Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance

 ,
,  ,
, 

Abstract
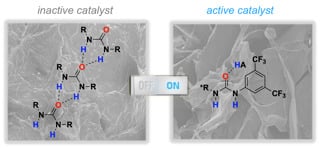
1. Introduction
2. Results and Discussion
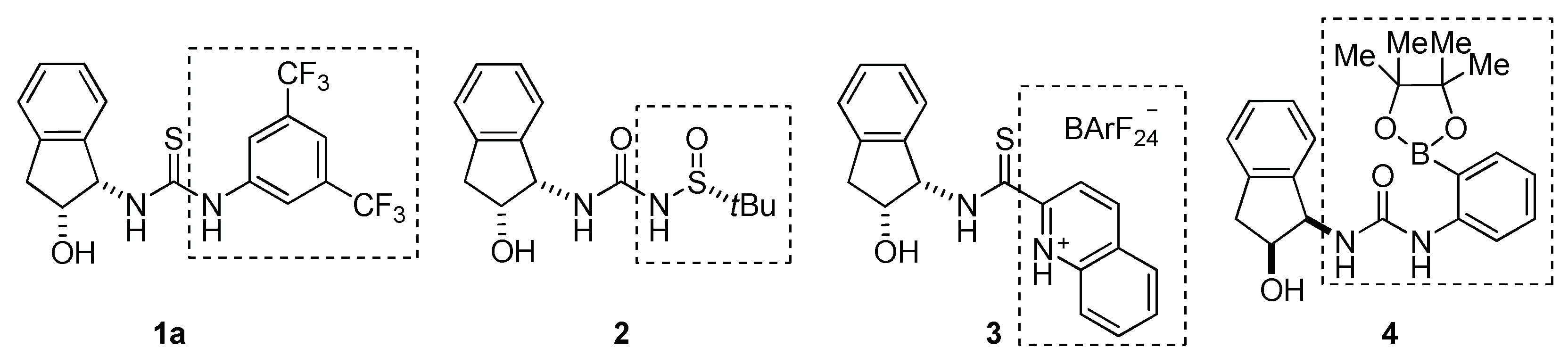
2.1. Cooperative Effect in the Mixture (Urea Catalyst + Brønsted Acid)
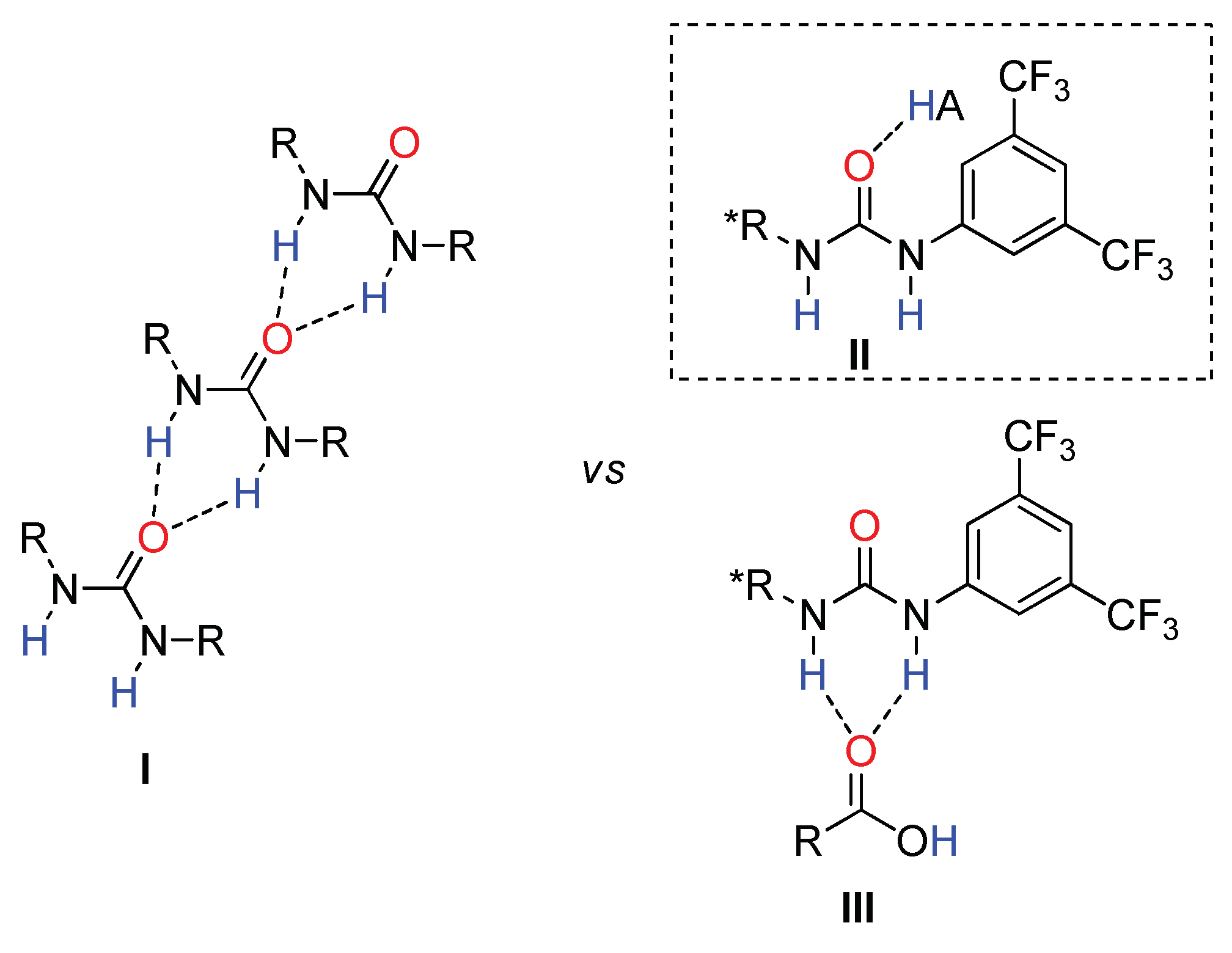
2.2. Effect of Brønsted Acid on Urea Aggregation and Mechanistic Hypothesis
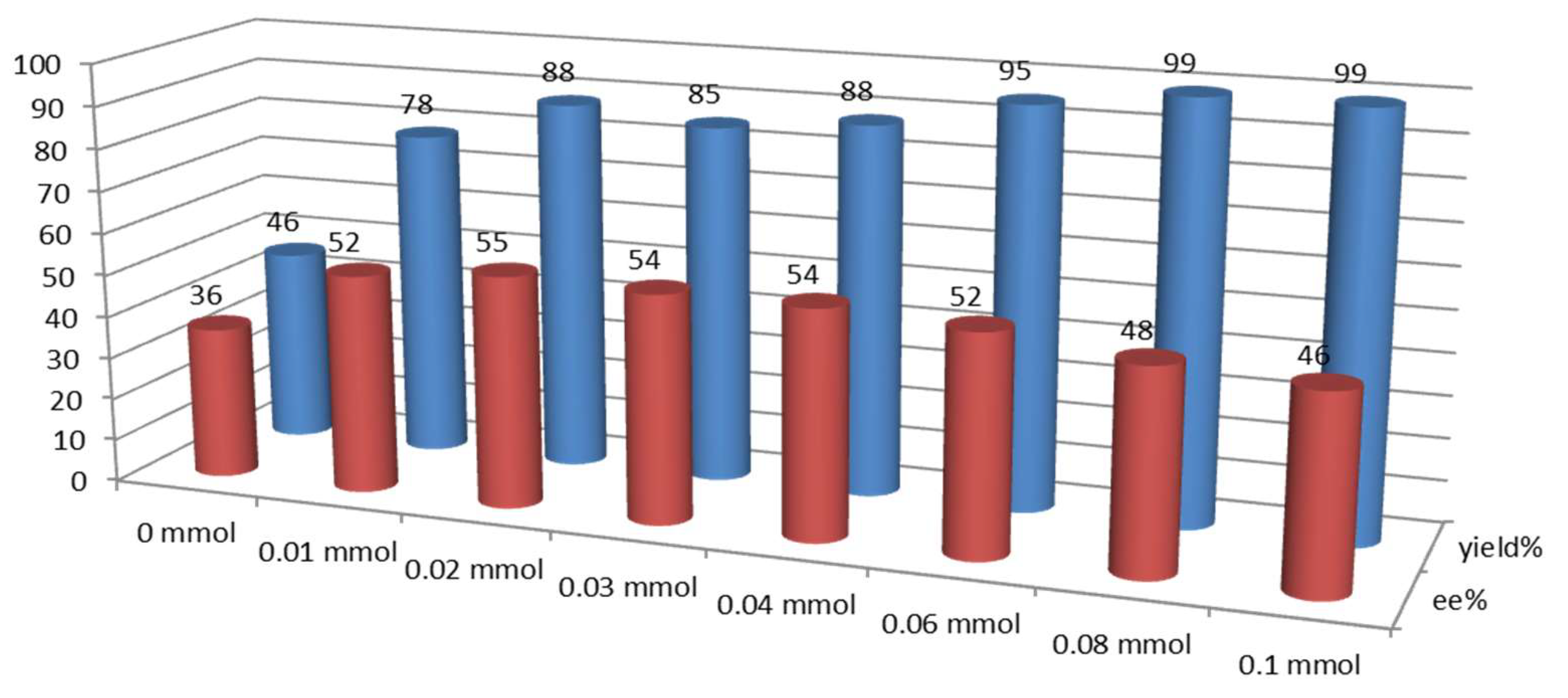
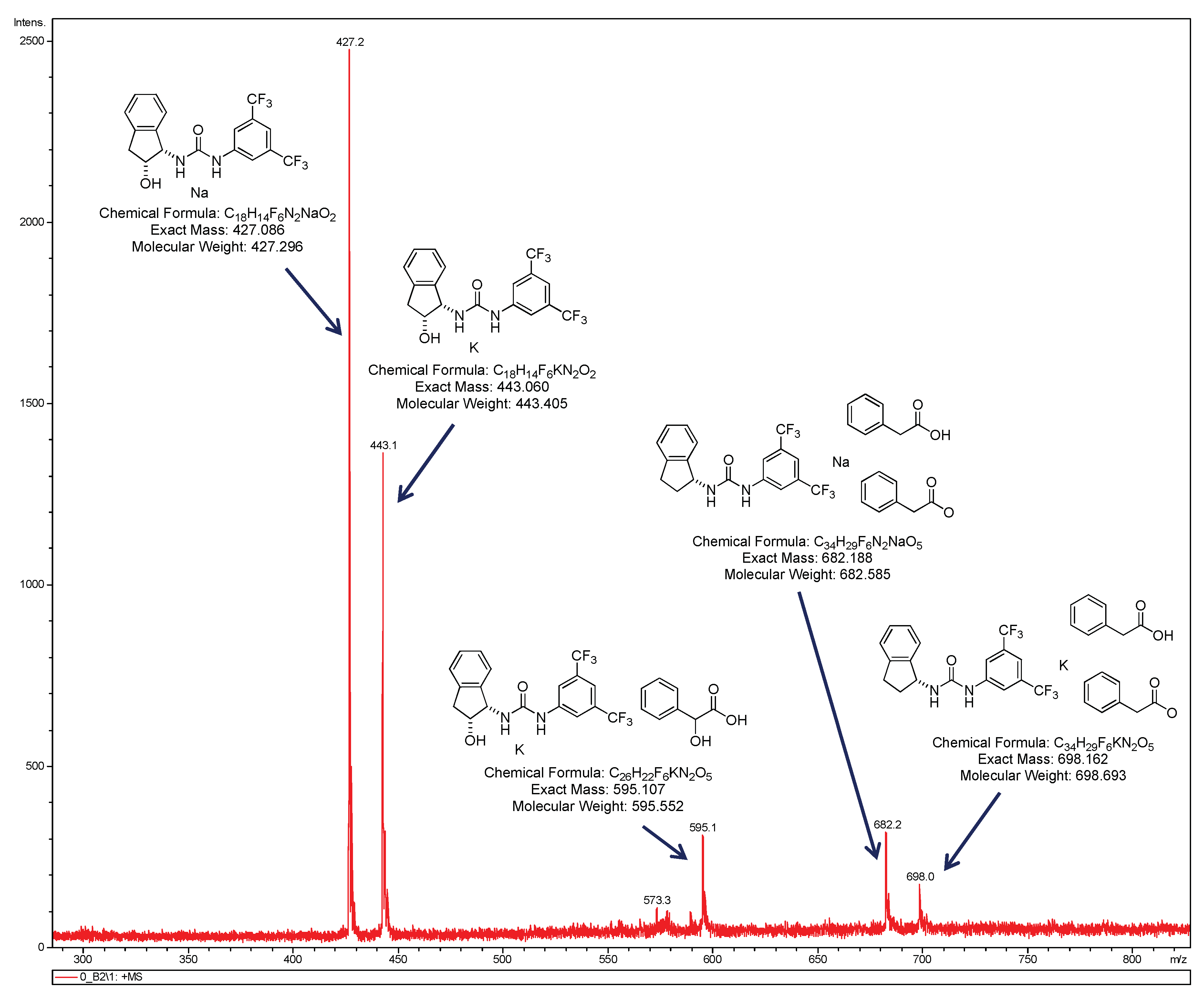
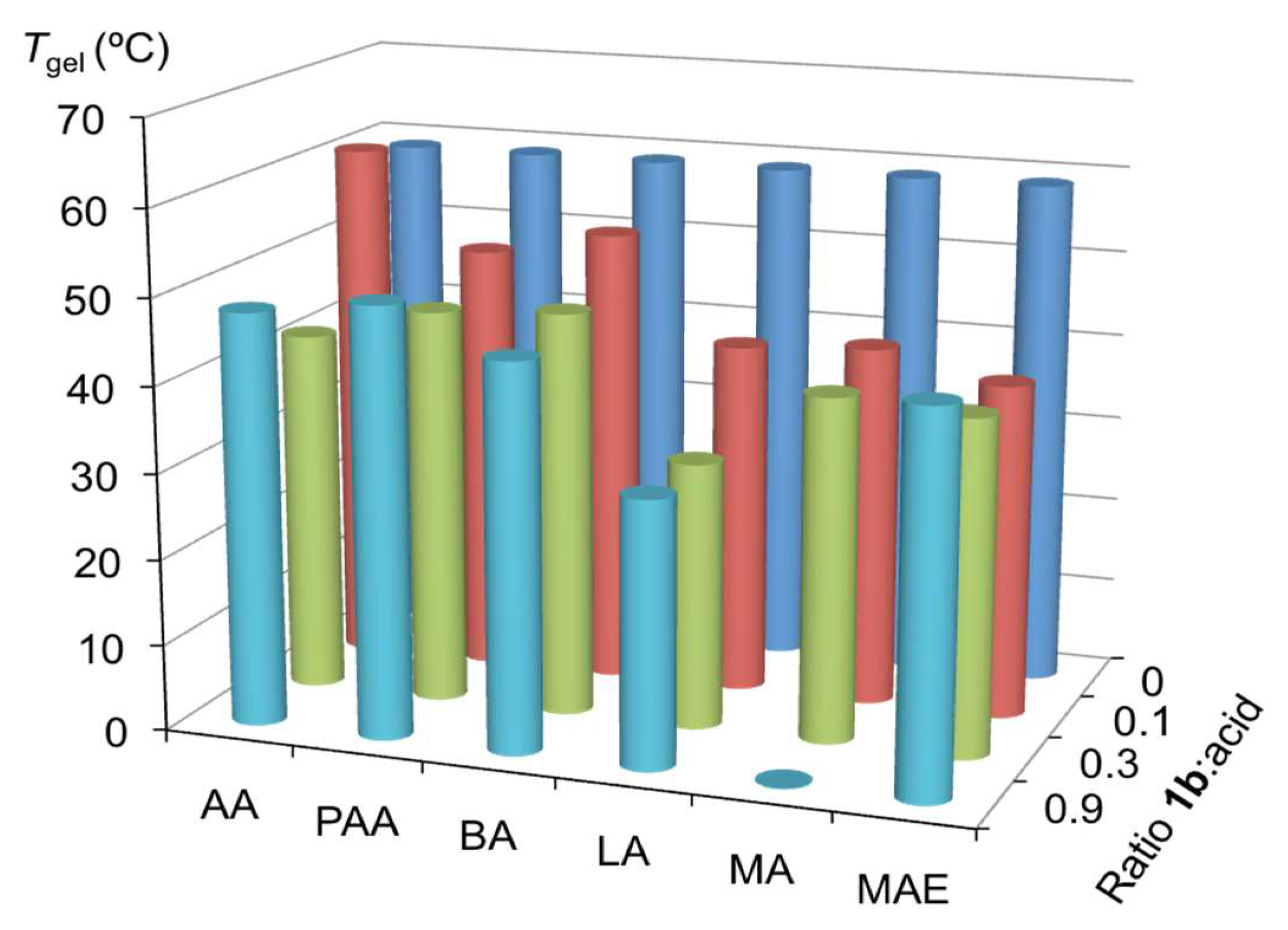
2.3. Effect of Brønsted Acids on the Stability of Urea Aggregates
3. Materials and Methods
3.1. Experimental Details
3.2. General Procedure for the 1b·HA-Catalyzed Friedel–Crafts Alkylation Reaction
3.3. Evaluation of the Stability of Urea Aggregates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Whiley-VHC: Weinheim, Germany, 2004. [Google Scholar]
- Acc. Chem. Res. 2004, 37 (8). Special Issue on Organocatalysis; Houk, K.N., List, B., Eds.; American Chemical Society: Washington, DC, USA, 2004. [Google Scholar]
- Enantioselective Organocatalysis; Dalko, P.I., Ed.; Whiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Chem. Rev. 2007, 107 (12). Special Issue on Organocatalysis; List, B., Ed.; American Chemical Society: Washington, DC, USA, 2007. [Google Scholar]
- Recent Developments in Asymmetric Organocatalysis; Pellissier, H., Ed.; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Asymmetric Organocatalysis; List, B., Ed.; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Current Organic Chemistry, Volume 15, Number 13; Herrera, R.P., Ed.; Bentham Science: Soest, The Netherlands, 2011. [Google Scholar]
- Science of Synthesis Asymmetric Organocatalysis; List, B., Maruoka, K., Eds.; Thieme: Sttutgart, Germany, 2012. [Google Scholar]
- Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Application; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- De Figueiredo, R.M.; Christmann, M. Organocatalytic Synthesis of Drugs and Bioactive Natural Products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Marqués-López, E.; Herrera, R.P.; Christmann, M. Asymmetric organocatalysis in total synthesis—A trial by fire. Nat. Prod. Rep. 2010, 27, 1138–1167. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. Comprehensive Enantioselective Organocatalysis; Dalko, P., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1359–1383. [Google Scholar]
- Dalko, P.I.; Mosan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A. Organocatalysis by Hydrogen Bonding Networks in Organocatalysis; Reetz, M.T., List, B., Jaroch, H., Weinmann, H., Eds.; Springer: Berlin, Germany, 2008; Volume 2, pp. 281–297. [Google Scholar]
- Hydrogen Bonding in Organic Synthesis; Pihko, P.M., Ed.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Kerstin, E.-E.; Berkessel, A. Noncovalent organocatalysis based on hydrogen bonding: elucidation of reaction paths by computational methods. Top. Curr. Chem. 2010, 291, 1–27. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y. Recognition and activation by ureas and thioureas: stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem. 2005, 3, 4299–4306. [Google Scholar] [CrossRef] [PubMed]
- Connon, S.J. Organocatalysis Mediated by (Thio)urea Derivatives. Chem. Eur. J. 2006, 12, 5418–5429. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, H.; Takemoto, Y. Discovery and Application of Asymmetric Reaction by Multi-Functional Thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. El renacer de un nuevo campo: La Organocatálisis Asimétrica. Tioureas como organocatalizadores. An. Quím. 2009, 105, 5–12. (In Spanish) [Google Scholar]
- Takemoto, Y. Development of Chiral Thiourea Catalysts and Its Application to Asymmetric Catalytic Reactions. Chem. Pharm. Bull. 2010, 58, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Sohtome, Y.; Nagasawa, K. The Design of Chiral Double Hydrogen Bonding Networks and Their Applications to Catalytic Asymmetric Carbon-Carbon and Carbon-Oxygen Bond-Forming Reactions. Synlett 2010, 1–22. [Google Scholar] [CrossRef]
- Sohtome, Y.; Nagasawa, K. Dynamic asymmetric organocatalysis: Cooperative effects of weak interactions and conformational flexibility in asymmetric organocatalysts. Chem. Commun. 2012, 48, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. New Strategies in Chemical Synthesis and Catalysis; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp. 175–199. [Google Scholar]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shi, X. Cinchonine and thiourea. Chem. Commun. 2013, 49, 8583–8585. [Google Scholar] [CrossRef] [PubMed]
- Narayanaperumal, S.; Rivera, D.G.; Silva, R.C.; Paixão, M.W. Terpene-Derived Bifunctional Thioureas in Asymmetric Organocatalysis. ChemCatChem. 2013, 5, 2756–2773. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, Z.; Xing, H. N,N′-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea: a privileged motif for catalyst development. Org. Biomol. Chem. 2014, 12, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, C.-J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine–thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; Kuo, L.H. Altering the Stereochemistry of Allylation Reactions of Cyclic. alpha.-Sulfinyl Radicals with Diarylureas. J. Org. Chem. 1994, 59, 3259–3261. [Google Scholar] [CrossRef]
- Curran, D.P.; Kuo, L.H. Acceleration of a dipolar Claisen rearrangement by Hydrogen bonding to a soluble diaryl^urea. Tetrahedron Lett. 1995, 36, 6647–6650. [Google Scholar] [CrossRef]
- Etter, M.C.; Panunto, T.W. 1,3-Bis(m-nitrophenyl)urea: An exceptionally good complexing agent for proton acceptors. J. Am. Chem. Soc. 1988, 110, 5896–5897. [Google Scholar] [CrossRef]
- Etter, M.C.; Urbañczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T.W. Hydrogen bond-directed cocrystallization and molecular recognition properties of diarylureas. J. Am. Chem. Soc. 1990, 112, 8415–8426. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Kelly, T.R.; Kim, M.H. Relative Binding Affinity of Carboxylate and Its Isosteres: Nitro, Phosphate, Phosphonate, Sulfonate, and. delta.-Lactone. J. Am. Chem. Soc. 1994, 116, 7072–7080. [Google Scholar] [CrossRef]
- Auvil, T.J.; Schafer, A.G.; Mattson, A.E. Design Strategies for Enhanced Hydrogen-Bond Donor Catalysts. Eur. J. Org. Chem. 2014, 2633–2646. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Low-loading asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 2406–2447. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Catalytic enantioselective Friedel-Crafts alkylation of indoles with nitroalkenes by using a simple thiourea organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 6734–6737. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Monge, D.; Martín-Zamora, E.; Fernández, R.; Lassaletta, J.M. Organocatalytic Conjugate Addition of Formaldehyde N,N-Dialkylhydrazones to β,γ-Unsaturated α-Keto Esters. Org. Lett. 2007, 9, 3303–3306. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Alcaine, A.; Tejero, T.; Herrera, R.P. Enhanced Efficiency of Thiourea Catalysts by External Brønsted Acids in the Friedel—Crafts Alkylation of Indoles. Eur. J. Org. Chem. 2011, 3700–3705. [Google Scholar] [CrossRef]
- Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R.P. A Friedel–Crafts alkylation mechanism using an aminoindanol-derived thiourea catalyst. Org. Biomol. Chem. 2014, 12, 4503–4510. [Google Scholar] [CrossRef] [PubMed]
- Schön, E.-M.; Marqués-López, E.; Herrera, R.P.; Alemán, C.; Díaz, D.D. Exploiting Molecular Self-Assembly: From Urea-Based Organocatalysts to Multifunctional Supramolecular Gels. Chem. Eur. J. 2014, 20, 10720–10731. [Google Scholar] [CrossRef] [PubMed]
- Juste-Navarro, V.; Marqués-López, E.; Herrera, R.P. Thiourea-Catalyzed Addition of Indoles to Aliphatic β,γ-Unsaturated α-Ketoesters. Asian J. Org. Chem. 2015, 4, 884–889. [Google Scholar] [CrossRef]
- Schiller, J.; Pérez-Ruiz, R.; Sampedro, D.; Marqués-López, E.; Herrera, R.P.; Díaz, D.D. Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study. Sensors 2016, 16, 658. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.C.; Herrera, R.P. Hydrogen Bonding Networks in Chiral Thiourea Organocatalysts: Evidence on the Importance of the Aminoindanol Moiety. Cryst. Growth Des. 2016, 16, 5091–5099. [Google Scholar] [CrossRef]
- Izaga, A.; Herrera, R.P.; Gimeno, M.C. Gold(I)-Mediated Thiourea Organocatalyst Activation: A Synergic Effect for Asymmetric Catalysis. ChemCatChem 2017, 9, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Robak, M.T.; Trincado, M.; Ellman, J.A. Enantioselective Aza-Henry Reaction with an N-Sulfinyl Urea Organocatalyst. J. Am. Chem. Soc. 2007, 129, 15110–15111. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, M.; Seidel, D. Catalytic Enantioselective Additions of Indoles to Nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Burkett, J.A.; Mattson, A.E. Internal Lewis Acid Assisted Hydrogen Bond Donor Catalysis. Org. Lett. 2011, 13, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Sonsona, I.G.; Marqués-López, E.; Herrera, R.P. The aminoindanol core as a key scaffold in bifunctional organocatalysts. Beilstein J. Org. Chem. 2016, 12, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Kass, S.R. Enantioselective Friedel–Crafts Alkylation between Nitroalkenes and Indoles Catalyzed by Charge Activated Thiourea Organocatalysts. J. Org. Chem. 2017, 82, 13288–13296. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Sigman, M.S. Systematically Probing the Effect of Catalyst Acidity in a Hydrogen-Bond-Catalyzed Enantioselective Reaction. Angew. Chem. Int. Ed. 2007, 46, 4748–4750. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Diez-Martinez, A.; Merino, P.; Herrera, R.P. The Role of the Indole in Important Organocatalytic Enantioselective Friedel-Crafts Alkylation Reactions. Curr. Org. Chem. 2009, 13, 1585–1609. [Google Scholar] [CrossRef]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2210. [Google Scholar] [CrossRef] [PubMed]
- Terrasson, V.; de Figueiredo, R.M.; Campagne, J.M. Organocatalyzed Asymmetric Friedel–Crafts Reactions. Eur. J. Org. Chem. 2010, 2635–2655. [Google Scholar] [CrossRef]
- Zeng, M.; You, S.-L. Asymmetric Friedel-Crafts Alkylation of Indoles: The Control of Enantio- and Regioselectivity. Synlett 2010, 1289–1301. [Google Scholar] [CrossRef]
- Dessole, G.; Herrera, R.P.; Ricci, A. H-Bonding Organocatalysed Friedel-Crafts Alkylation of Aromatic and Heteroaromatic Systems with Nitroolefins. Synlett 2004, 2374–2378. [Google Scholar] [CrossRef]
- Pettersen, D.; Herrera, R.P.; Bernardi, L.; Fini, F.; Sgarzani, V.; Fernández, R.; Lassaletta, J.M.; Ricci, A. A Broadened Scope for the Use of Hydrazones as Neutral Nucleophiles in the Presence of H-Bonding Organocatalysts. Synlett 2006, 239–242. [Google Scholar] [CrossRef]
- Bernardi, L.; Fini, F.; Herrera, R.P.; Ricci, A.; Sgarzani, V. Enantioselective aza-Henry reaction using cinchona organocatalysts. Tetrahedron 2006, 62, 375–380. [Google Scholar] [CrossRef]
- Alcaine, A.; Marqués-López, E.; Merino, P.; Tejero, T.; Herrera, R.P. Thiourea catalyzed organocatalytic enantioselective Michael addition of diphenyl phosphite to nitroalkenes. Org. Biomol. Chem. 2011, 9, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Auria-Luna, F.; Marqués-López, E.; Mohammadi, S.; Heiran, R.; Herrera, R.P. New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′-(1′,4′-dihydropyridine)] Derivatives. Molecules 2015, 20, 15807–15826. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G.; Algrim, D.J.; Harrelson, J.A., Jr. The relative ease of removing a proton, a hydrogen atom, or an electron from carboxamides versus thiocarboxamides. J. Am. Chem. Soc. 1988, 110, 5903–5904. [Google Scholar] [CrossRef]
- This behavior can be also rationalized in terms of ab initio methods, which support differences in energy for the N-H···S compared with N-H···O interaction in the chain dimers, being weaker in thioureas than in ureas (Ref. 69 and 70).
- Masunov, A.; Dannenberg, J.J. Theoretical Study of Urea. I. Monomers and Dimers. J. Phys. Chem. A 1999, 103, 178–184. [Google Scholar] [CrossRef]
- Masunov, A.; Dannenberg, J.J. Theoretical Study of Urea and Thiourea. 2. Chains and Ribbons. J. Phys. Chem. B 2000, 104, 806–810. [Google Scholar] [CrossRef]
- Berkessel, A.; Cleemann, F.; Mukherjee, S.; Müller, T.N.; Lex, J. Highly Efficient Dynamic Kinetic Resolution of Azlactones by Urea-Based Bifunctional Organocatalysts. Angew. Chem. Int. Ed. 2005, 44, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Scheerder, J.; Engbersen, J.F.J.; Casnati, A.; Ungaro, R.; Reinhoudt, D.N. Complexation of Halide Anions and Tricarboxylate Anions by Neutral Urea-Derivatized p-tert-Butylcalix[6]arenes. J. Org. Chem. 1995, 60, 6448–6454. [Google Scholar] [CrossRef]
- Weil, T.; Kotke, M.; Kleiner, C.M.; Schreiner, P.R. Cooperative Brønsted Acid-Type Organocatalysis: Alcoholysis of Styrene Oxides. Org. Lett. 2008, 10, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Companyó, X.; Valero, G.; Crovetto, L.; Moyano, A.; Rios, R. Highly Enantio- and Diastereoselective Organocatalytic Desymmetrization of Prochiral Cyclohexanones by Simple Direct Aldol Reaction Catalyzed by Proline. Chem. Eur. J. 2009, 15, 6564–6568. [Google Scholar] [CrossRef] [PubMed]
- Reis, Ö.; Eymur, S.; Reis, B.; Demir, A.S. Direct enantioselective aldol reactions catalyzed by a proline–thiourea host–guest complex. Chem. Commun. 2009, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Substrate-Dependent Nonlinear Effects in Proline–Thiourea-Catalyzed Aldol Reactions: Unraveling the Role of the Thiourea Co-Catalyst. Chem. Eur. J. 2010, 16, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted Acid-Thiourea Co-catalysis: Enantioselective, Catalytic Protio-Pictet-Spengler Reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric Cooperative Catalysis of Strong Brønsted Acid–Promoted Reactions Using Chiral Ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-L.; Shi, M. Chiral Thiourea-Phosphine Organocatalysts in the Asymmetric Aza-Morita–Baylis–Hillman Reaction. Adv. Synth. Catal. 2007, 349, 2129–2135. [Google Scholar] [CrossRef]
- Escuder, B.; LLusar, M.; Miravet, J.F. Insight on the NMR Study of Supramolecular Gels and Its Application to Monitor Molecular Recognition on Self-Assembled Fibers. J. Org. Chem. 2006, 71, 7747–7752. [Google Scholar] [CrossRef] [PubMed]
- Schoonbeek, F.S.; van Esch, J.H.; Hulst, R.; Kellogg, R.M.; Feringa, B.L. Geminal Bis-ureas as Gelators for Organic Solvents: Gelation Properties and Structural Studies in Solution and in the Gel State. Chem. Eur. J. 2000, 6, 2633–2643. [Google Scholar] [CrossRef]
- Jung, J.H.; Shinkai, S.; Shimizu, T. Spectral Characterization of Self-Assemblies of Aldopyranoside Amphiphilic Gelators: What is the Essential Structural Difference Between Simple Amphiphiles and Bolaamphiphiles? Chem. Eur. J. 2002, 8, 2684–2690. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S., Jr. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational iffusion: Part 1. Basic theory. Conc. Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Reactive Intermediates; Santos, L.S., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Liu, X.-G.; Jiang, J.-J.; Shia, M. Development of axially chiral bis(arylthiourea)-based organocatalysts and their application in the enantioselective Henry reaction. Tetrahedron: Asymmetry 2007, 18, 2773–2781. [Google Scholar] [CrossRef]
- Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. Asymmetric Friedel-Crafts Alkylations of Indoles with Nitroalkenes Catalyzed by Zn(II)-Bisoxazoline Complexes. J. Org. Chem. 2006, 71, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Meshram, H.M.; Rao, N.N.; Kumar, G.S. Boric Acid–Mediated Mild and Efficient Friedel–Crafts Alkylation of Indoles with Nitro Styrenes. Synth. Commun. 2010, 40, 3496–3500. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Wu, F.; Wan, B. A New Type of Bis(sulfonamide)-Diamine Ligand for a Cu(OTf)2-Catalyzed Asymmetric Friedel-Crafts Alkylation Reaction of Indoles with Nitroalkenes. Org. Lett. 2011, 13, 4834–4837. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral Phosphoric Acid Catalyzed Enantioselective Friedel–Crafts Alkylation of Indoles with Nitroalkenes: Cooperative Effect of 3 Å Molecular Sieves. Angew. Chem. Int. Ed. 2008, 47, 4016–4018. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Solvent | Time (Days) | Yield (%) b | ee (%) c |
|---|---|---|---|---|
| 1 d | CHCl3 | 3 | 46 | 36 |
| 2 | CHCl3 | 3 | 88 | 55 |
| 3 d | Toluene | 3 | 10 | 18 |
| 4 | Toluene | 3 | 38 | 42 |
| 5d | Xylene | 4 | 18 | 28 |
| 6 | Xylene | 4 | 95 | 47 |
| 7 d | CH3CN | 10 | n.r. e | _ |
| 8 | CH3CN | 4 | n.d. f | 8 |
| 9 d | THF | 10 | n.r. e | _ |
| 10 | THF | 10 | n.r. e | _ |
| 11 d | AcOEt | 10 | n.r. e | _ |
| 12 | AcOEt | 10 | n.r. e | _ |

| Entry | Acid | T (°C) | Time (Days) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|
| 1 | - | 15 | 5 | 24 | 46 |
| 2 | 9ad | 15 | 4 | 60 | 62 |
| 3 | 9ae | 15 | 4 | 77 | 60 |
| 4 | 9af | 15 | 4 | 82 | 57 |
| 5 | - | -25 | 5 | 15 | 57 |
| 6 | 9ae | -25 | 5 | 23 | 68 |

| Entry | Catalyst | Acid | Yield (%) b | ee (%) c |
|---|---|---|---|---|
| 1 | 1b | - | 46 | 36 |
| 2 | 1b | (±)-9a | 88 | 55 |
| 3 | 1b |  | 83 | 48 |
| 4 | 1b |  | 57 | 40 |
| 5 | 1b |  | 55 | 37 |
| 6 | 1b | MeOH | 68 | 40 |
| 7 | 1c | (±)-9a | 18 (13) d | Rac. (Rac.) d |
| 8 | 1d | (±)-9a | 25 (7) d | Rac. (Rac.) d |

| Entry | (±)-9a (mmol) | δNHa (ppm) | δHb (ppm) | δHc (ppm) | δHd (ppm) | δHd’ (ppm) | Ratio Integrals Urea:Standard |
|---|---|---|---|---|---|---|---|
| 1 | 0 | 7.937 | 5.553 | 4.691 | 3.238 | 2.976 | 0.22:1 |
| 2 | 0.01 | 7.888 | 5.818 | 4.670 | 3.219 | 2.949 | 0.37:1 |
| 3 | 0.02 | 7.852 | 5.971 | 4.635 | 3.177 | 2.923 | 0.62:1 |
| 4 | 0.03 | 7.852 | 6.030 | 4.629 | 3.173 | 2.926 | 0.70:1 |
| 5 | 0.04 | 7.844 | 6.032 | 4.630 | 3.176 | 2.923 | 0.79:1 |
| 6 | 0.06 | 7.845 | 6.034 | 4.625 | 3.167 | 2.920 | 0.78:1 |
| 7 | 0.08 | 7.770 | 5.960 | 4.554 | 3.093 | 2.820 | 0.82:1 |
| 8 | 0.1 | 7.844 | 6.035 | 4.631 | 3.169 | 2.920 | 0.88:1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonsona, I.G.; Marqués-López, E.; Häring, M.; Díaz, D.D.; Herrera, R.P. Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance. Catalysts 2018, 8, 305. https://doi.org/10.3390/catal8080305
Sonsona IG, Marqués-López E, Häring M, Díaz DD, Herrera RP. Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance. Catalysts. 2018; 8(8):305. https://doi.org/10.3390/catal8080305
Chicago/Turabian StyleSonsona, Isaac G., Eugenia Marqués-López, Marleen Häring, David Díaz Díaz, and Raquel P. Herrera. 2018. "Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance" Catalysts 8, no. 8: 305. https://doi.org/10.3390/catal8080305
APA StyleSonsona, I. G., Marqués-López, E., Häring, M., Díaz, D. D., & Herrera, R. P. (2018). Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance. Catalysts, 8(8), 305. https://doi.org/10.3390/catal8080305