Assessing the Potential of Co-Pt Bronze for Electrocatalysis in Acidic Media


Abstract
1. Introduction
2. Results and Discussion
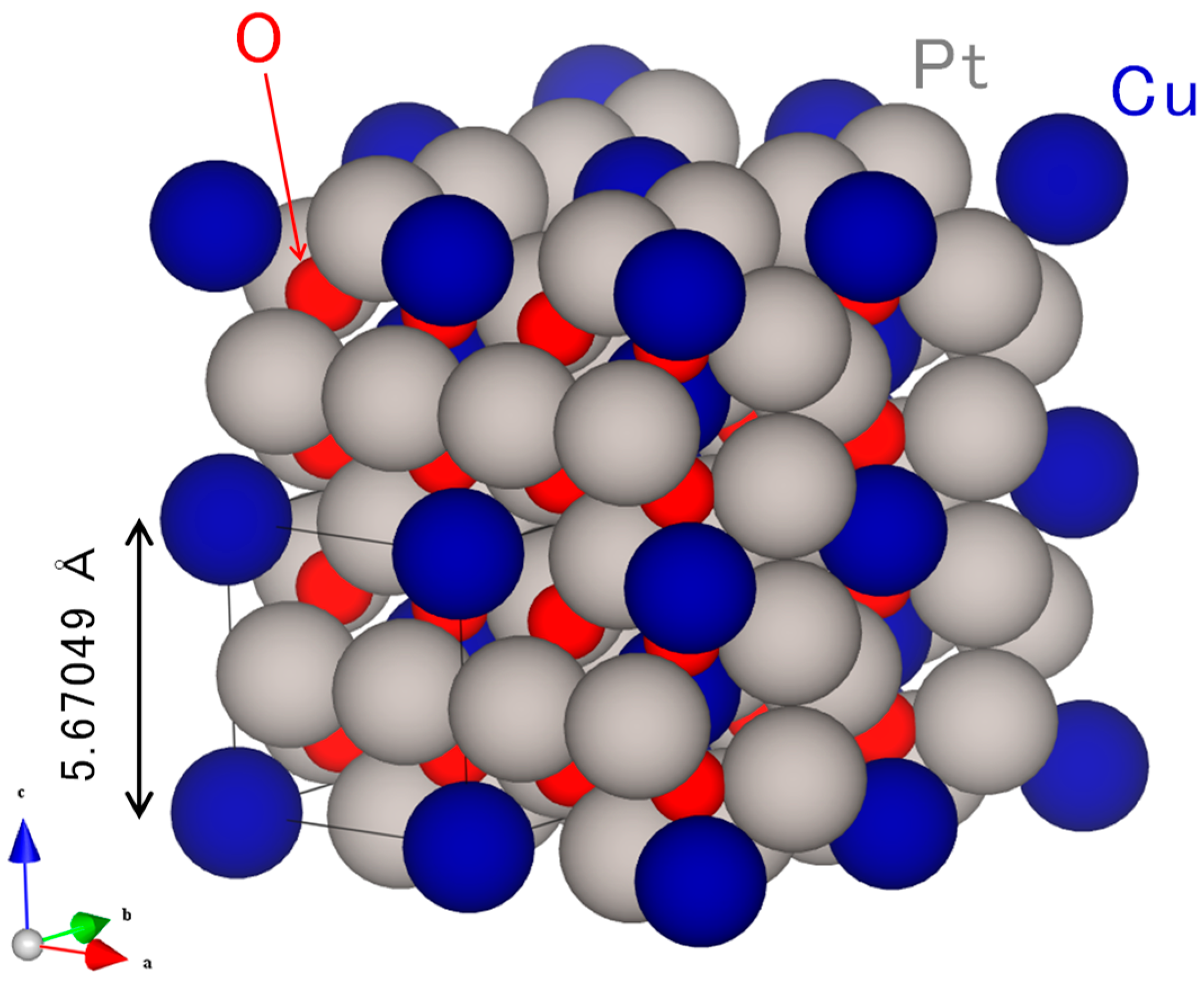
2.1. Original Co-Pt Bronze
2.1.1. Characterization
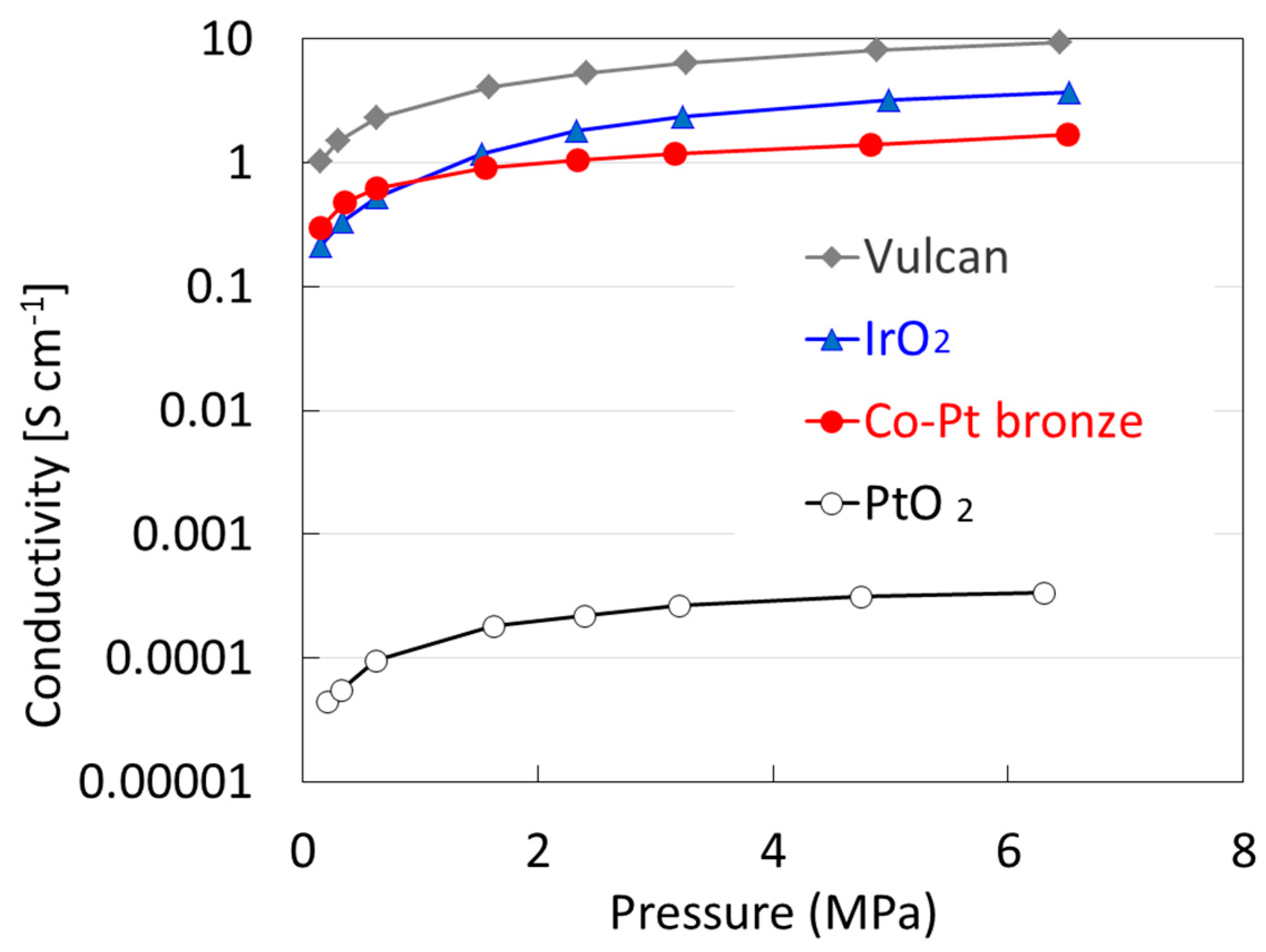
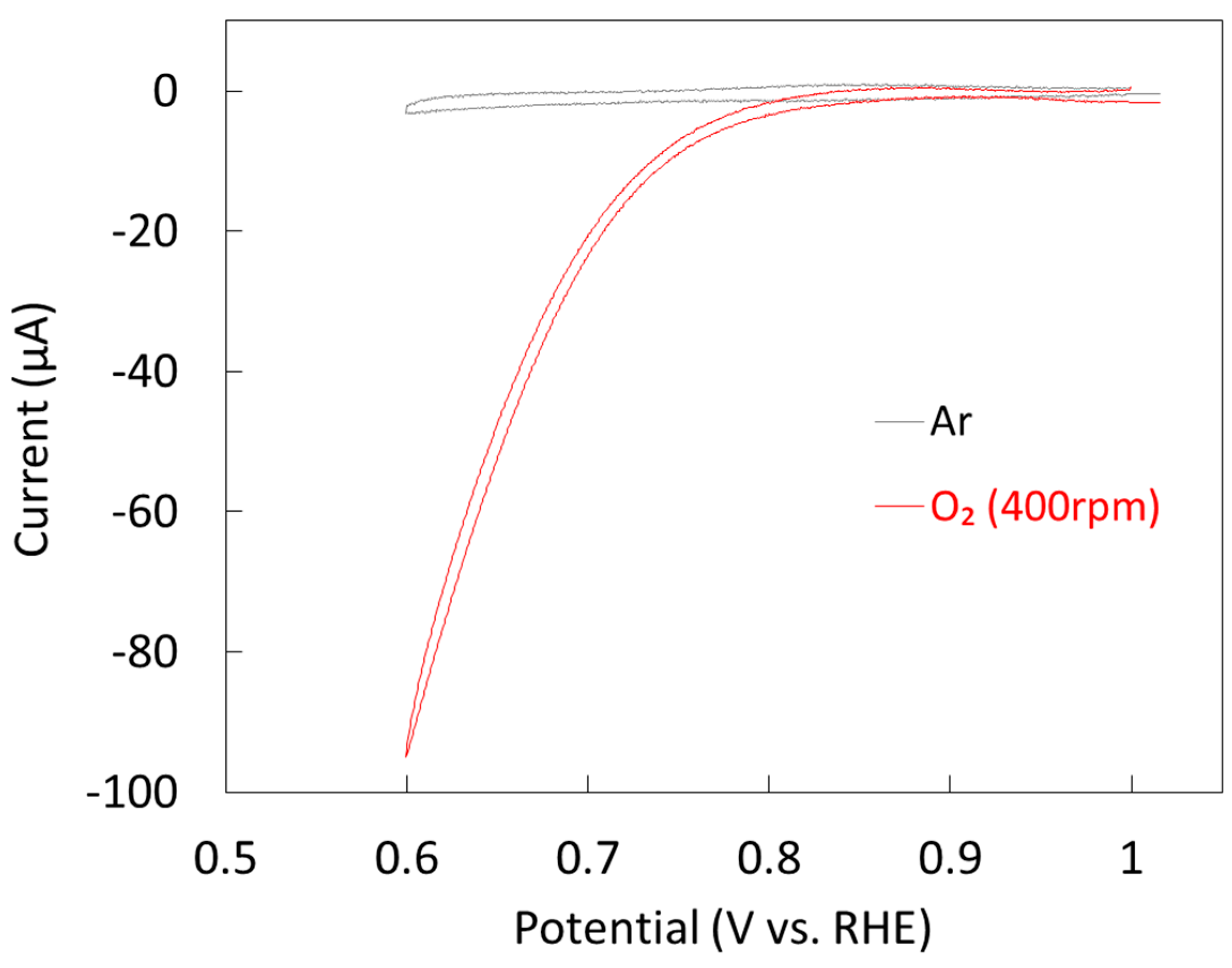
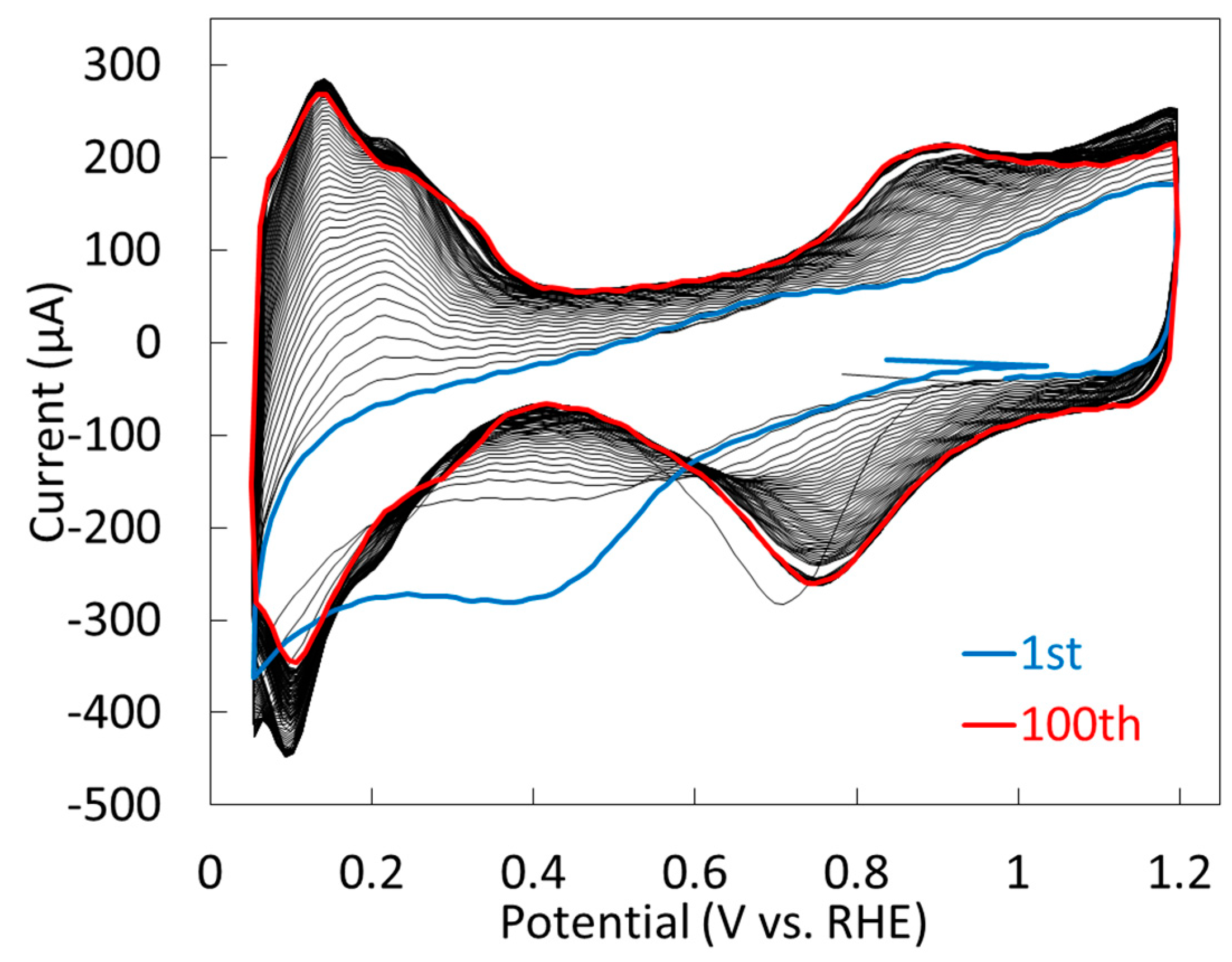
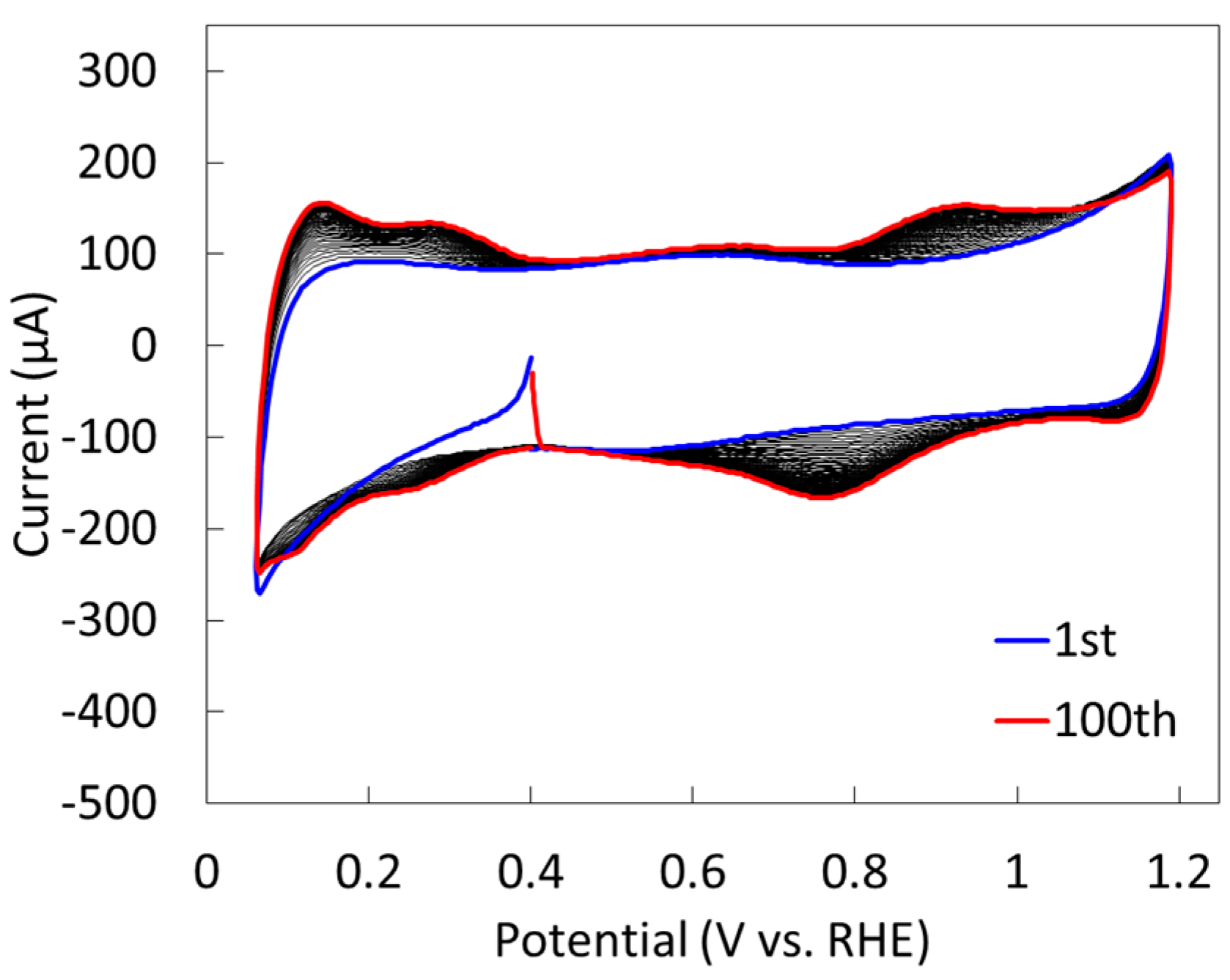
2.1.2. Electrochemical Properties
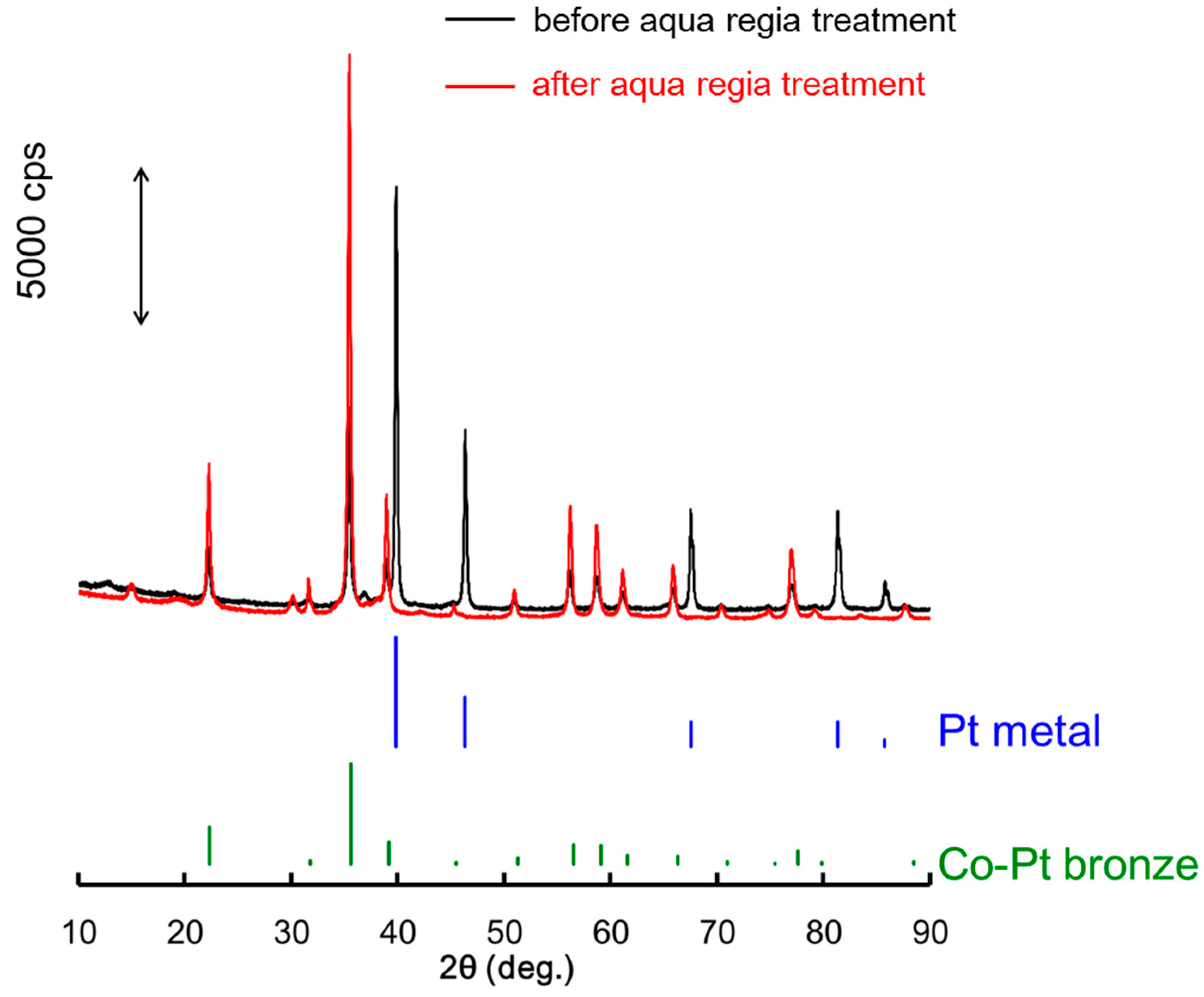
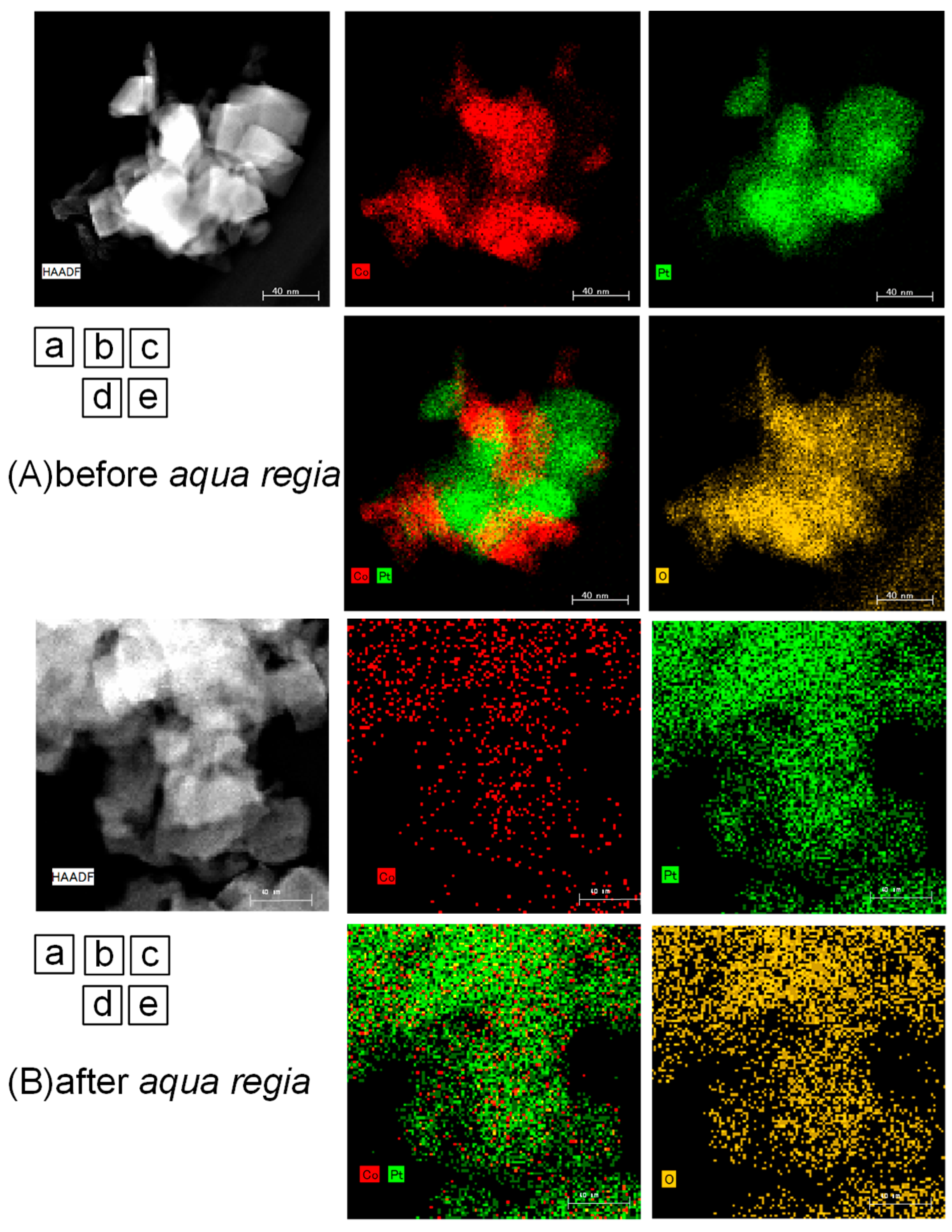
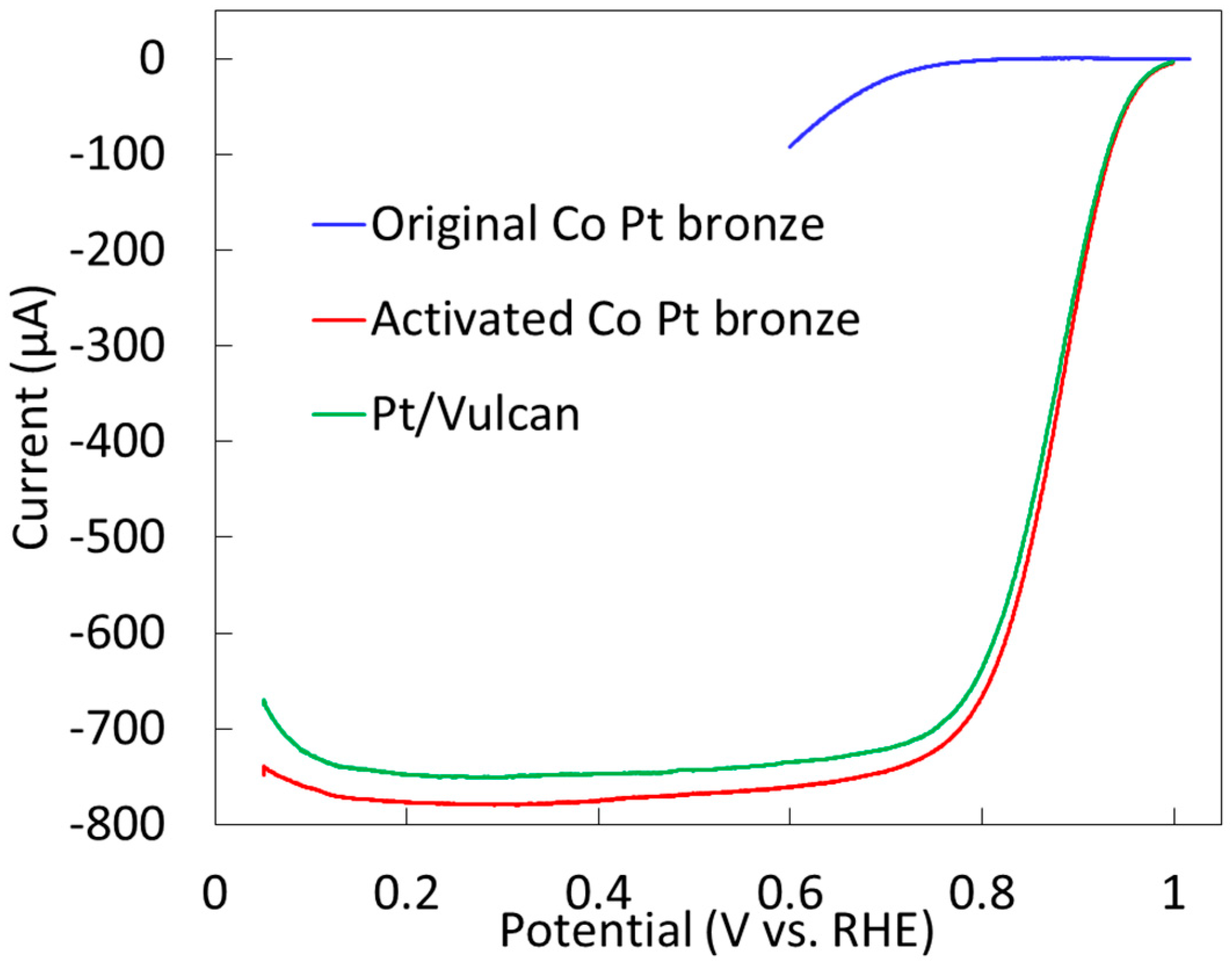
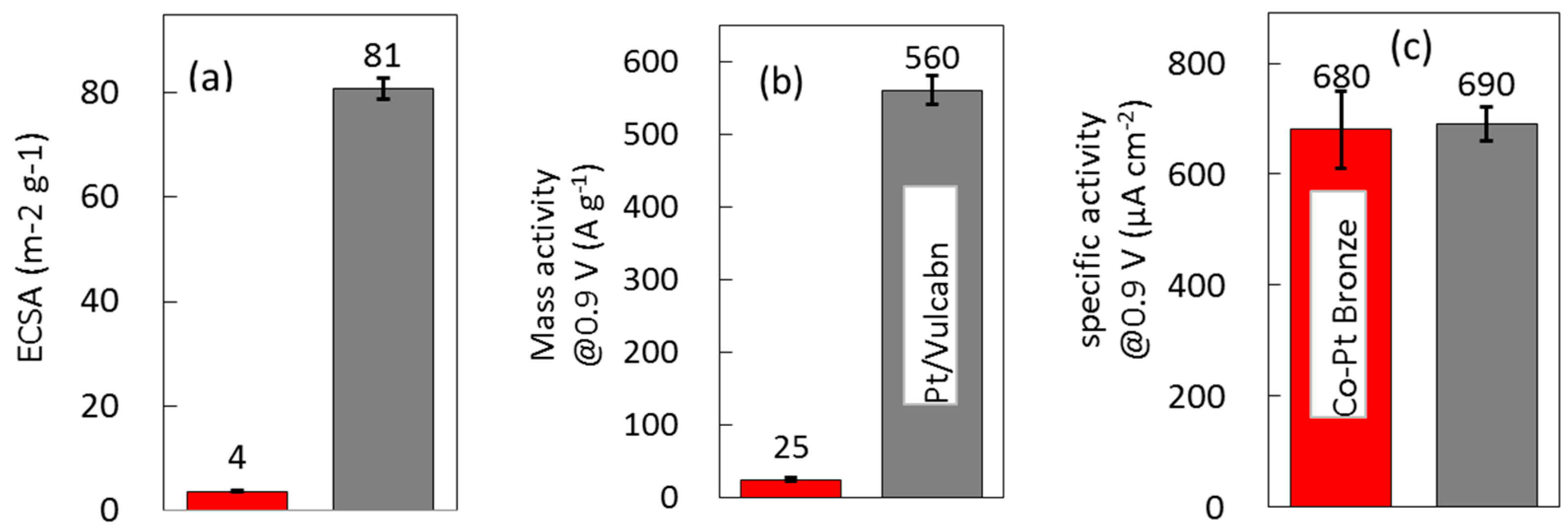
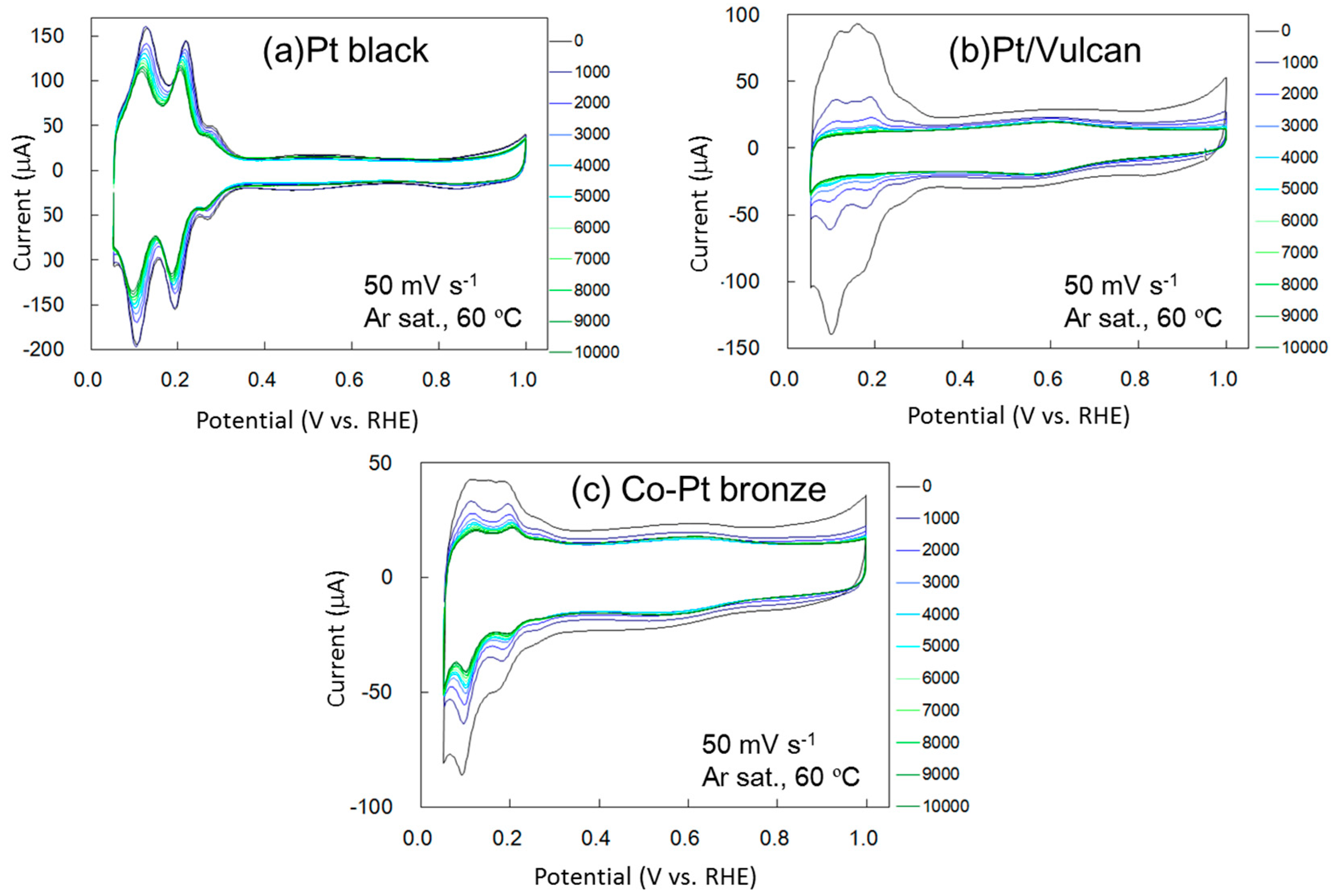
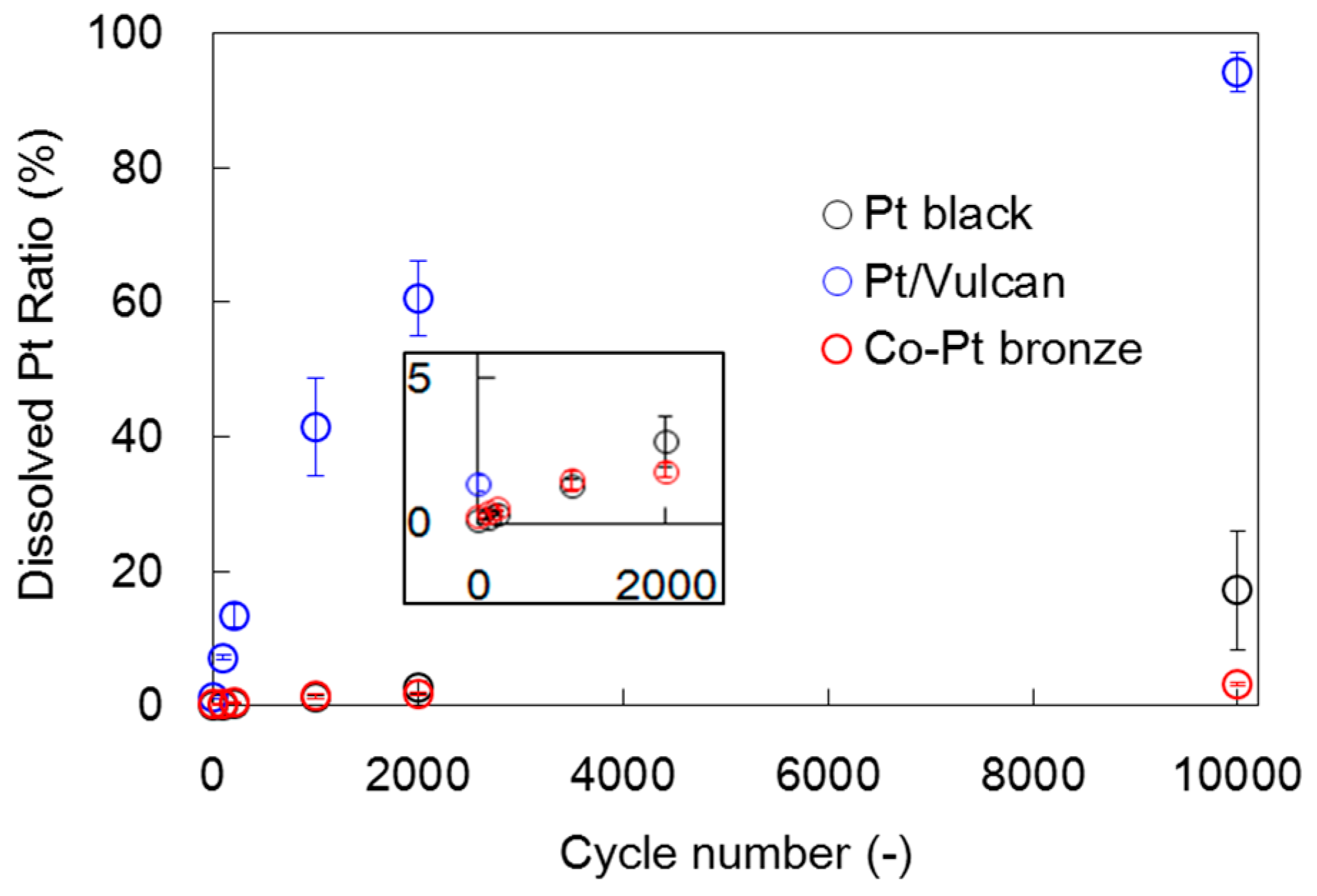
2.2. Surface Treated Co-Pt Bronze
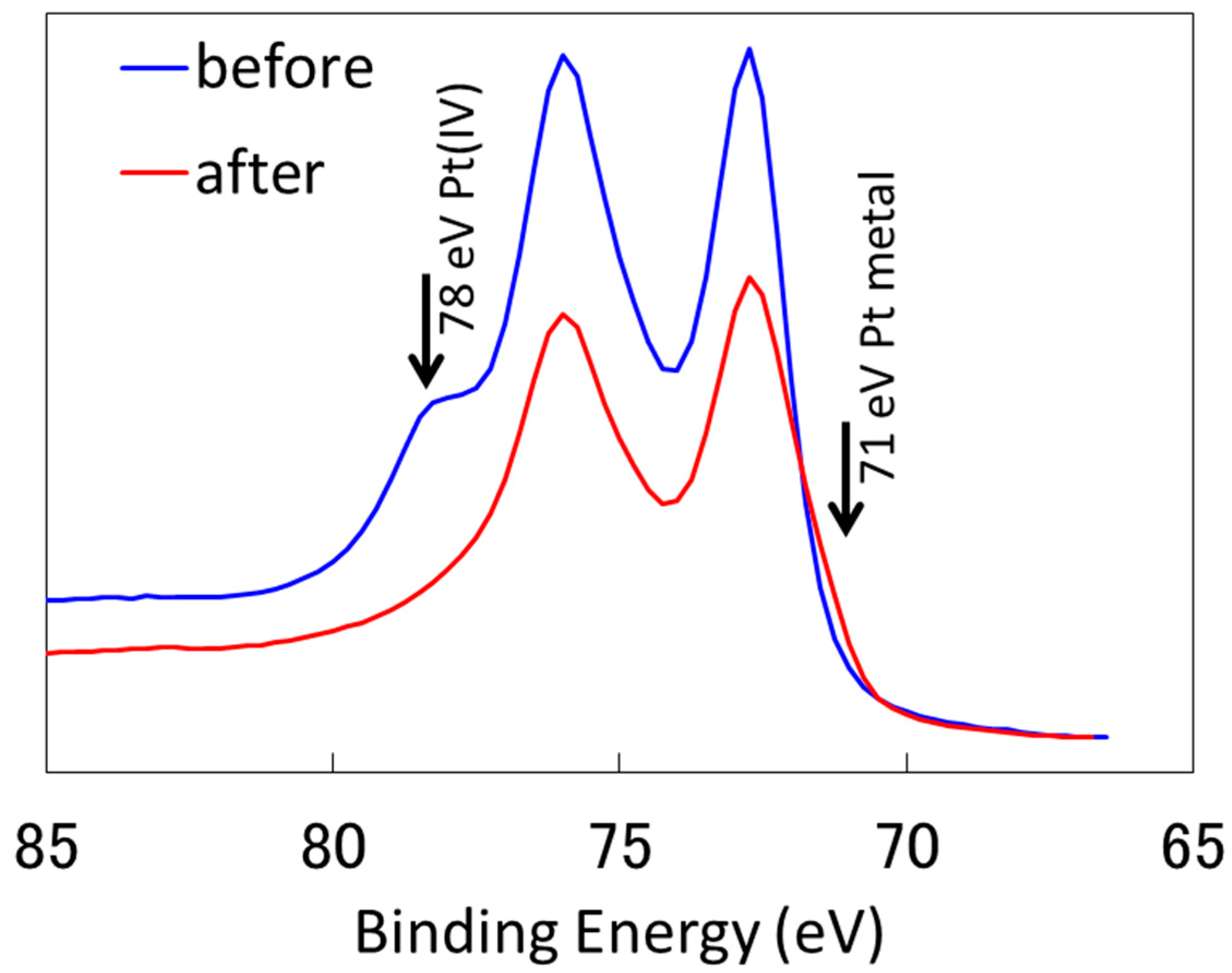
2.2.1. Characterization
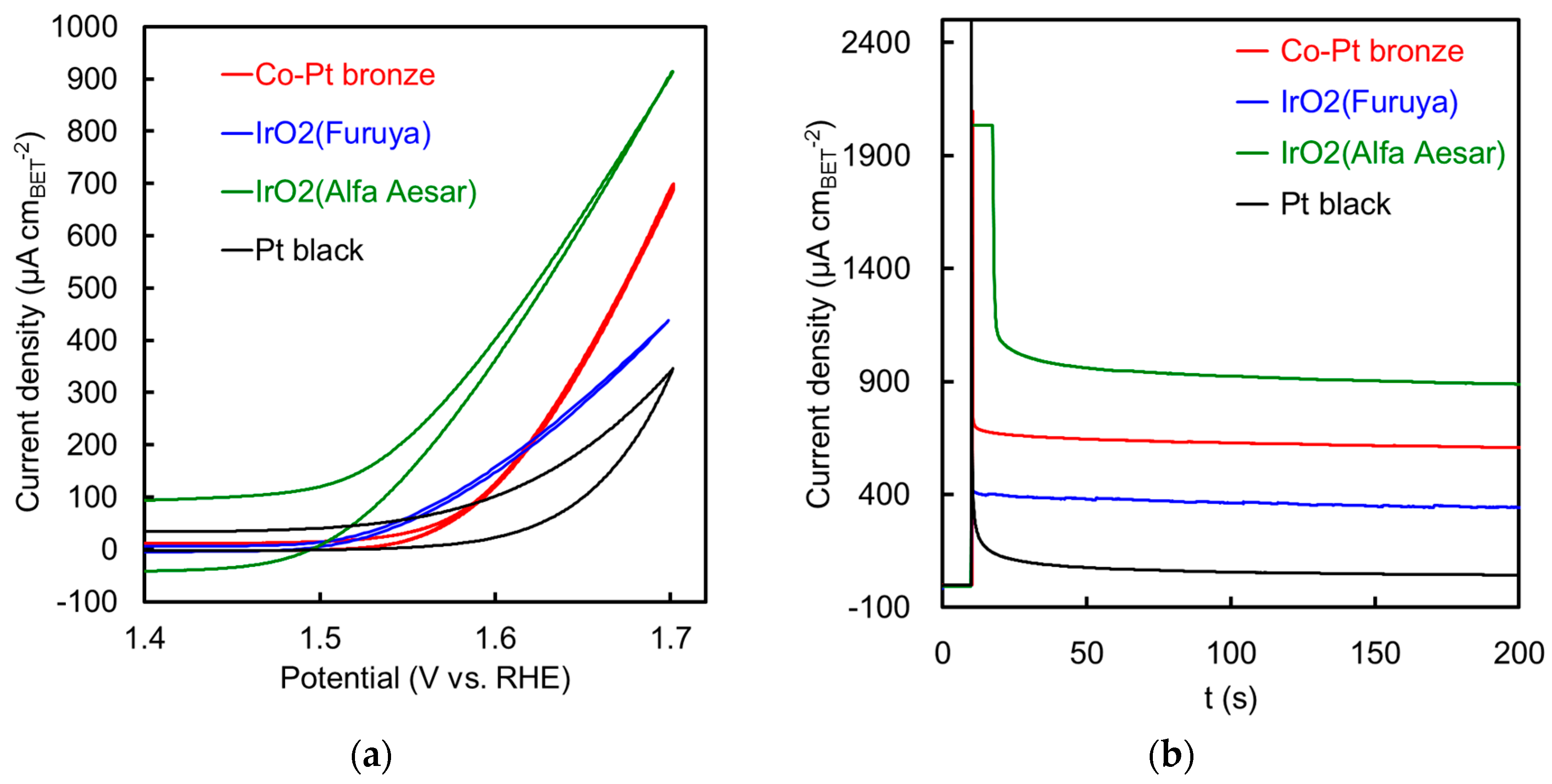
2.2.2. Electrochemical Properties
3. Experimental
3.1. Materials
3.2. Synthesis
3.3. Characterization
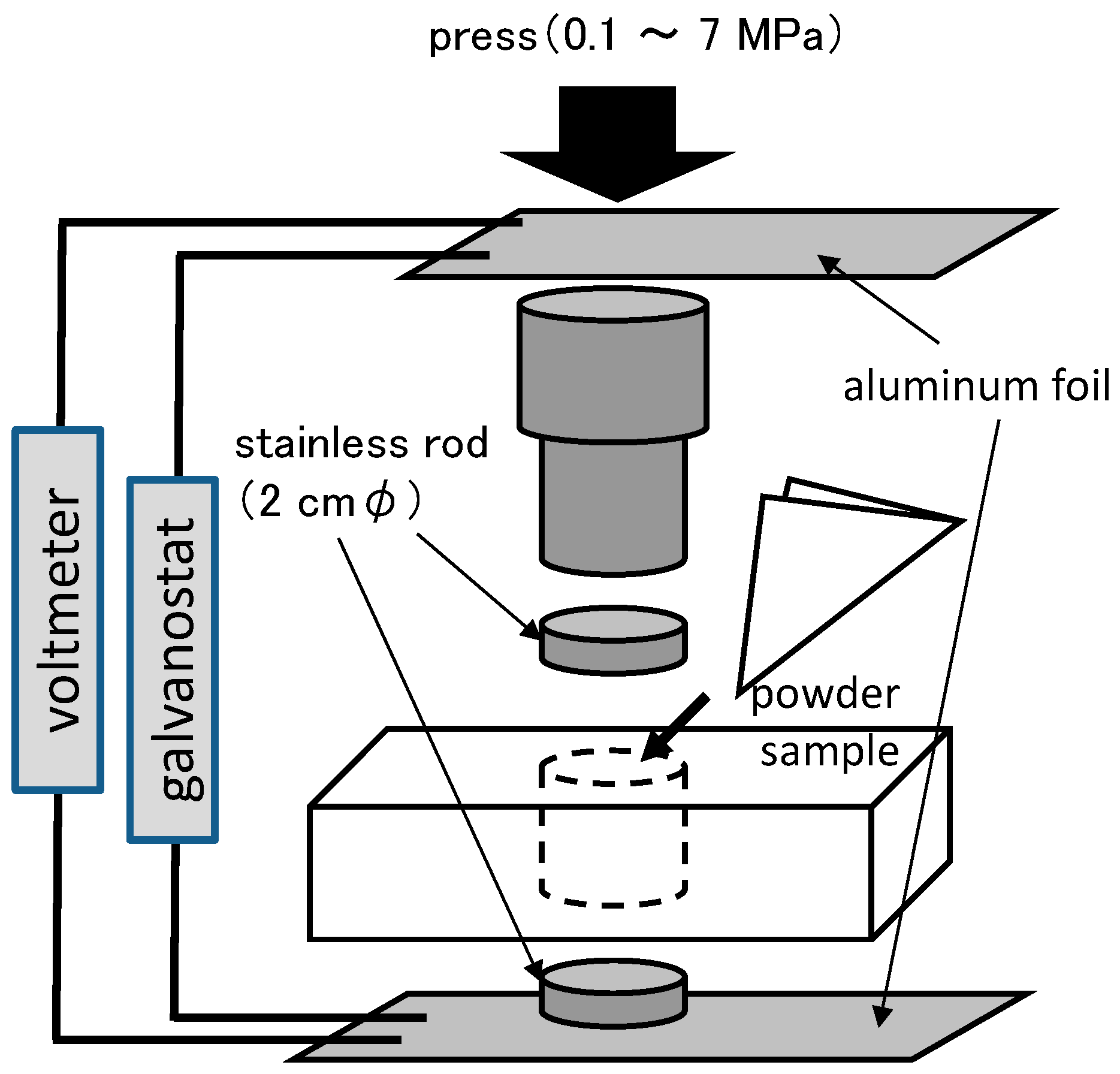
3.4. Electrochemical Tests
4. Conclusions
- Water electrolyzer anode with high OER activity without Ir or Ru,
- PEFC cathode catalyst with high load-cycle durability and high start-up shutdown durability due to the high OER activity [7],
- PEFC anode catalyst with high potential-reversal durability due to the high OER activity [7],
- Regenerative fuel cell oxygen-side catalyst.
- Examining long time durability as a water electrolyzer anode,
- Synthesizing smaller particles to improve the mass-activity,
- Synthesizing and testing other metal-platinum bronzes rather than Co-Pt bronze.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Adzic, R.R.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Shao, M.; Wang, J.X.; Nilekar, A.U.; Mavrikakis, M.; Valerio, J.A.; Uribe, F. Platinum monolayer fuel cell electrocatalysts. Top. Catal. 2007, 46, 249–262. [Google Scholar] [CrossRef]
- Chung, D.Y.; Jun, S.W.; Yoon, G.; Kwon, S.G.; Shin, D.Y.; Seo, P.; Yoo, J.M.; Shin, H.; Chung, Y.H.; Kim, H.; et al. Highly Durable and Active PtFe Nanocatalyst for Electrochemical Oxygen Reduction Reaction. J. Am. Chem. Soc. 2015, 137, 15478–15485. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Xie, S.F.; Shao, M.H.; Odell, J.H.; Lu, N.; Peng, H.C.; Protsailo, L.; Guerrero, S.; Park, J.H.; Xia, X.H.; et al. Synthesis and Characterization of 9 nm Pt-Ni Octahedra with a Record High Activity of 3.3 A/mg(Pt) for the Oxygen Reduction Reaction. Nano Lett. 2013, 13, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Pivovar, B. Extended, Continuous PtNanostructures in Thick, Dispersed Electrodes; 2015 Annual Merit Review; DOE: Washington, DC, USA, 2015; p. 8. [Google Scholar]
- Meyers, J.P.; Darling, R.M. Model of Carbon Corrosion in PEM Fuel Cells. J. Electrochem. Soc. 2006, 153, A1432–A1442. [Google Scholar] [CrossRef]
- Gallagher, K.G.; Darling, R.M.; Fuller, T.F. Carbon-support corrosion mechanisms and models. In Handbook of Fuel Cells; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; Volume 6, pp. 819–828. [Google Scholar]
- Han, L.; Dong, S.; Wang, E. Transition-Metal (Co, Ni, and Fe)-Based Electrocatalysts for the Water Oxidation Reaction. Adv. Mater. 2016, 28, 9266–9291. [Google Scholar] [CrossRef] [PubMed]
- Reier, T.; Nong, H.N.; Teschner, D.; Schlögl, R.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction in Acidic Environments—Reaction Mechanisms and Catalysts. Adv. Energy Mater. 2017, 7, 1601275. [Google Scholar] [CrossRef]
- Alia, S.M.; Shulda, S.; Ngo, C.; Pylypenko, S.; Pivovar, B.S. Iridium-Based Nanowires as Highly Active, Oxygen Evolution Reaction Electrocatalysts. ACS Catal. 2018, 8, 2111–2120. [Google Scholar] [CrossRef]
- Alia, S.M.; Rasimick, B.; Ngo, C.; Neyerlin, K.C.; Kocha, S.S.; Pylypenko, S.; Xu, H.; Pivovar, B.S. Activity and Durability of Iridium Nanoparticles in the Oxygen Evolution Reaction. J. Electrochem. Soc. 2016, 163, F3105–F3112. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Rudnick, R.L.; Gao, S. 3.01—Composition of the Continental Crust A2—Holland, Heinrich D. In Treatise on Geochemistry; Turekian, K.K., Ed.; Pergamon: Oxford, UK, 2003; pp. 1–64. [Google Scholar]
- Park, S.; Shao, Y.; Liu, J.; Wang, Y. Oxygen electrocatalysts for water electrolyzers and reversible fuel cells: Status and perspective. Energy Environ. Sci. 2012, 5, 9331–9344. [Google Scholar] [CrossRef]
- Shannon, R.D.; Gier, T.E.; Carcia, P.F.; Bierstedt, P.E.; Flippen, R.B.; Vega, A.J. Synthesis and properties of platinum metal-oxides of the type MXPT3O4. Inorg. Chem. 1982, 21, 3372–3382. [Google Scholar] [CrossRef]
- Schwartz, K.B.; Parise, J.B.; Prewitt, C.T.; Shannon, R.D. Analysis of structural distortions in nonstoichiometric ternary platinum oxides—Li0.64Pt3O4 and Co0.37Na0.14Pt3O4. Acta Crystallogr. Sect. B Struct. Sci. 1982, 38, 2109–2116. [Google Scholar] [CrossRef]
- Swette, L.; Kackley, N.; McCatty, S.A. Oxygen electrodes for rechargeable alkaline fuel cells. III. J. Power Sources 1991, 36, 323–339. [Google Scholar] [CrossRef]
- Swette, L.; Kackley, N. Oxygen electrodes for rechargeable alkaline fuel cells—II. J. Power Sources 1990, 29, 423–436. [Google Scholar] [CrossRef]
- Schwartz, K.B.; Prewitt, C.T. Structural and electronic-properties of binary and ternary platinum oxides. J. Phys. Chem. Solids 1984, 45, 1–21. [Google Scholar] [CrossRef]
- Tryk, D.A.; Zhang, Z.; Aldred, W.H.; Kim, S.; Scherson, D.A.; Yeager, E.B. Nickel-Platinum Bronzes as Electrocatalysts for Oxygen Reduction in Acid Electrolytes. In Proceedings of the Symposium on Electrode Materials and Processes for Energy Conversion and Storage, Pennington, NJ, USA, 1994; Supramaniam Srinivasan, D.D.M., Khandkar, A.C., Eds.; Electrochemical Society: Pennington, NJ, USA, 1994. [Google Scholar]
- Marković, N.M.; Schmidt, T.J.; Stamenković, V.; Ross, P.N. Oxygen Reduction Reaction on Pt and Pt Bimetallic Surfaces: A Selective Review. Fuel Cells 2001, 1, 105–116. [Google Scholar] [CrossRef]
- Anderson, A.B.; Roques, J.; Mukerjee, S.; Murthi, V.S.; Markovic, N.M.; Stamenkovic, V. Activation Energies for Oxygen Reduction on Platinum Alloys: Theory and Experiment. J. Phys. Chem. B 2005, 109, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Choi, R.; Han, S.W.; Park, J.T. Shape-Controlled Synthesis of Pt3Co Nanocrystals with High Electrocatalytic Activity toward Oxygen Reduction. Chem. Eur. J. 2011, 17, 12280–12284. [Google Scholar] [CrossRef] [PubMed]
- Pantea, D.; Darmstadt, H.; Kaliaguine, S.; Sümmchen, L.; Roy, C. Electrical conductivity of thermal carbon blacks: Influence of surface chemistry. Carbon 2001, 39, 1147–1158. [Google Scholar] [CrossRef]
- Zalitis, C.M.; Sharman, J.; Wright, E.; Kucernak, A.R. Properties of the hydrogen oxidation reaction on Pt/C catalysts at optimised high mass transport conditions and its relevance to the anode reaction in PEFCs and cathode reactions in electrolysers. Electrochim. Acta 2015, 176, 763–776. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Richards, R.M.; Pivovar, B.S.; Kocha, S.S. Oxygen reduction reaction measurements on platinum electrocatalysts utilizing rotating disk electrode technique I. Impact of impurities, measurement protocols and applied corrections. J. Electrochem. Soc. 2015, 162, F1144–F1158. [Google Scholar] [CrossRef]
- Obata, H.; Yoshida, T.; Ogawa, H. Determination of picomolar levels of platinum in estuarine waters: A comparison of cathodic stripping voltammetry and isotope dilution-inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2006, 580, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zadeii, J.; Lin, M.S. Ultrasensitive voltammetric measurements based on coupling hydrogen catalytic systems with controlled interfacial accumulation of the catalyst. J. Electroanal. Chem. Interfacial Electrochem. 1987, 237, 281–287. [Google Scholar] [CrossRef]
- Zhao, Z.; Freiser, H. Differential pulse polarographic determination of trace levels of platinum. Anal. Chem. 1986, 58, 1498–1501. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specific Surface Area (BET) (m2·g−1) | Mean Diameter * (nm) | Loading/GC (µg) | Loaded Surface Area/GC (cm2) | |
|---|---|---|---|---|
| Co-Pt bronze | 19 | 25 | 16 | 2.9 |
| Pt black | 13 | 22 | 11 | 1.4 |
| IrO2 (Furuya) | 54 | 9.6 | 13 | 7.2 |
| IrO2 (Alfa Aesar) | 34 | 15 | 16 | 5.4 |
| Specific Surface Area (m2 g−1) | Specific Activity@1.55 V (µA cm−2) | Mass Activity@1.55 V (A gPGM−1) | Specific Activity@1.60 V (µA cm−2) | Mass Activity@1.60 V (A gPGM−1) | |
|---|---|---|---|---|---|
| Co-Pt bronze | 19 *1 | 31 | 7.8 | 125 | 31 |
| IrO2 (Furuya) | 54 *1 | 61 | 38 | 158 | 98 |
| IrO2 (Alfa Aesar) This work | 34 *1 | 212 | 85 | 402 | 161 |
| IrO2 (Alfa Aesar [10,11]) | 28.7 *2 | 480 | 138 | 2536 | 729 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamitaka, Y.; Taguchi, N.; Morimoto, Y. Assessing the Potential of Co-Pt Bronze for Electrocatalysis in Acidic Media. Catalysts 2018, 8, 258. https://doi.org/10.3390/catal8070258
Kamitaka Y, Taguchi N, Morimoto Y. Assessing the Potential of Co-Pt Bronze for Electrocatalysis in Acidic Media. Catalysts. 2018; 8(7):258. https://doi.org/10.3390/catal8070258
Chicago/Turabian StyleKamitaka, Yuji, Noboru Taguchi, and Yu Morimoto. 2018. "Assessing the Potential of Co-Pt Bronze for Electrocatalysis in Acidic Media" Catalysts 8, no. 7: 258. https://doi.org/10.3390/catal8070258
APA StyleKamitaka, Y., Taguchi, N., & Morimoto, Y. (2018). Assessing the Potential of Co-Pt Bronze for Electrocatalysis in Acidic Media. Catalysts, 8(7), 258. https://doi.org/10.3390/catal8070258