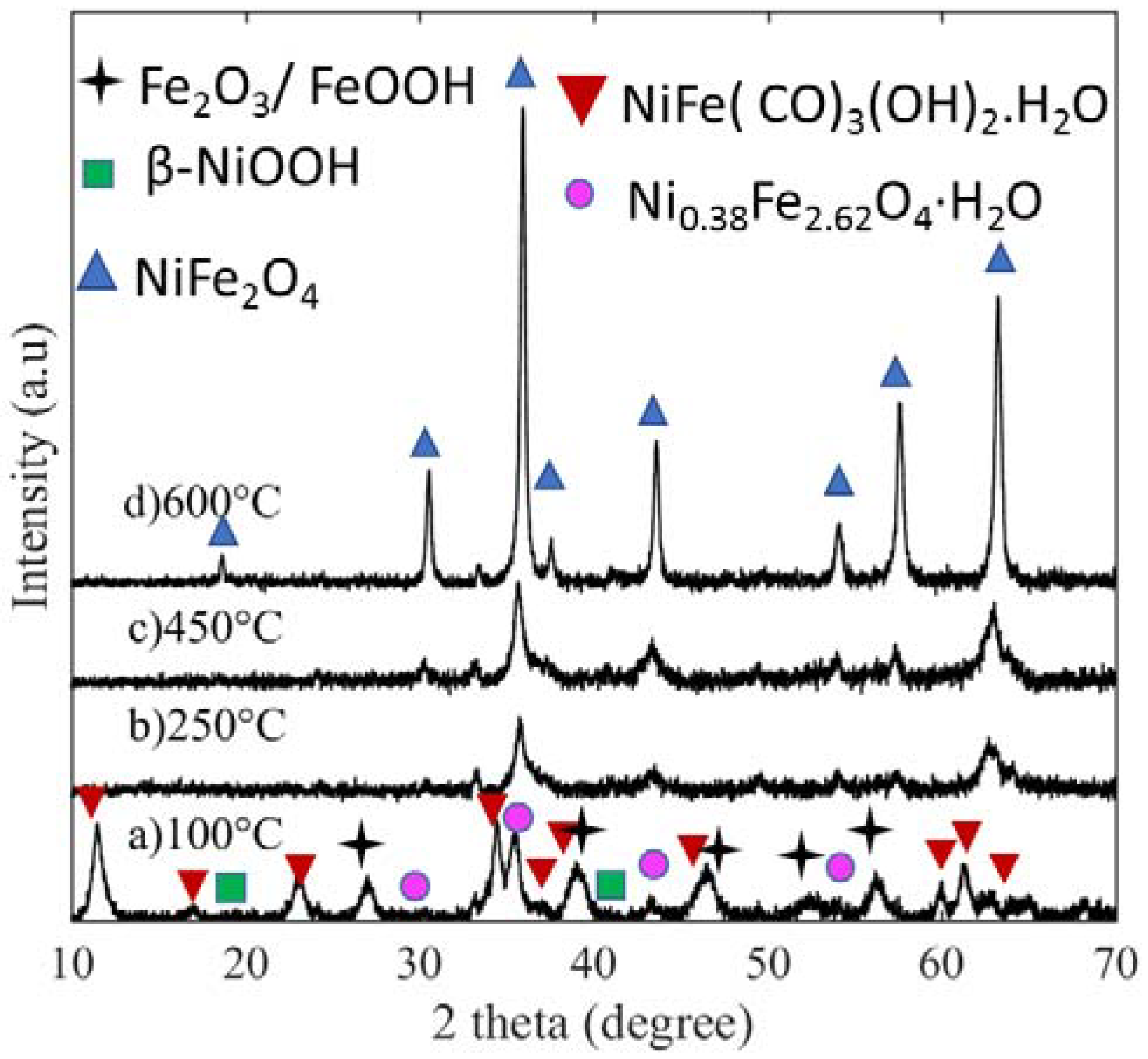
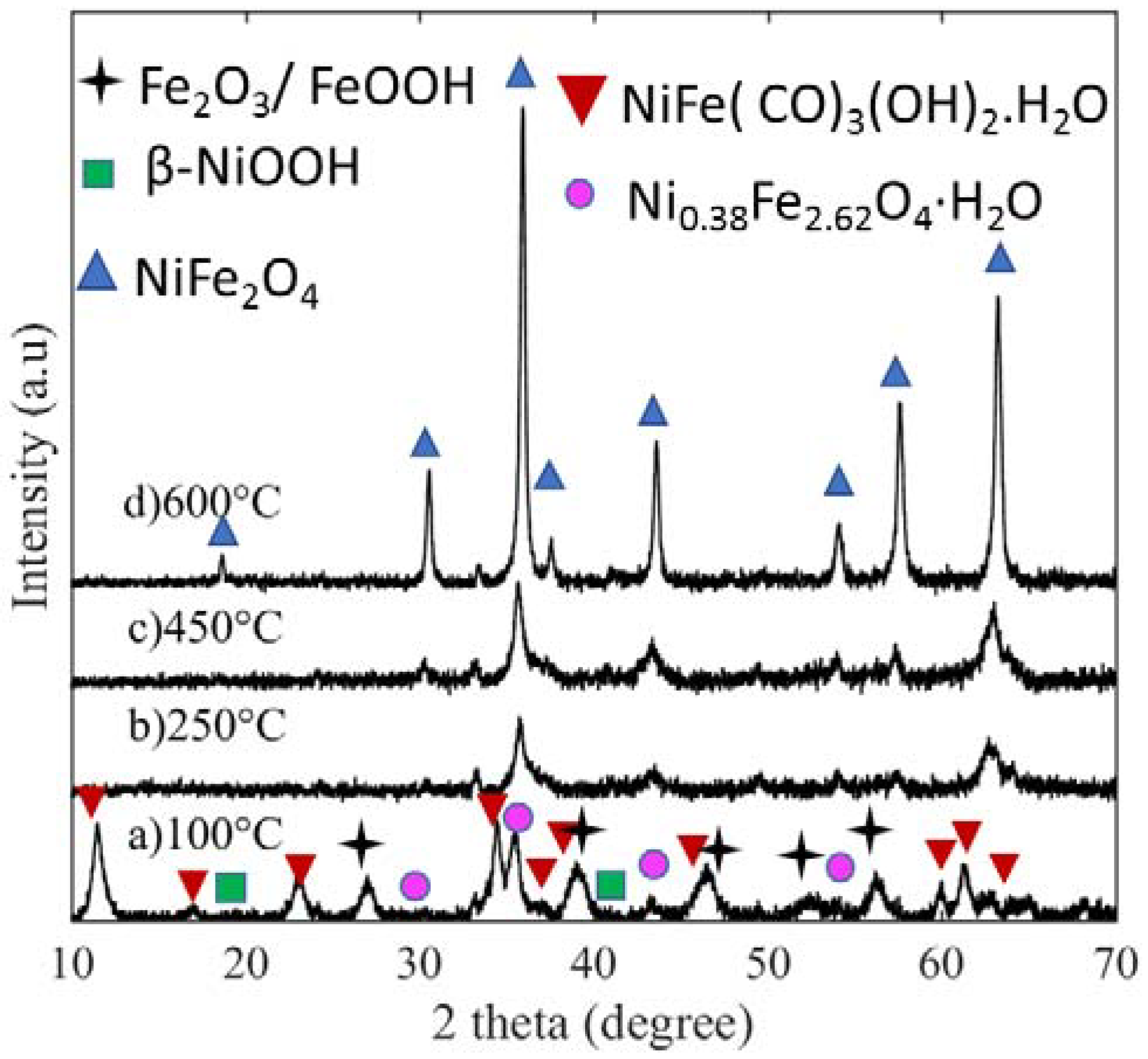
The crystalline phase evolution of NiFe oxides is shown in
Figure 1 as a function of calcination temperature, in which the patterns listed from (a) to (d) correspond to 100, 250, 450, and 600 °C calcined samples, respectively. At 100 °C, the sample has three major phases: namely, nickel iron hydroxide hydrate (Ni
0.38Fe
2.62O
4·H
2O, PDF 00-056-1344), layered double hydroxide carbonate phase (Ni
0.75Fe
0.25(CO
3)
0.125(OH)
2. 0.38 H
2O, PDF 00-040-0215), and Iron Oxide Hydrate phase (β-Fe
2O
3H
2O, PDF 00-003-0440). Further, there are two minor phases: NiOOH and FeOOH. When the temperature is further raised to 250 °C, water and carbon dioxide are liberated, as shown in Equations (1) and (2) from the layered carbonate and hydroxide (LDH) phases leading to the formation of spinel nickel ferrite (NiFe
2O
4 JCPDS74-2081). Further calcination at 450 and 600 °C results in more crystalline NiFe
2O
4. The measured Scherrer crystallite size for 100, 250, 450, and 600 °C calcined samples is given in
Table 1. The crystallite size is measured at 11.52°, 003 orientation of LDH phase. For the remaining samples, crystallite sizes are measured at ≈35.90°, (311) orientation of nickel ferrite. The peaks at 35.9°and 65.3° correspond to (311) and (440) crystal orientations of inverse nickel ferrite (NiFe
2O
4) for those samples calcined at 250, 450, and 600 °C. The intensities of the peaks at (311) and (440) suggest that an increase in calcination temperature increased both the (311) and the (440) crystal facets. Thus, calcination temperature has a direct effect on phase, crystallinity, and crystallite size.
Using this synthesis approach, well-crystallized nanoparticles were prepared starting from 250 °C. To further understand the calcination temperature effect on the samples morphology, TEM measurement was performed, as shown in
Figure 2a–d. As observed from
Figure 2a–c, the majority of the particles are in nanorod morphology and some of them in thin platelet and oblong structure at 100 to 450 °C. However, at 250 and 450 °C the particles start forming cavities and agglomeration to form bigger particles. At 600 °C, the particle morphology is completely changed into clunches having both spherical and cubic structures with different edges, confirming the XRD results, where the crystallite size changes from 13.85 to 10.89 nm for the samples at 100 and 250 °C, respectively and increases to 13.53 nm at 450 °C and then finally, it grows to 26.66 nm at 600 °C. The anomalous change especially at 250 °C is due to the decarbonization and dehydration causes the crystallite size to shrink which is also responsible for the observed cavities in the TEM image (
Figure 2b). The TEM result shows that in most of the samples, the particles have well dispersed-nanorod structures which are favourable catalytic active sites for oxygen redox reaction [
23].
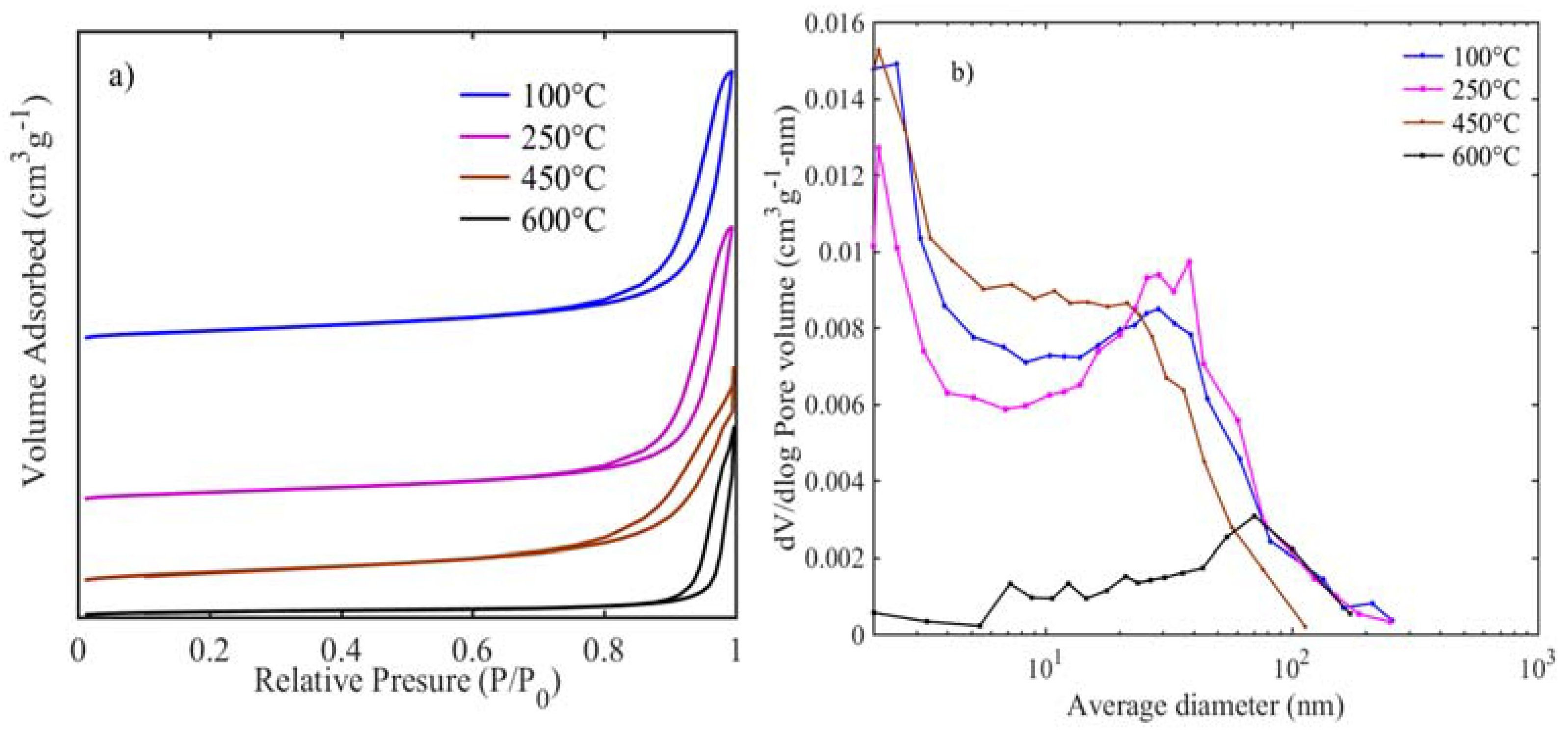
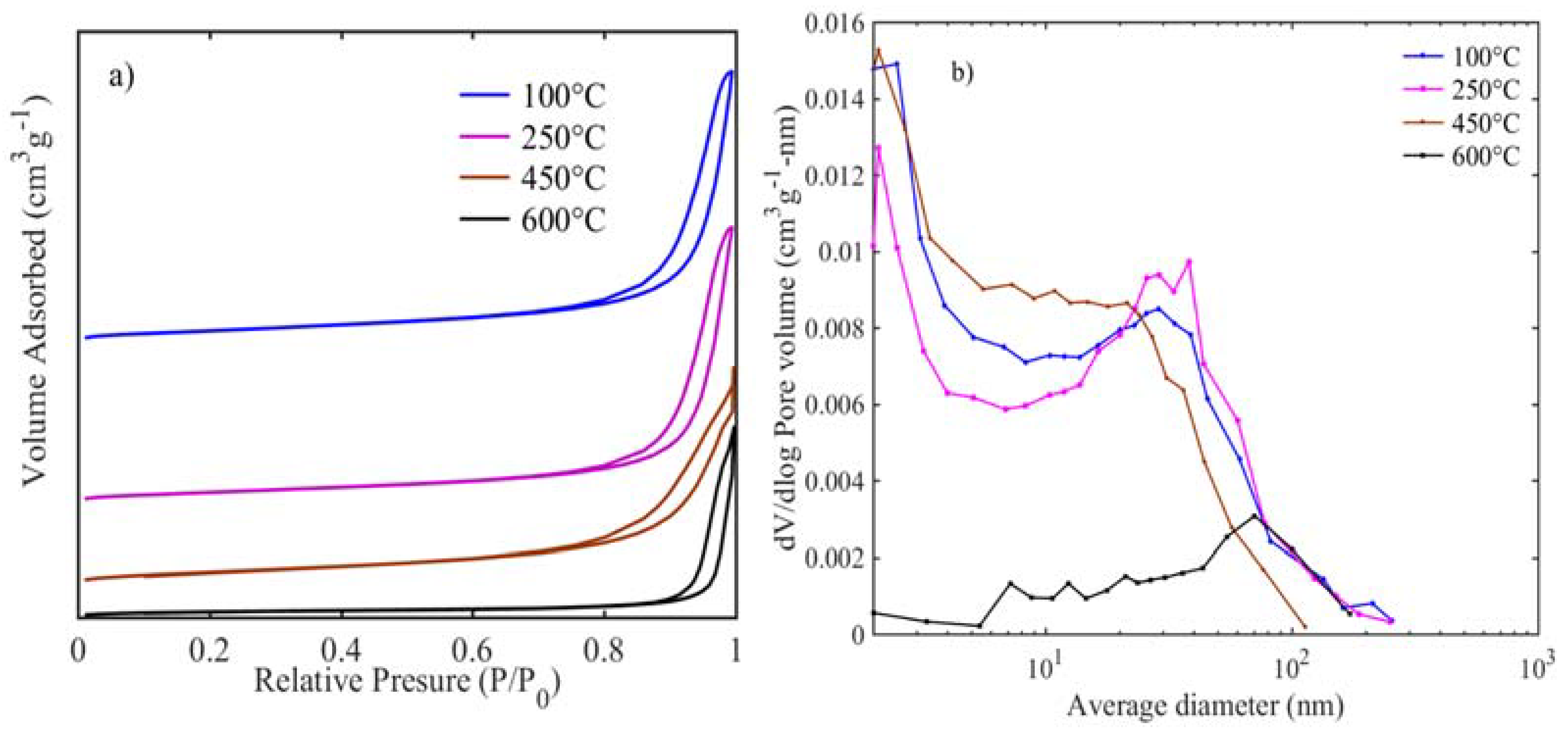
Usually, electrocatalytic activity is directly related to the accessible surface area for the oxygen redox reaction [
24]. BET surface area, pore volume, and pore size distribution results are presented in
Figure 3 and
Table 1. All the samples are of the type-IV isotherm (
Figure 3a) with H3 hysteresis loop, which is typical for aggregates of micro/mesoporous particles [
25]. Upon increasing the calcination temperature, an increase in hysteresis between adsorption-desorption is observed with relative pressures moving more towards unity. The highest surface area (131 m
2 g
−1) is exhibited for the sample calcined at 100 °C, and when the calcination temperature increases to 250 °C, it results in a similar surface area of 127 m
2 g
−1. Thus, the decarbonization and dehydration have not altered the surface and porous nature of the samples. Further increase in calcination temperature results in a surface area of 118 m
2 g
−1 at 450 °C, which is dramatically reduced to 40 m
2 g
−1 at 600 °C. This confirms that the increase in crystallite size as observed in the TEM and decrease in BET areas are consistent with the calcination-induced nanoparticle growth [
26]. Average pore size distribution for samples at 100–450 °C (
Figure 3b) shows pores are in a bi-modal size that have less than 2 nm micropores and mesopores between 10–50 nm, while for the sample heat-treated at 600 °C, most of the pores are in the meso to macropore range (10–60 nm). The pore size distributions for those samples calcined at 100–450 are similar to layered and pillared solids, while the sample at 600 °C is similar to porous ceramics [
27]. Likewise, the pore volumes tend to decrease substantially with the increase in heat-treatment. Therefore XRD, TEM, and BET results reveal that the nano-sized, rod-like, and high surface area NiFeO
x catalyst surfaces are easily accessible by the oxygen and hydroxide ion for the desirable gas-liquid interface oxygen redox reactions.
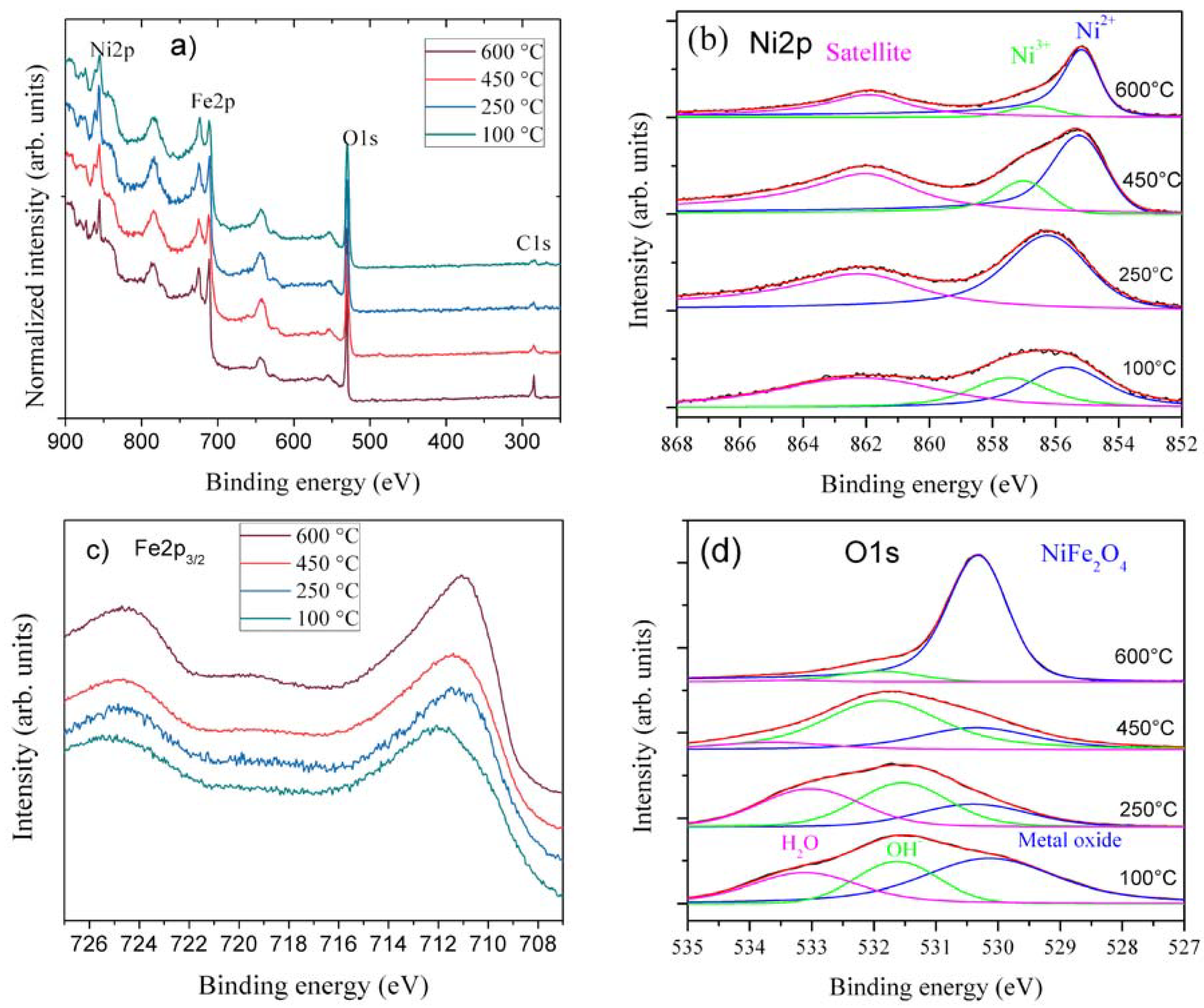
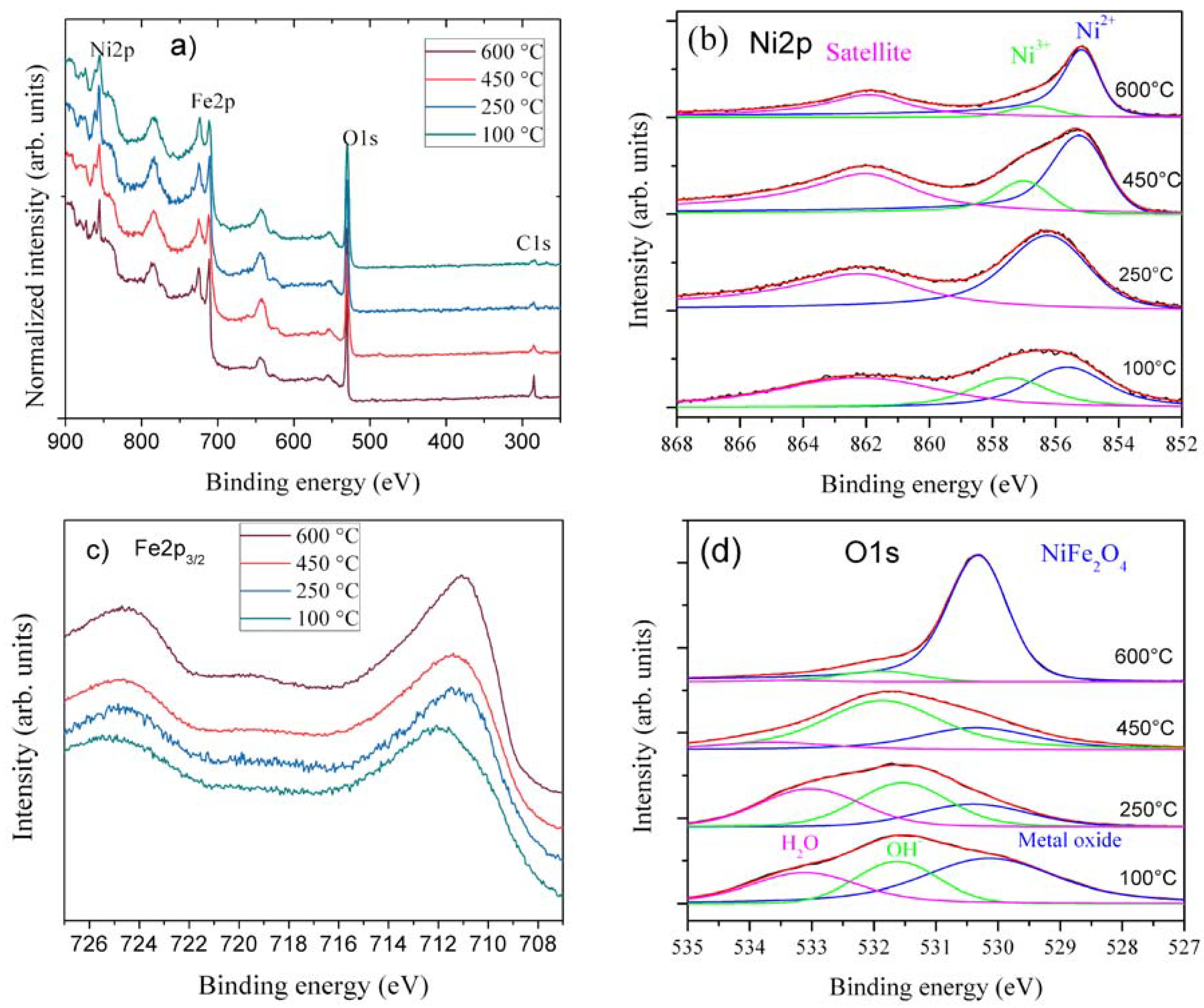
The surface oxidation state of the samples is analyzed using XPS with focus on Ni2p, Fe2p, O1s, and C1s regions.
Figure 4a–d shows the wide spectra and close up spectra of Fe2p, Ni2p, and O1s from all four samples. Peak deconvolutions of Ni2p and O1s are shown in
Figure 4b,d. The wide spectra show minimal carbon presence, except at 600 °C (
Figure 4a). The relative Ni2p intensity decreases at higher temperatures. O1s, Fe2p, and Ni2p are broad at lower temperature calcinations, while they sharpen at higher temperatures. This is in agreement with the sharper and pure phase formation at higher temperatures in XRD. Ni2p spectra show broad and multiple split peaks spanning 859 to 854 eV, confirming different metal oxidation states and co-ordinations in the lattice. However, at higher temperatures, Ni peaks narrow and move to lower binding energy. At 100 °C, samples have both octahedral arising from Ni(OH)
2 and NiFe(CO)
3(OH)
2, and tetrahedral coordination from NiOOH, while at 250–450 °C, calcination leads to more tetrahedral coordination in the lattice. This indicates that in low-temperature spinel, Ni
2+ ions tend to be both in the octahedral and tetrahedral coordination, as they are in inverse spinels, while at 600 °C, Ni ions solely go to octahedral coordination [
28], as shown in
Figure 4b. The relative intensities of peak deconvoluted-ration (
Table S1) of Ni
2+/Ni
3+ and O1S also confirm that more metal oxide with spinel structure forms at high temperatures. The Ni peak shapes are similar to those reported by the authors of [
29]. Fe2p
3/2 spectra from the four samples are presented in
Figure 4c. There are no dramatic variations in line shape and peak position. At 100 °C, spectra are closer to Fe
2O
3 [
30,
31]; they also have slight additional intensity at higher binding energy from the NiFe(CO)
3(OH)
2. The 250 and 450 °C samples spectra are gradually shifted from the 100 °C spectrum towards the 600 °C spectrum. The 600 °C calcined sample spectrum resembles the Fe2p spectrum from NiFe
2O
4 [
28]. O1s spectra of different samples are shown in
Figure 4d, which substantially supports the distinction between the calcined samples. At 100 °C, there are three peaks: metal oxide, hydroxide, and H
2O [
30]. The metal oxide peak can be assigned to Fe
2O
3, since it is one of the starting materials and is observed in XRD. Both OH
− and H
2O groups are also part of the phases observed in XRD. Further heating up to 250 °C decreases the metal oxide peak and coincides with Fe
2O
3 conversion into the spinel phase. At 400–650 °C, there is only metal-oxide peak confirming that almost all water and hydroxide has been removed and the NiFe
2O
4 phase dominates. However, the peak position shift to low binding energy at low temperatures coincides with the observed peak shift in Fe and Ni spectra. Therefore, the XPS results suggest multiple oxidation state and surface functional groups are present at low temperatures, particularly at 250 °C. A surface rich in oxygen functional groups will enhance the electrocatalytic activity of the water oxidation [
32,
33].
To understand the type of surface groups present in the NiFeO
x, Raman spectroscopy was performed and shown in
Figure S4.
Figure S4a shows the Raman spectra of 100–250 °C heat-treated samples. At 100 °C, samples have shown the characteristic peaks of CO
32−, M-OH, and M
II-O-M
III [
34]. When the heat treatment increased to 250 °C, it resulted in characteristic signals of NiFe
2O
4 and LDH phases. Noticeably, the highly intensive M-OH signal is observed at 295 cm
−1. This observation, together with XPS results, confirms that more Ni hydroxide phases are formed on the surface of 250 °C sample.
Figure S4b shows the 450–600 °C heat-treated samples’ Raman spectra. The temperature increase causes the disappearance of M-OH and CO
32−. The mixture of Ni and iron oxide phases is observed at 450 °C. The XPS Ni2p spectra showed two types of Ni oxidation states at 450 °C, whereas at 600 °C the Raman spectra shown in
Figure S4b is similar to commercial NiFe
2O
4 [
35]. Thus, the low-temperature samples have a wide range of surface functional groups, which is beneficial for redox reactions.
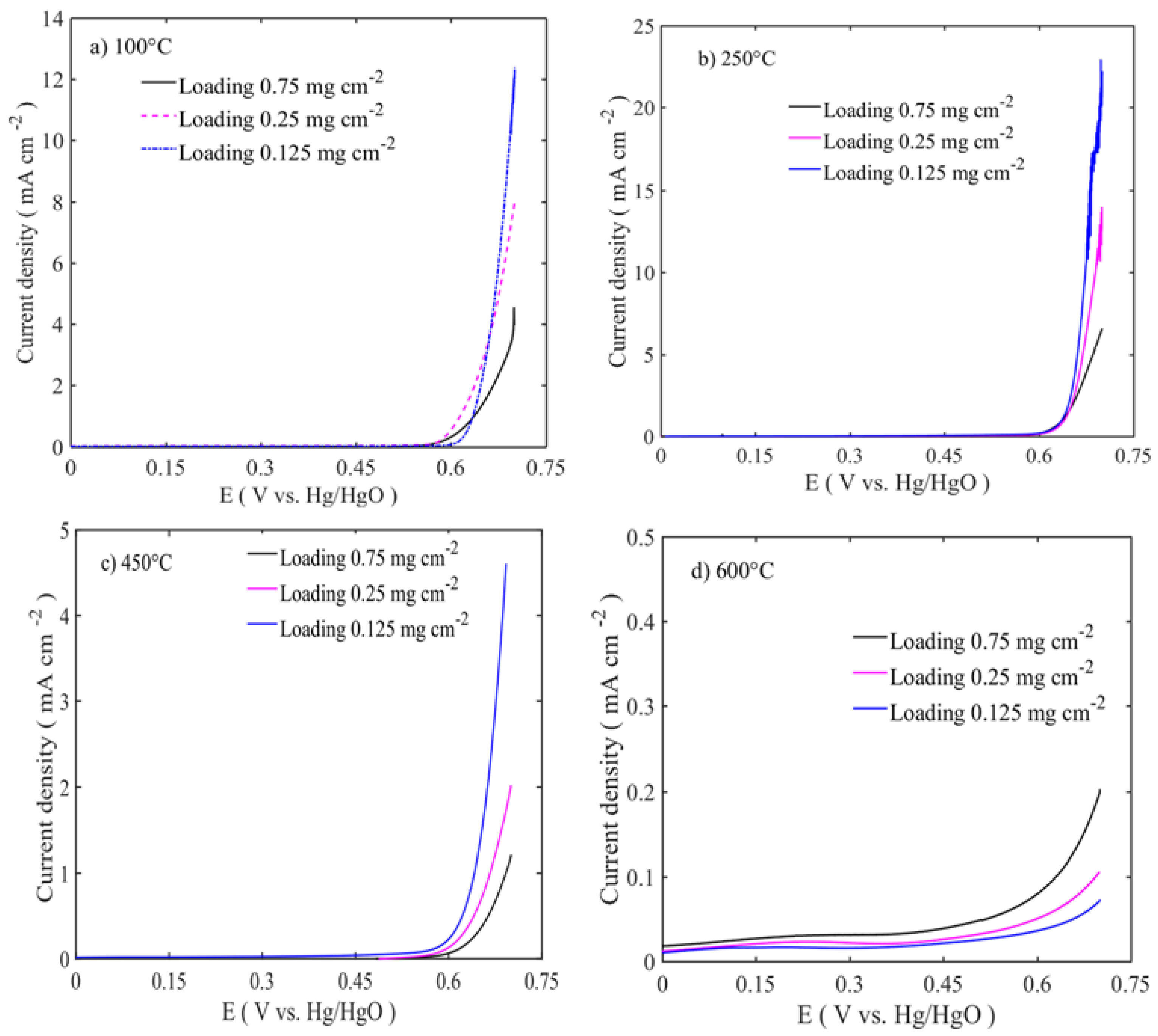
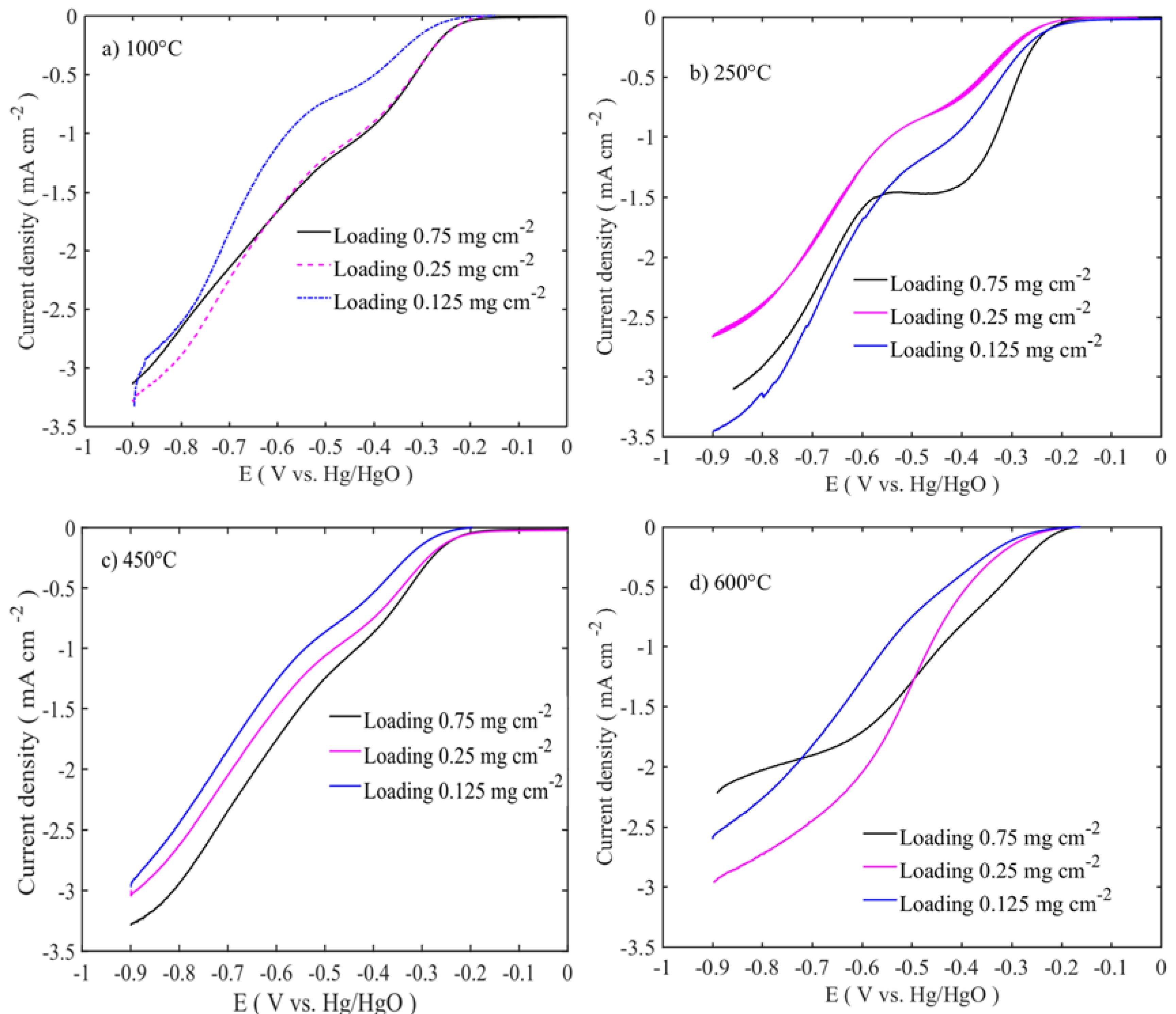
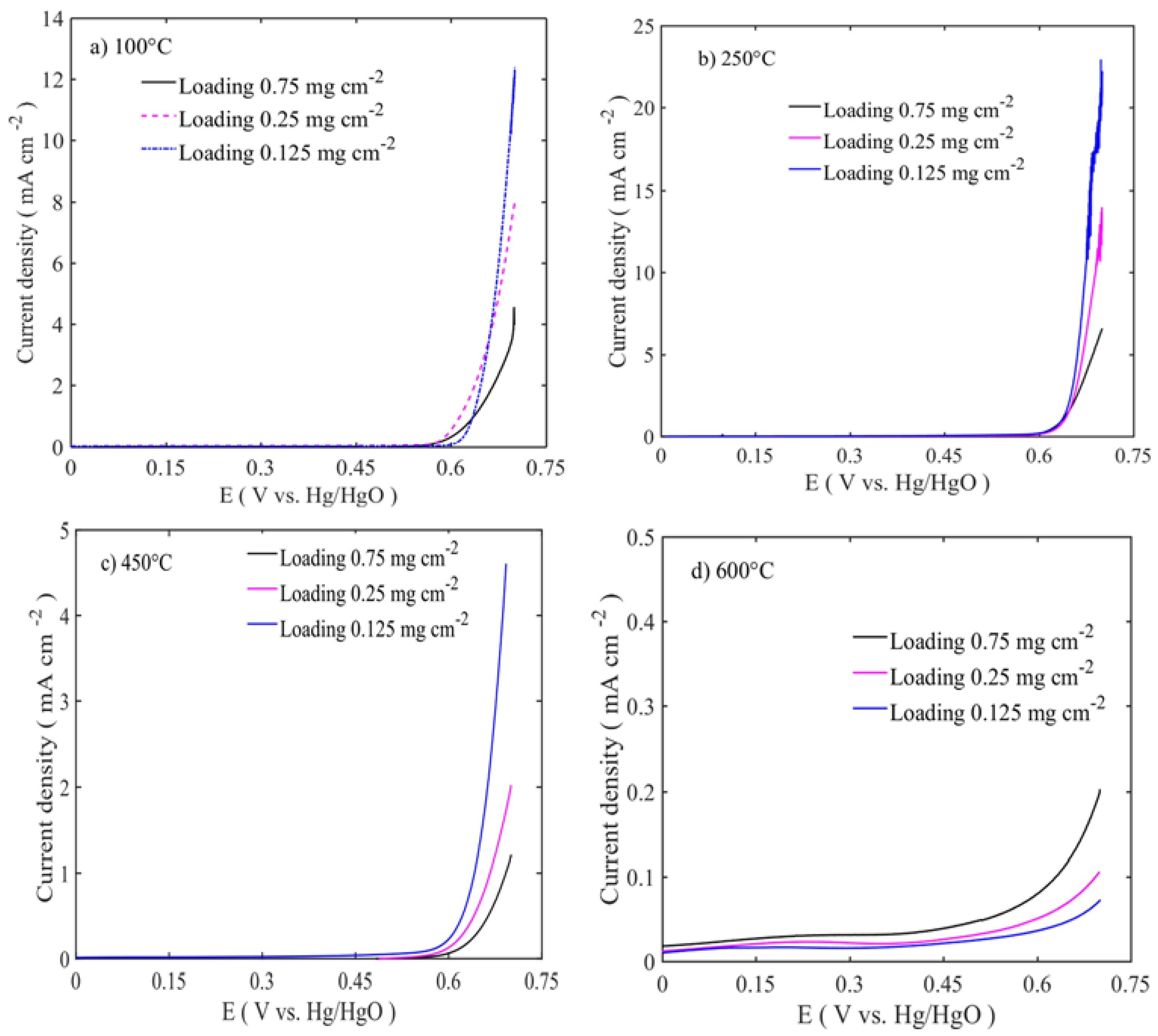
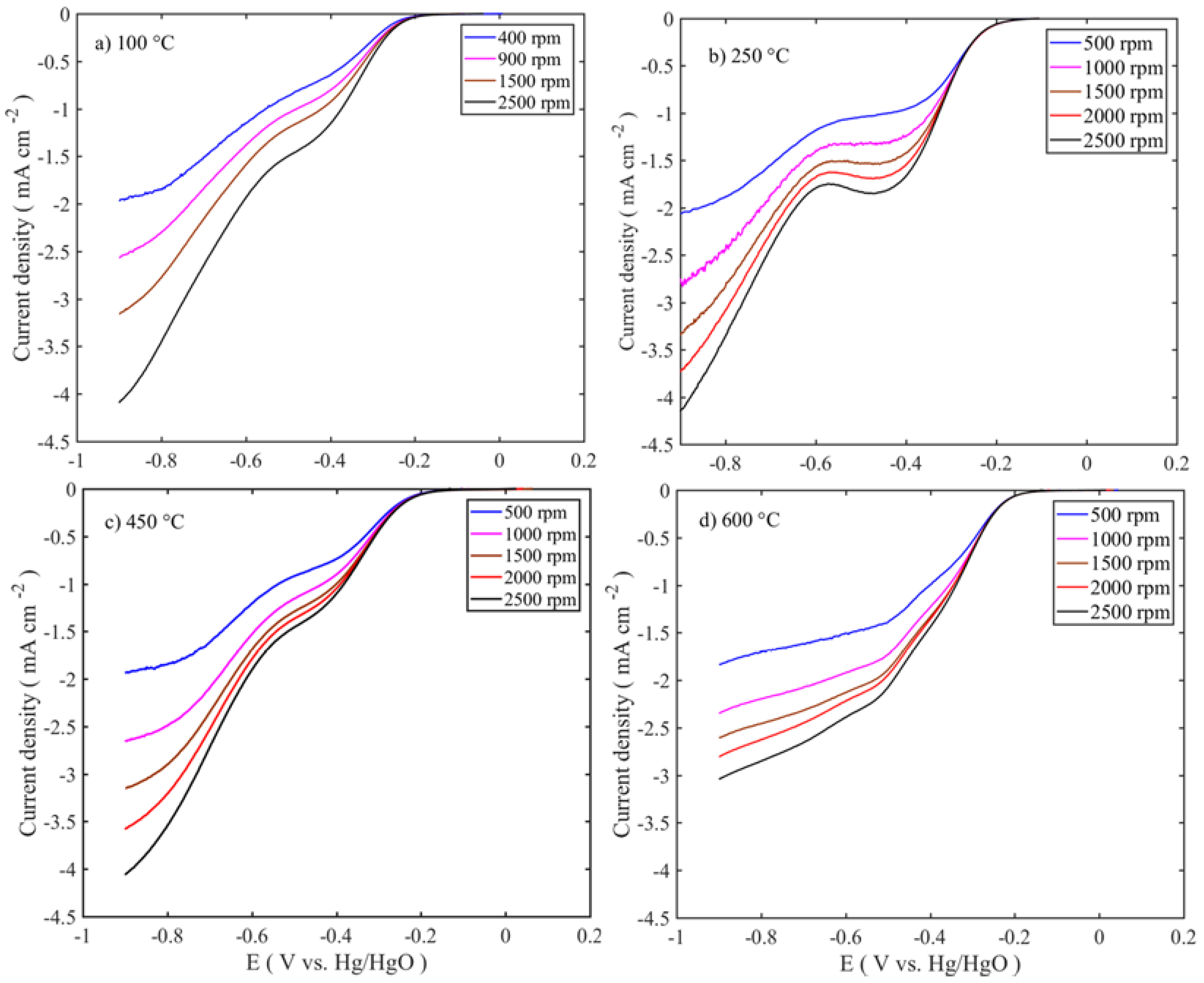
The electrocatalytic activities of the catalysts were assessed by linear sweep voltammetry (LSV) with the rotation rate of 1600 rpm, as shown in
Figure 5 and
Figure 6. The polarization curves were iRu corrected using the current interruption method. In order to observe the effect of the catalyst loading on the electrochemical activity, three different catalyst loadings were chosen: 0.125, 0.25, and 0.75 mg cm
−2. The best performing catalyst for OER is the sample calcined at 250 °C with a catalyst loading of 0.125 mg cm
−2. The activity trends are in the following order: 250 °C > 100 °C > 450 °C > 600 °C, as shown in
Figure 4. The increased catalyst loading has greatly influenced the OER activity and the onset potential. The OER onset potentials of the samples calcined between 100 and 450 °C are low for the ones with the highest loadings. However, in the case of 600 °C calcined sample, the OER onset potential is high for the lowest loading. This might be due to the different nature of the sample surface, morphology, and phase formations. As shown in the TEM image in
Figure 2a,b, low temperature-treated particles have the large number of edges and cavities, where these active sites are easily accessible, while at high temperatures, the particles are agglomerated with the active sites that are blocked and have the lower number of edge sites. At the same time, thin catalyst layer accommodates better metal center accessibility, resulting in enhanced OER activity. The highest catalyst activity on the OER side is 23 mA cm
−2 at 0.7 V vs. Hg/HgO is for the lowest catalyst loading at 0.125 mg cm
−2. The activity at 0.7 V vs. Hg/HgO changes from 23 to 8 and 5 mA cm
−2, respectively, when the loading is increased to 0.25 and 0.75 mg cm
−2.
Figure S1 shows the cyclic voltammetry of the sample at 250 °C, where there are two well-defined peaks associated with Ni metal center oxidation to 3
+. The following oxidation reactions are given [
34,
36]:
Step 1(surface oxidation)
NiII − FeIII + OH−sol ↔ (OH−)ie − NiIII − FeIII + e−
Step 2 (hydroxide adsorption)
(OH−)ie − NiIII − Fe III + OH−sol ↔ (OH−)ie − NiIII − FeIII − OH−ads + e−
Step 3 (charge transfer)
(OH−)ie − NiIII − FeIII − OH−ads + OH−sol ↔ (OH−)ie − NiIII − FeIII+ ½ O2 + H2O + e−
During the first step, Ni
2+ is oxidized to Ni
3+ accompanied with hydroxide ion intercalation into inter-layer space, followed by partial charge shift to Fe
3+ from Ni
3+, resulting in more than 3
+ oxidation states [
37]. It was reported that Fe-O bond contraction occurs at the mixed hydroxide (NiFeOOH) after nickel hydroxide oxidation to oxyhydroxide state, which gives optimal adsorption energies for OER intermediaries (OH. O, OOH) [
38]. This results in the hydroxide adsorption on the iron active site (step 2). After this, one electron transfer is followed by oxygen evolution (rate determining step-rds). As also shown by the XPS results, the different Ni ions are easily oxidized to higher states, while Fe
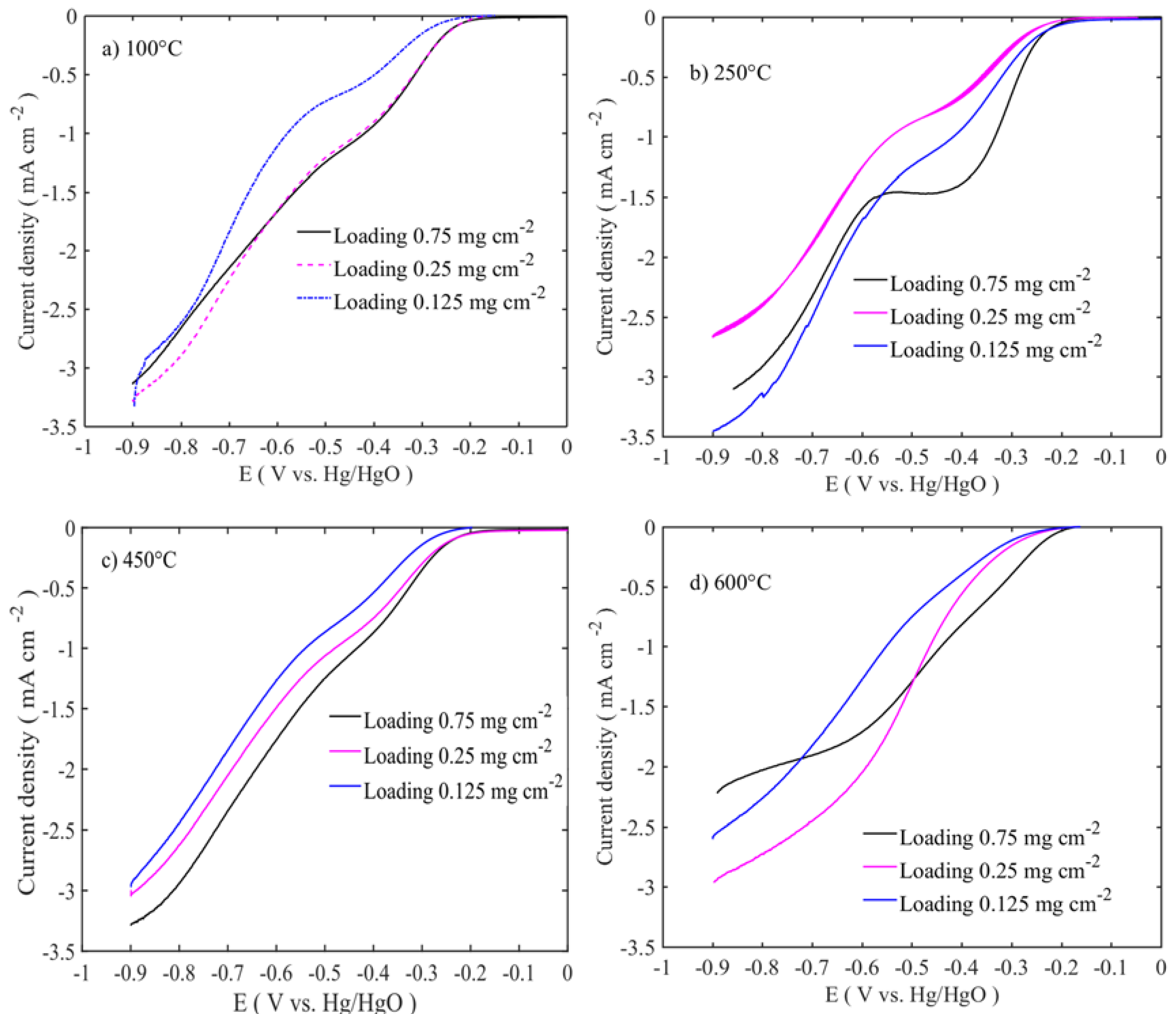
3+ enhances hydroxide adsorption and thus results in high OER activities at 100–250 °C. On the ORR side, catalyst loading has a diametrically opposite effect, since the activity reduces as the loading is decreased and the ORR onset potential starts at more negative potentials (
Figure 6a,d). Apart from this, different heat-treated catalysts have similar current density performances at the ORR side. Except for 600 °C calcined catalysts, all the catalysts show dual sigmoidal potential-current characteristics. This arises due to the differences in the underlining oxygen reduction mechanisms. Catalyst activity and selectivity are altered by catalyst loadings. Lower loading prefers more HO
2− generation than higher loadings [
39]. At 450 °C and 600 °C, heated catalysts have shown poorer performance on both OER and ORR activities due to the reduced surface area and accessible volume. Comparing the different loadings, lowest loadings for OER and the highest loadings for ORR are favourable for better catalytic activity. This suggests that on the OER side, thin catalyst layer exposes more active catalyst sites to the electrolyte and increases the mass transport of electroactive species. On the ORR side, catalyst conductivity and reaction mechanism might be altered based on catalyst loading.
To further understand the reaction mechanism, Tafel slopes are calculated from the polarization curve of
Figure 5 and
Figure 6 and the potential region between 0.6 to 0.63 V vs. Hg/HgO, which are shown in
Table 2. The Tafel slope trends are similar to the catalyst activity trends in
Figure 5. The lowest OER Tafel slope is 39.4 mV decade
−1, which corresponds to the 250 °C calcined sample, while the highest slope is 581 mV decade
−1, and it corresponds to 600 °C calcined sample. The lowest Tafel slope confirms the superior catalyst activity in the 100–250 °C calcined samples, and the highest Tafel slope at 600 °C heat treatment shows the poor catalytic activity. For the 250 °C calcined sample, the low Tafel slope (39–54 mV decade
−1), which is related to 2.3 (2RT/3F)-2.3RT/F and could be attributed to O-O bond formation, is the RDS for the OER. For the high temperature-treated samples (450–600 °C), the higher OER Tafel slope means that the water adsorption is the rds [
40]. These changes in Tafel slopes also confirm that the surfaces and electronic structures of the electrode interphases are different, and the adsorption-desorption kinetics of oxygen species involved in the reaction are also different, as evidenced in XPS and Raman study [
41]. The ORR Tafel slopes are calculated from the potential region between −0.35 to −0.45 V vs. Hg/HgO. The lower calcined samples (100–450 °C) show higher Tafel values (≈120 mV decade
−1), which are equivalent to 2.3 (2RT/F). For the 600 °C calcined sample, the Tafel slope is reduced to 70–100 mV decade
−1 for the different loadings. These low values in low-temperature calcined samples suggest the superior kinetics of the catalysts. The Tafel slope values, however, corresponding to different temperatures and loadings, do not follow uniform trends.
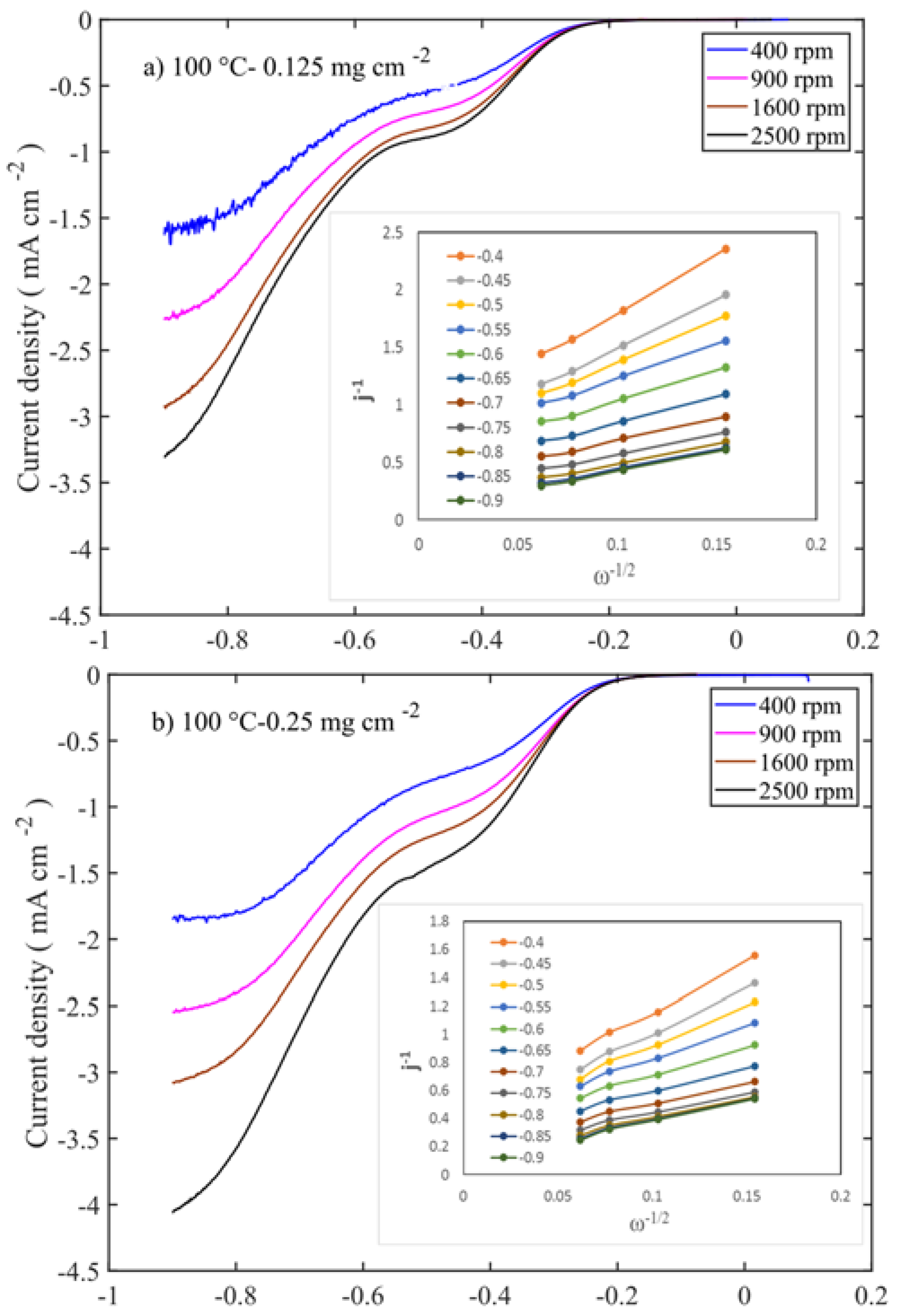
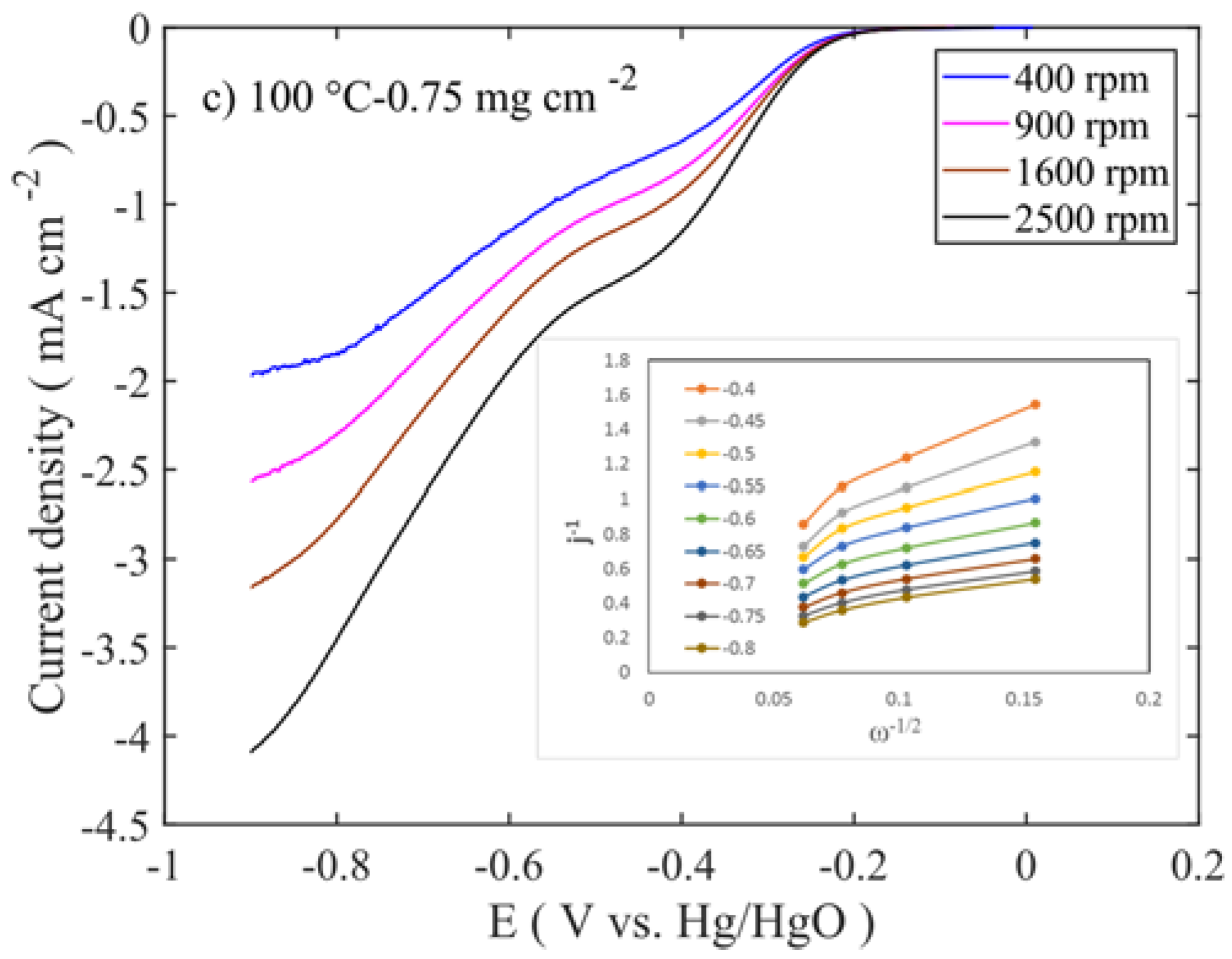
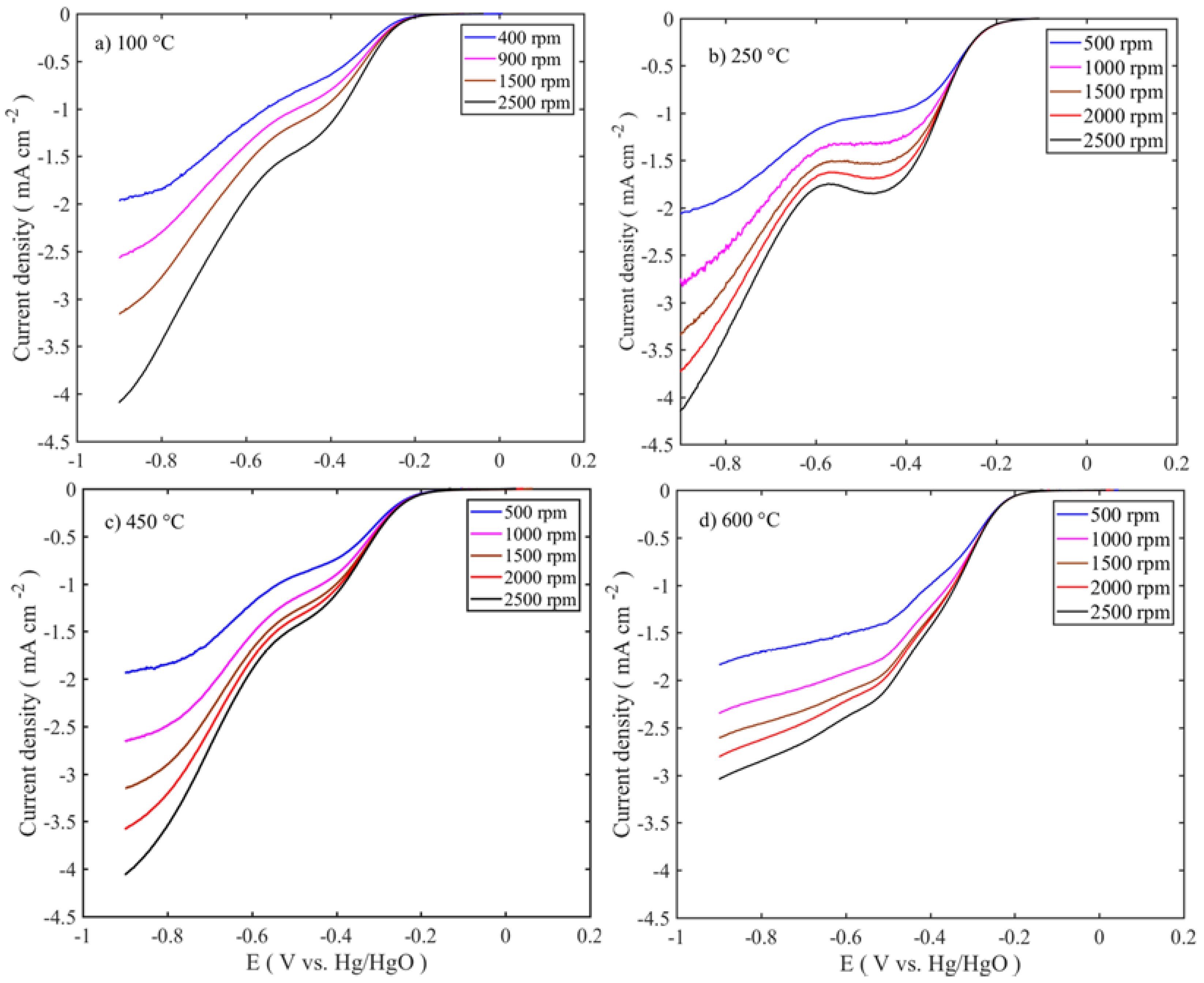
To evaluate the oxygen reduction activity of the catalysts, RDE experiments were carried out in 0.1 M KOH at different rotations (400–2500 rpm). The intrinsic activities of the catalysts are related to the number of electrons transferred during the oxygen reduction reaction, which were determined using the Koutecky-Levich equation.
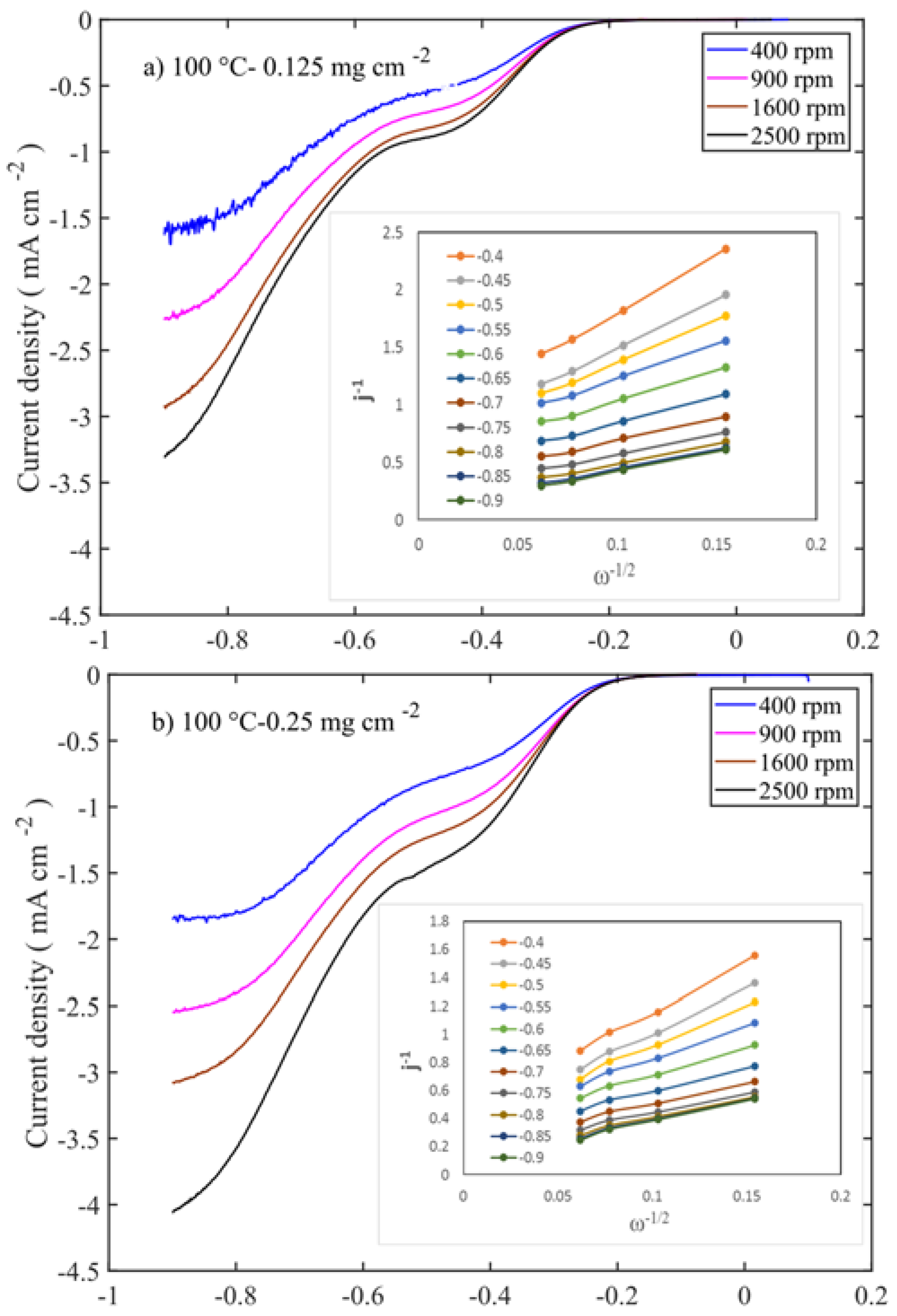
Figure 7 shows the LSV and Levich (inset Figure) curves for those at 100 °C calcined samples at different mass loadings.
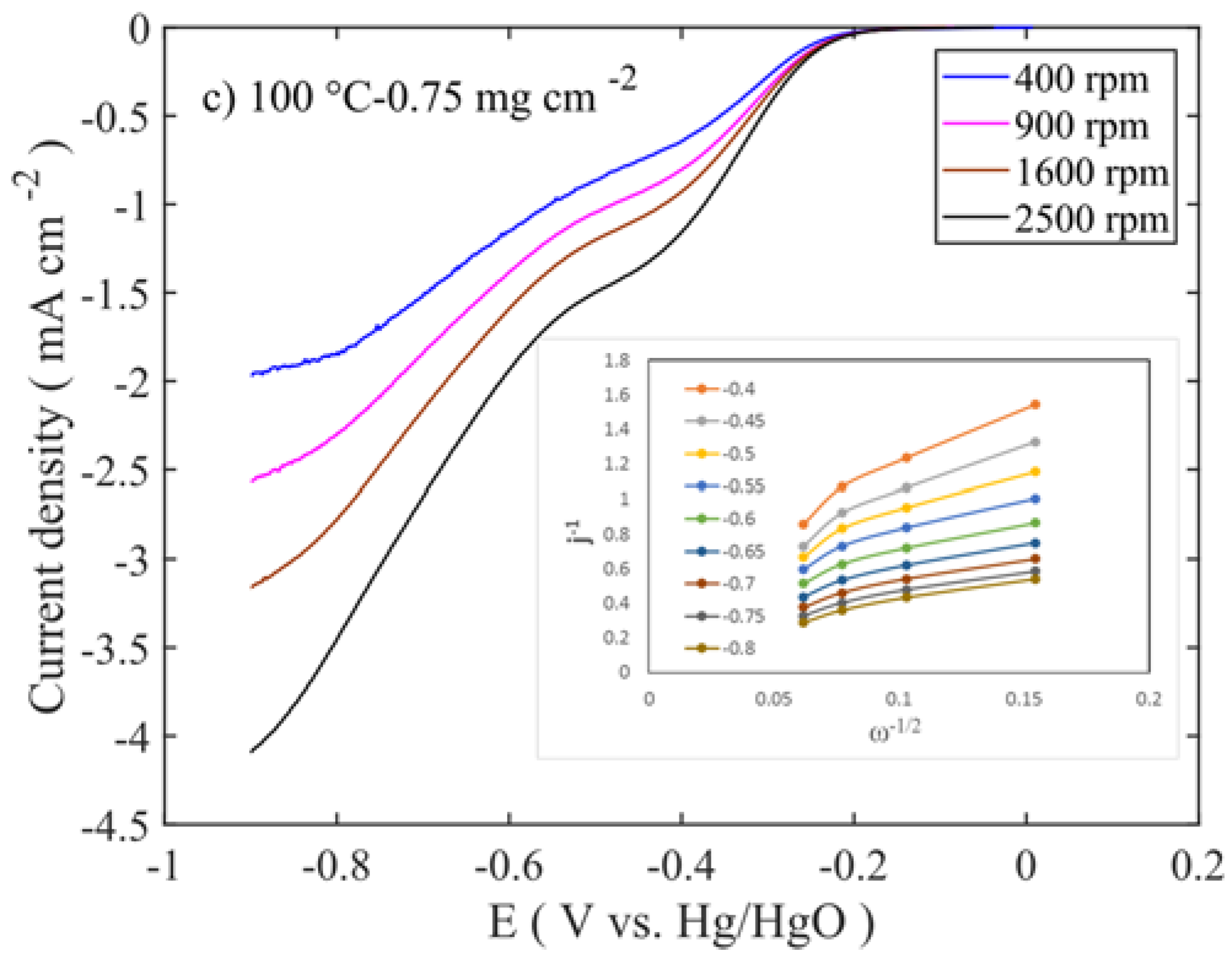
Figure 8 shows the LSV trend of the calcined samples at different temperatures and for single mass loadings (0.75 mg cm
−2). The other sample responses are given in
supplementary information (Figure S2). All the voltammograms exhibit the bi-sigmoidal shape. However, the polarization curve for the sample calcined at 600 °C tends to change to a single sigmoidal shape at higher loadings. The high ORR activities are observed at 100–250 °C sample, similar to those of the OER, and the activities are in the following order: 250 °C > 100 °C > 450 °C > 600 °C. The diffusion-limited current density (j
l) of the samples shows an increase with the rotation rate. The highest diffusion-limited current densities are observed for the samples with 0.25 and 0.75 mg cm
−2 mass loadings, which are calcined at 100 and 250 °C, respectively (
Figure 8a,b and
Figure S2e,h). Even though the former sample is a layered double hydroxide phase and the latter is a spinel oxide, their performances are at similar levels. This suggests that the properties like high surface area, smaller-crystallite size, and pore volume (≈130 m
2 g
−1 and 0.80 cm
3 g
−1) directly contribute to the enhancement of electrocatalytic activity. While the surface area and the pore volume for the 600 °C sample are 40 m
2 g
−1 and 0.40 cm
3 g
−1, which are one-third and half of the values of those corresponding to the samples calcined at 100–250 °C, this 450–600 °C catalyst activity is therefore much lower. Comparison of the different loadings shows that 0.75 mg cm
−2 delivers the highest diffusion-limited current density (
Figure 8), which is opposite to the OER performance. This suggests that thin layer enhances the OER, while in the ORR, thick layer is required to decompose the HO
2−. The linear curves of the Levich plots confirm that in all the samples, oxygen reduction proceeds through the first order reaction kinetics. Number of transferred electrons in ORR is calculated from the slope of the Levich plot. The number of transferred electrons in relation to the potential for all the samples is presented in
Supplementary Figure S5. In Levich plot, slopes are high in positive potential, whereas slopes are low in negative potential. This translates into a higher number of electrons being transferred in negative potential.
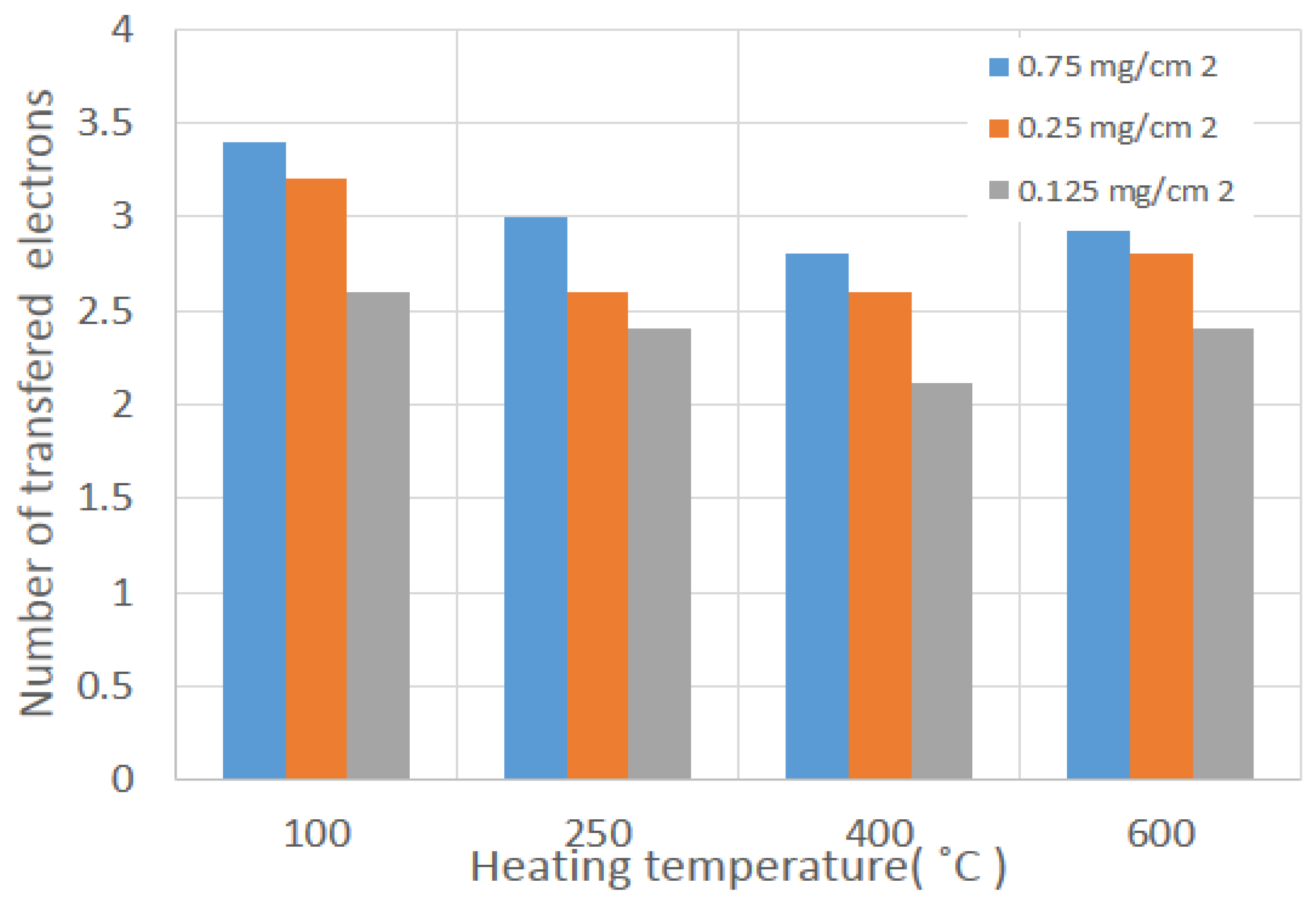
Figure 9 shows the number of transferred electrons, particularly at −0.7 V vs. Hg/HgO. The highest intrinsic activity is 3.4 e
− transfer, which corresponds to the 100 °C calcined sample with 0.75 mg cm
−2 mass loadings. Contrary to this, the lowest intrinsic activity is 2.7 e
− transfer, which corresponds to the 600 °C calcined sample with 0.75 mg·cm
−2. This trend confirms that lower catalyst loadings are more selective to hydrogen peroxide generation, as also reported in the literature [
39]. Thus, high surface area, smaller particles, and different phase formations enhance the ORR activities.
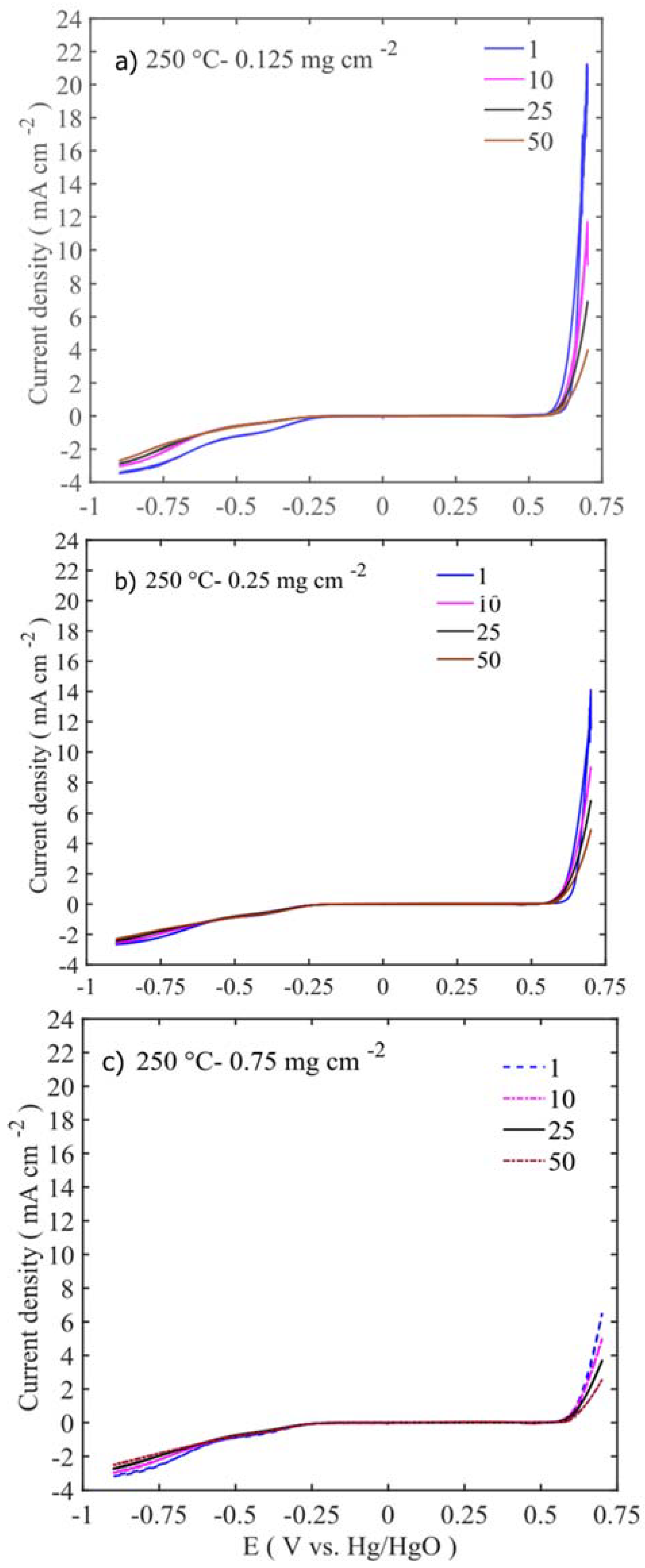
Electrochemical stability of the catalyst is very important and determines the cost and cycling behaviour of the metal-air battery. The catalysts were subjected to 50 consecutive potential sweeps in OER and ORR regions between −0.9 and 0.7 V vs. Hg/HgO, and the corresponding results are presented in
Figure 10 and
Figure S3. As discussed in the earlier section, the highest catalytic activities at the OER side are for 250 °C calcined samples at low catalyst loadings (
Figure 10a and
Figure S3), while at the ORR side, the same material exhibited high activity at high loadings (
Figure 10c). The ORR activities for all the samples are stable up to 50 cycles. The current densities are moreover stable around −2.5–3.5 mA cm
−2 at 50 cycles. However, on the OER side, each sample’s behaviour differs dramatically, as shown in
Figure S3. At low catalyst loadings (0.125 mg cm
−2), the performance drop is quite significant. In the 100 °C calcined samples, OER current density drops from 12 mA cm
−2 at the 1st cycle to 2.3 mA cm
−2 at 50th cycle (
Figure S3a). At 250 °C calcined sample, their activity drops from 21 mA cm
−2 at first cycle to 4 mA cm
−2 at 50th cycle (
Figure 9a). The samples calcined at high temperatures (450–600 °C) are shown in
Figure S3g,j and exhibit low OER current density and less dramatic current density drop, even at low catalyst loading (0.125 mg cm
−2). At high catalyst loadings (0.25–0.75 mg cm
−2), they show negligible OER activity and more constant current density throughout the 50 cycles.
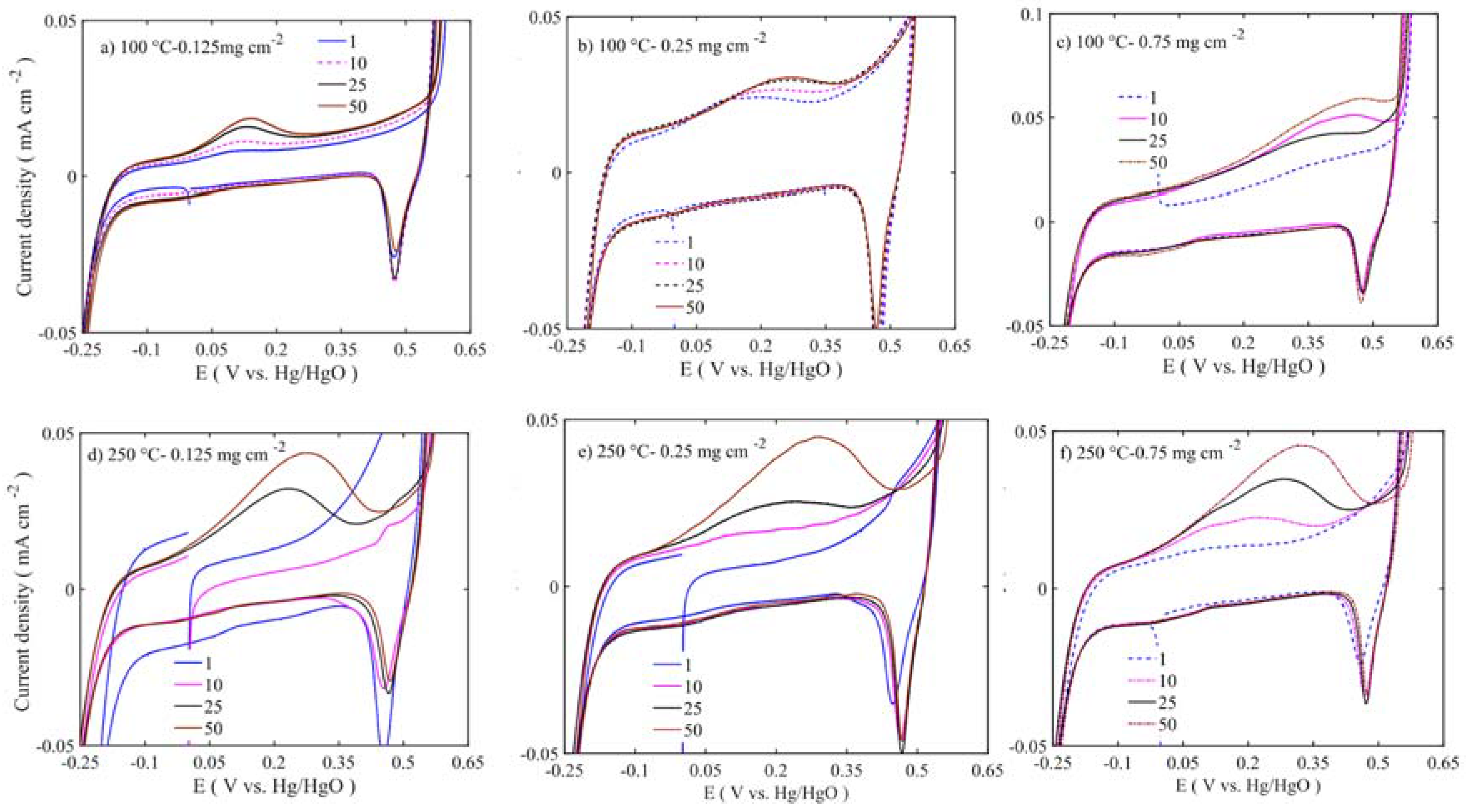
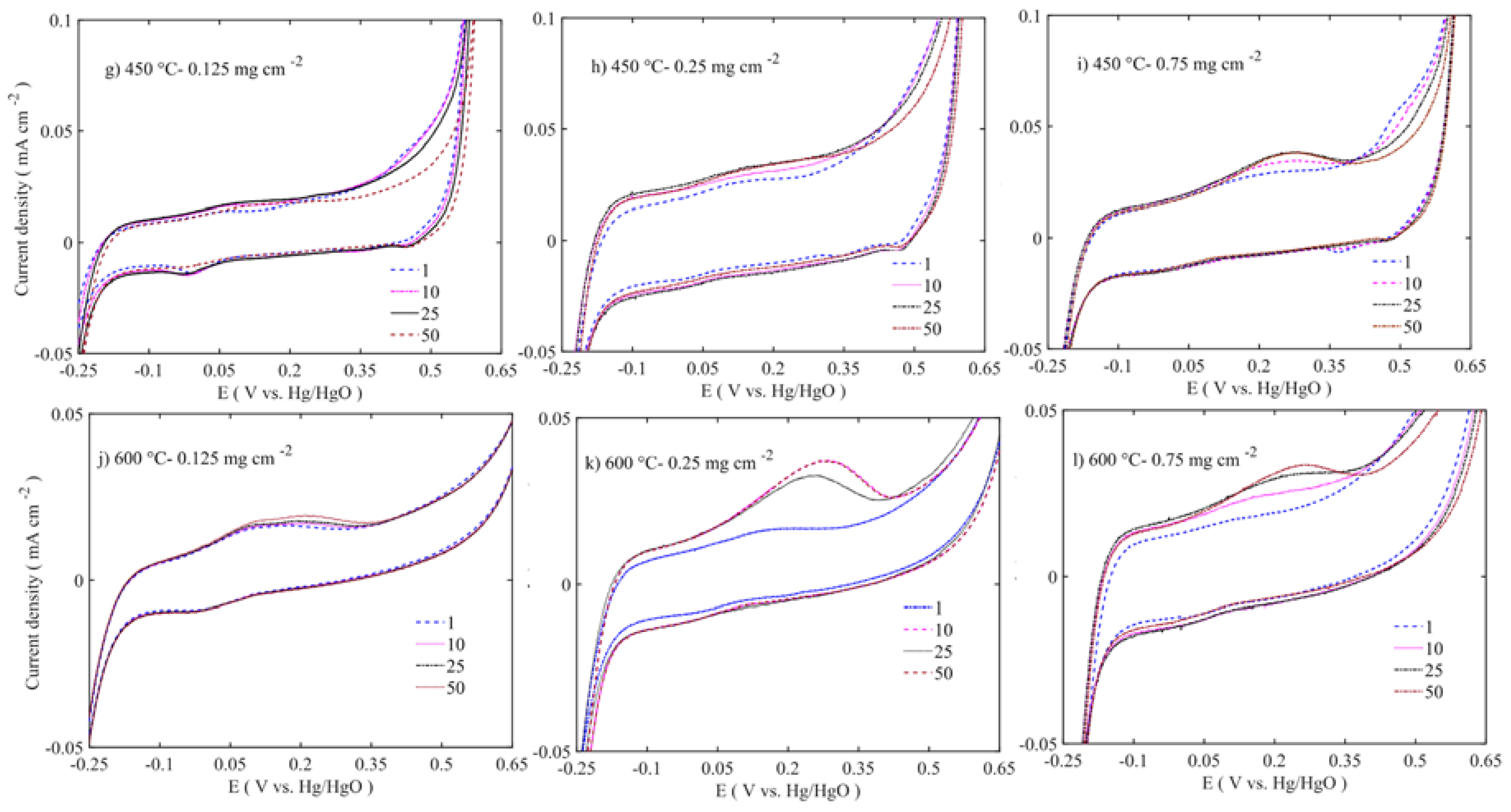
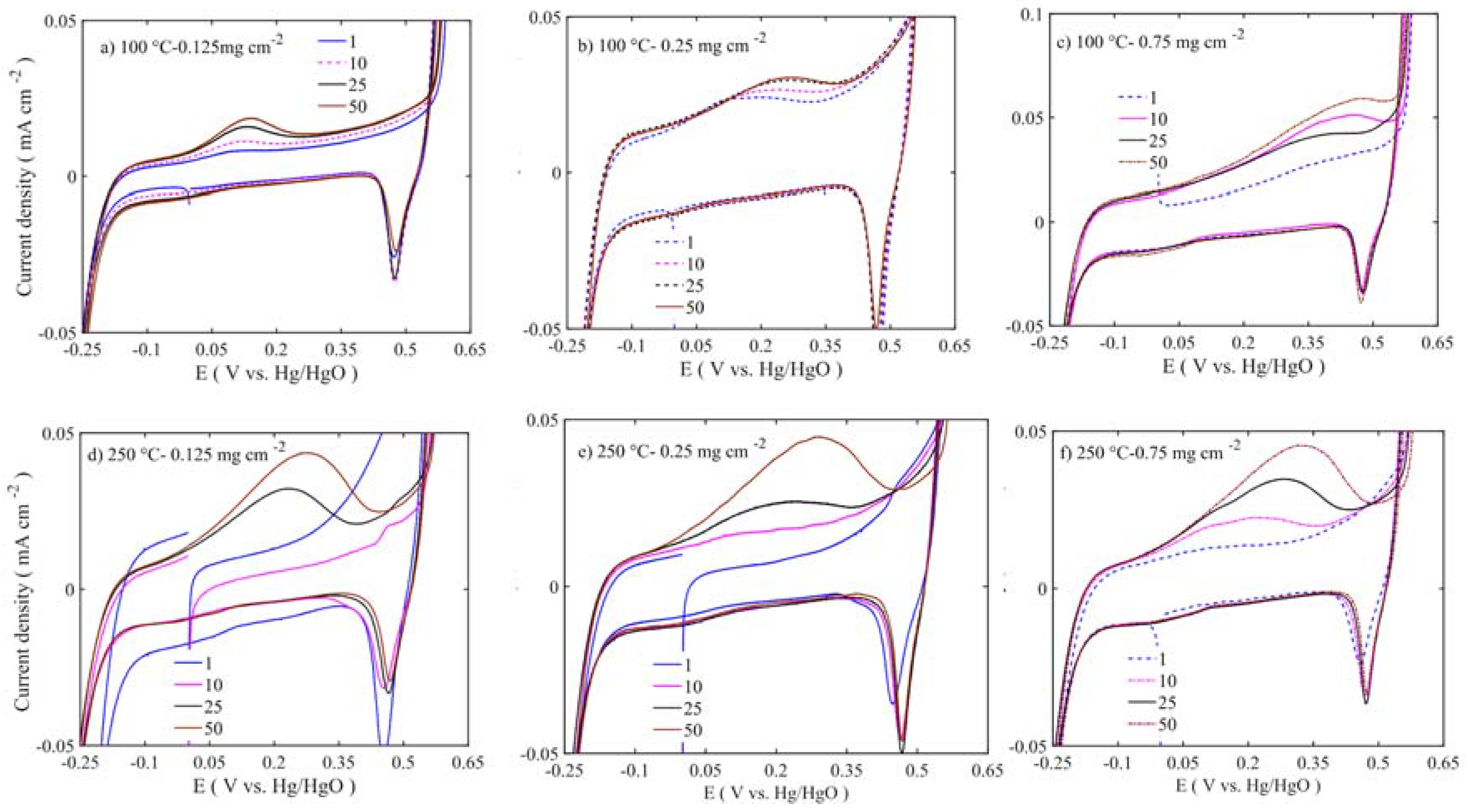
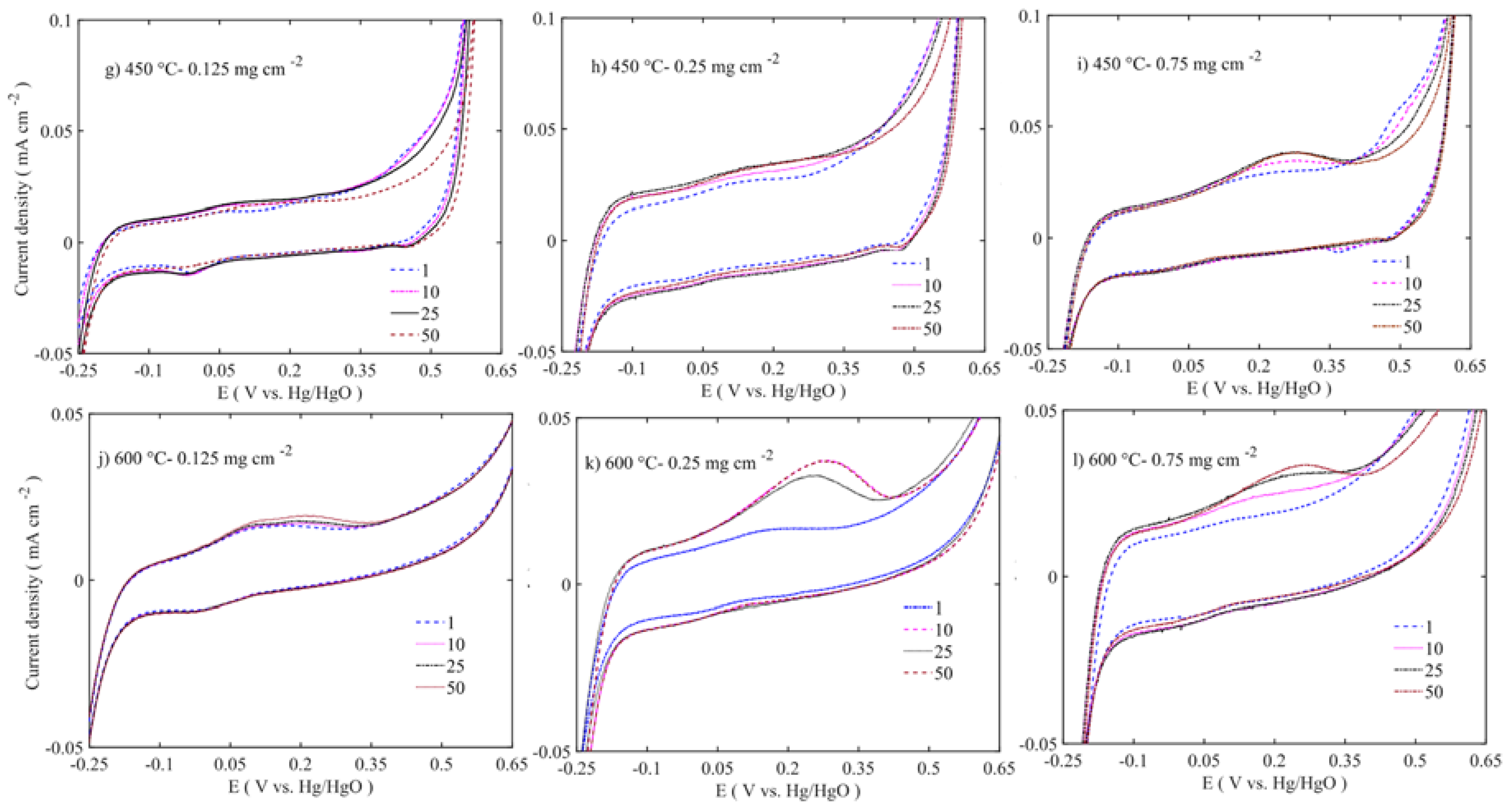
Figure 11 to
Figure 12 show enlargements of the Ni redox activity and stability. It is shown that the Ni
2+ oxidizes into Ni
3+ at 0.175 to 0.3 V vs. Hg/HgO [
42], while the reduction takes place at 0.475 V with depressed peak around 0 V. During the first cycle, the area under the curve is smaller. Regarding the 50 cycles, the area evolves and the oxidation peak shifts to higher potential. The increased oxidation peak does not translate into higher OER activity. At lower loadings, the oxidation peak is around 0.175 V vs. Hg/HgO, whereas at higher loading the peak position moves closer to 0.4 V vs. Hg/HgO. This suggests that the internal mass transfer resistance increase might cause this shift. However, all the Ni
3+ reduction peaks are at 0.475 V located at the same position across the different loadings. This peak shift moves the OER activity to higher overpotential. The samples calcined at 450–600 °C have shown depressed Ni oxidation and reduction peaks (
Figure 12). However, there are two consecutive oxidation and reduction peaks for the 450 °C calcined sample. At 600 °C, one oxidation peak is visible, with the reduction peak completely disappearing. Thus, in low-temperature samples (100–250 °C), more Ni ions are easily oxidized from nickel hydroxide to nickel oxyhydroxide phase, while in 450–600 °C samples there is not enough oxidizable nickel to contribute to the OER activity. As the XPS results also corroborated that multivalent nickel ions were not present at 600 °C, the Ni oxidation peak shift and area under peak increase are related to structural changes occurring along the potential cycles [
38]. These changes also alter the stability and activity of the catalyst.
Comparison of the activities of bifunctional oxygen electrodes with the noble and non-noble catalysts is shown in
Table 3. The oxygen electrode potential difference (Δ
E) is calculated from the difference between the potentials at 10 mA cm
−2 current density for OER and −1 mA cm
−2 for ORR. The oxygen electrode overpotential (0.33 V) on OER for the catalyst at low-temperatures (100–250 °C) is on par with noble metal catalyst and non-noble transition metal oxide catalysts in OER direction (
Table 3), while in the ORR direction, overpotential is very high due to the limited activities of the NiFeO
x. Most of the reported catalysts either have a large amount of carbon additives such as CNT and CNF or special additives such as some doping elements. However, our NiFeO
x catalysts still have shown superior performance, since they possess high OER current and better kinetics. The ORR side needs to be improved by engineering the electrode composition to get higher activities.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}