On the Impact of the Preparation Method on the Surface Basicity of Mg–Zr Mixed Oxide Catalysts for Tributyrin Transesterification

 ,
,  ,
,  and
and
Abstract
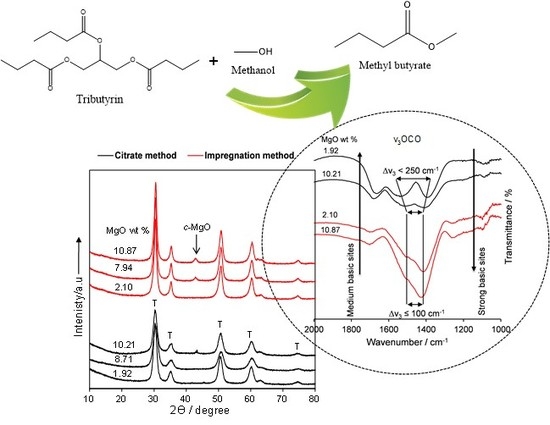
:
1. Introduction
2. Results and Discussion
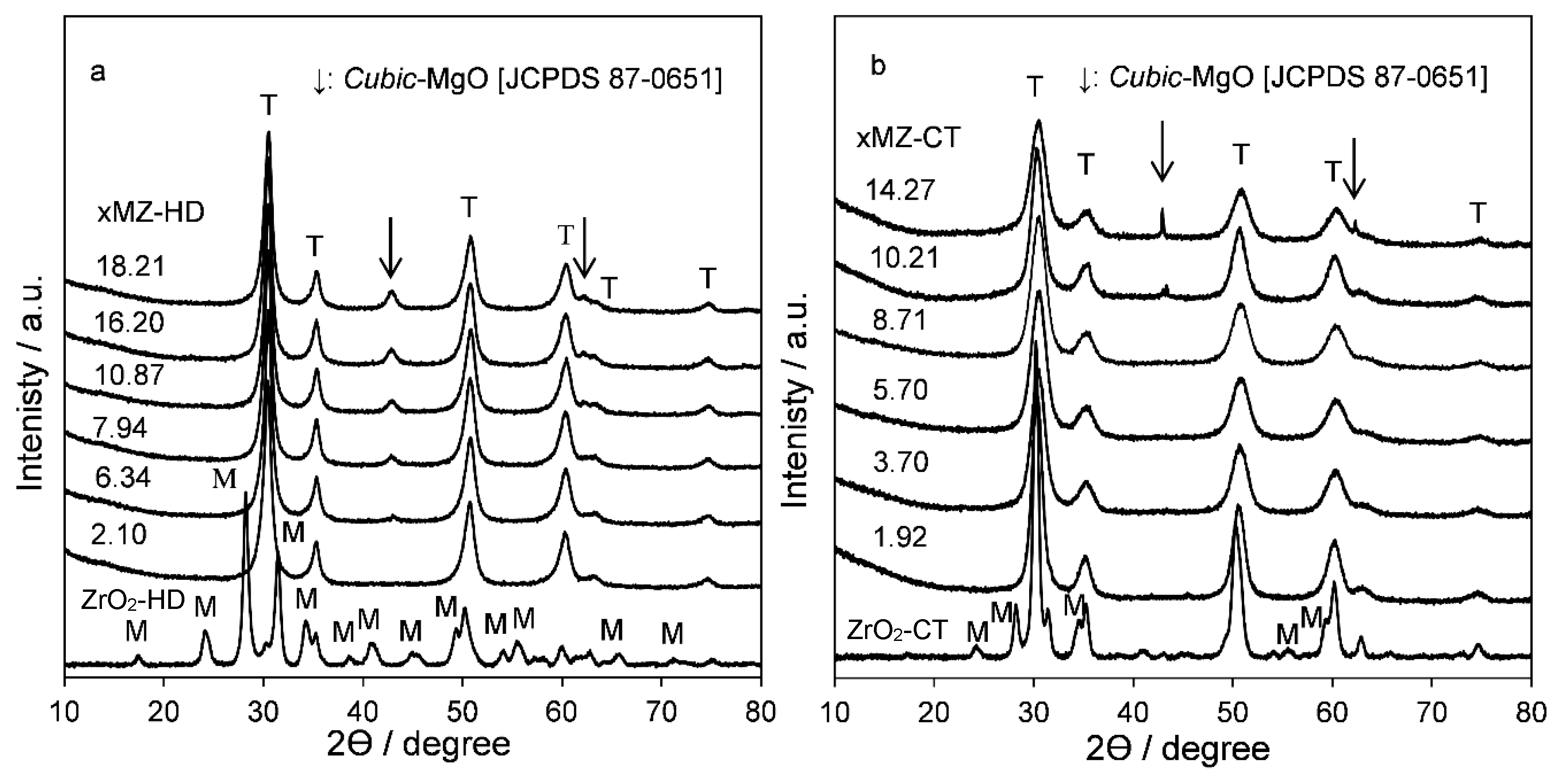
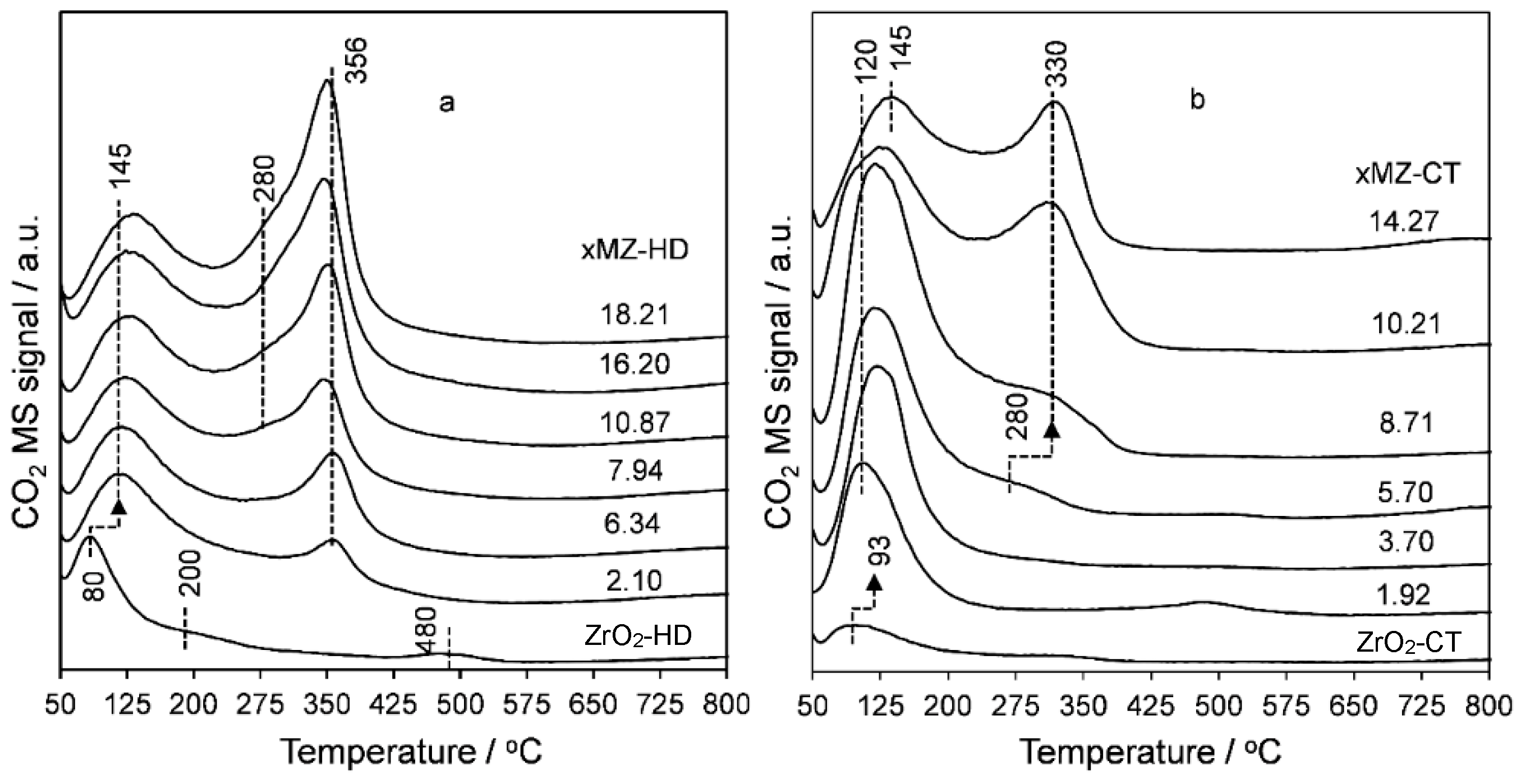
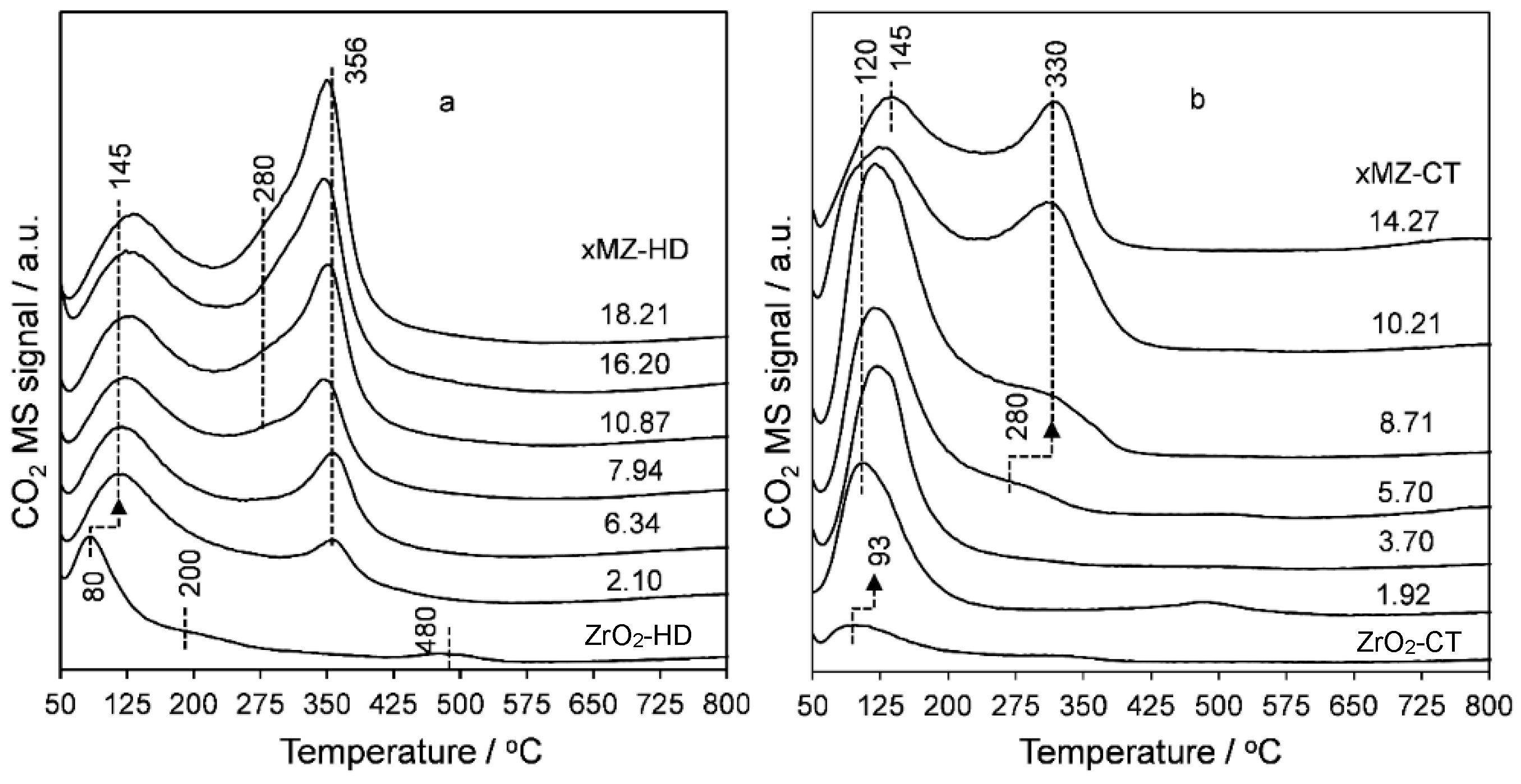
2.1. Thermal Analysis
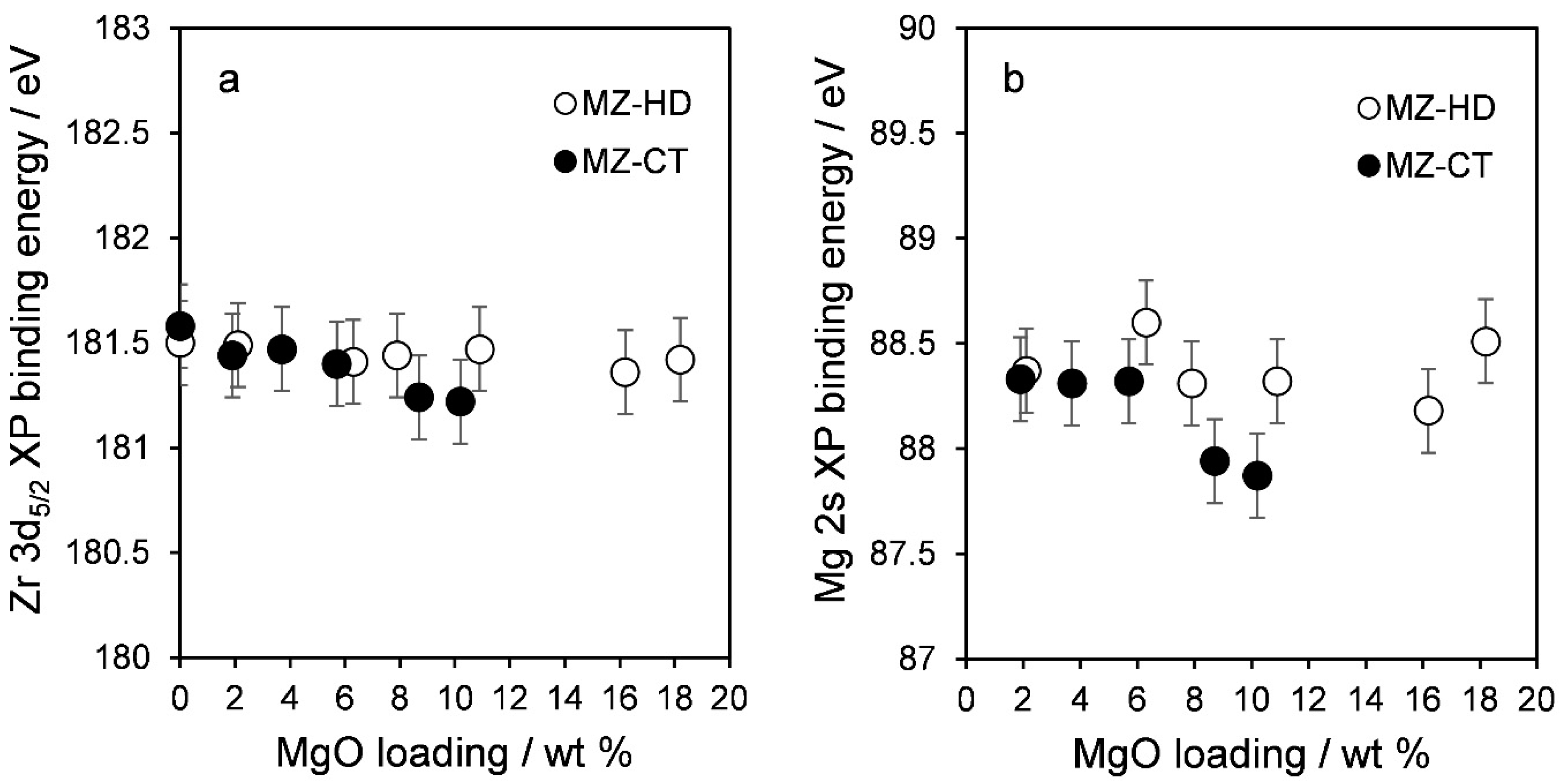
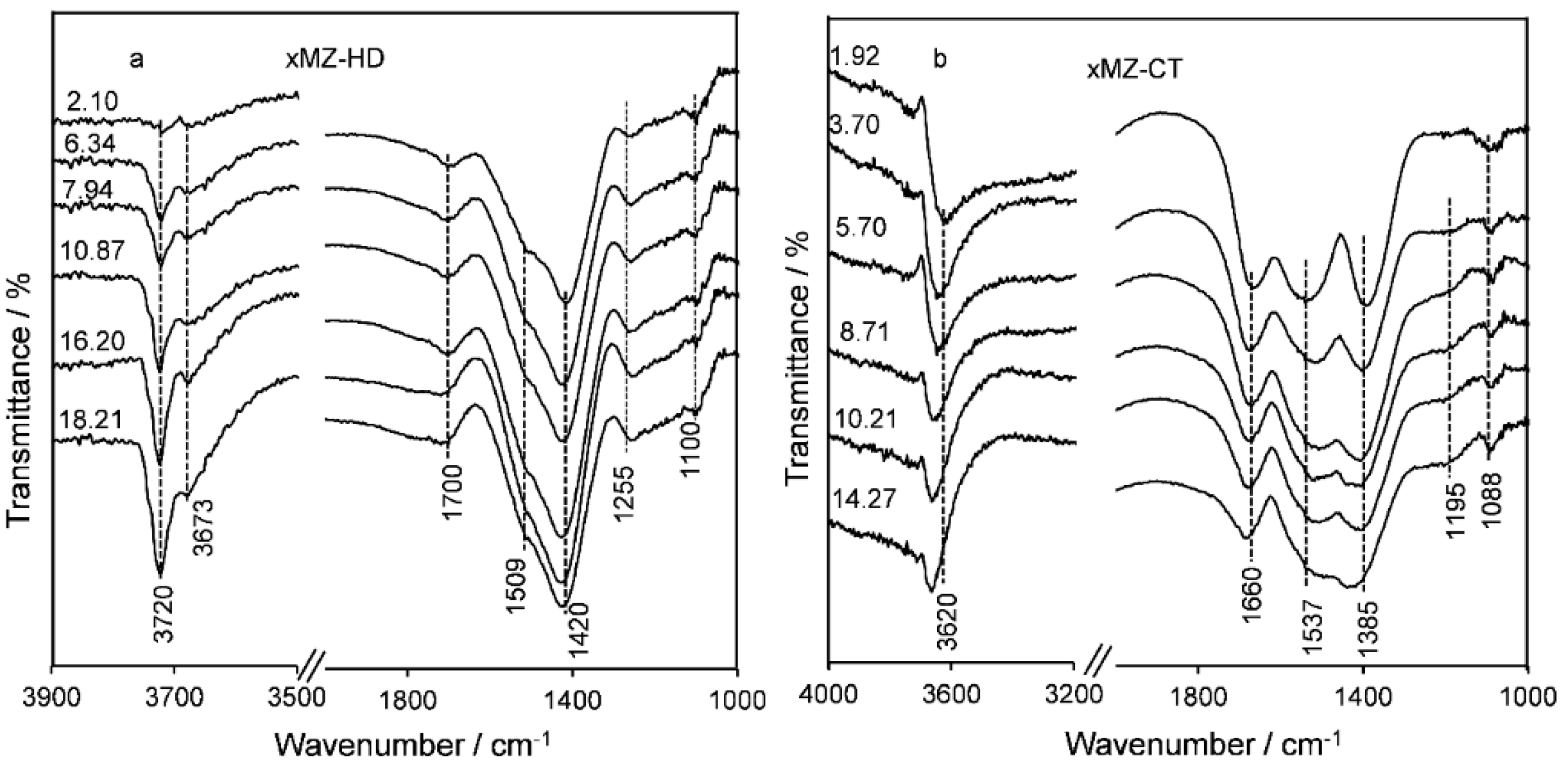
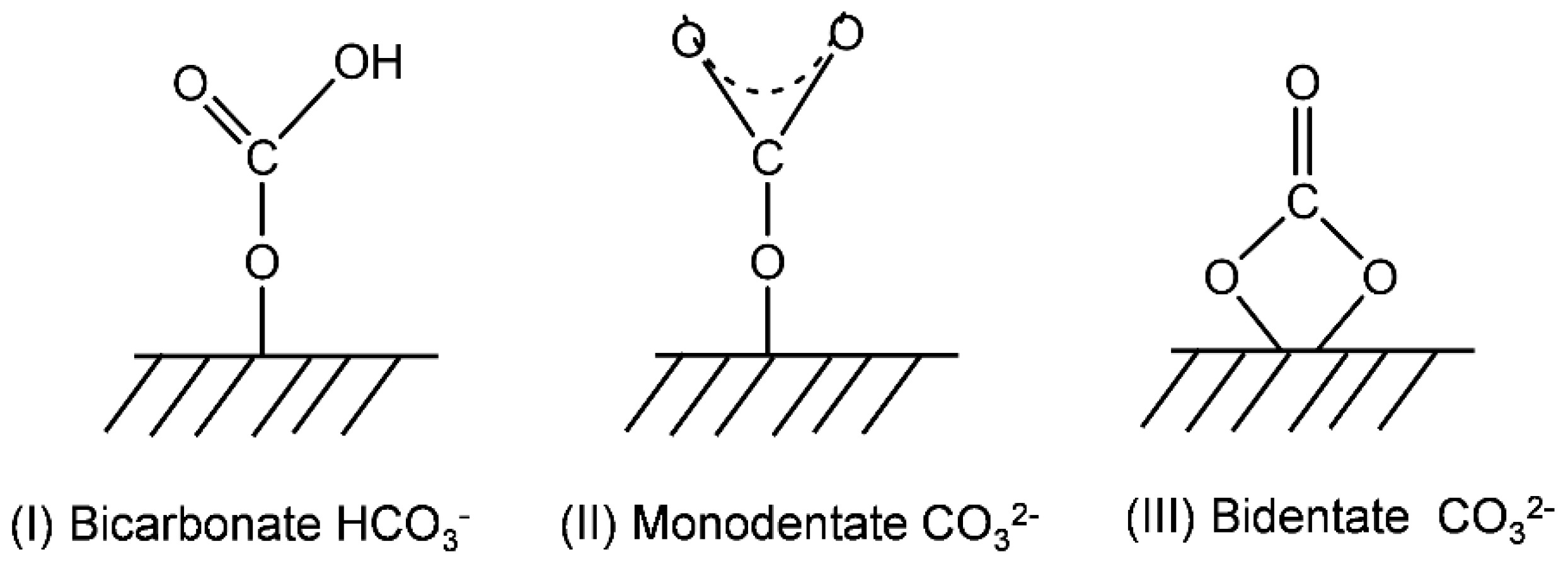
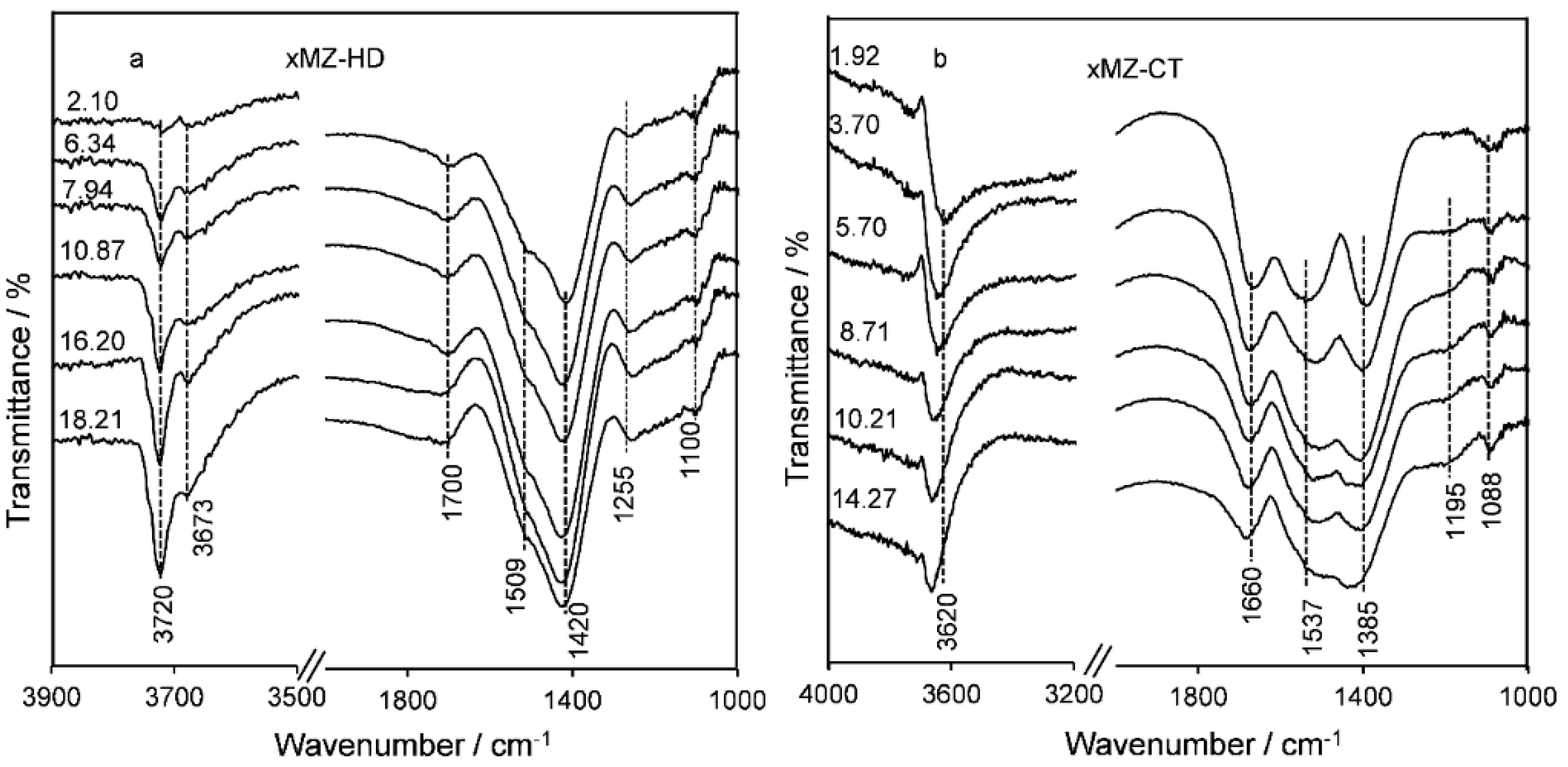
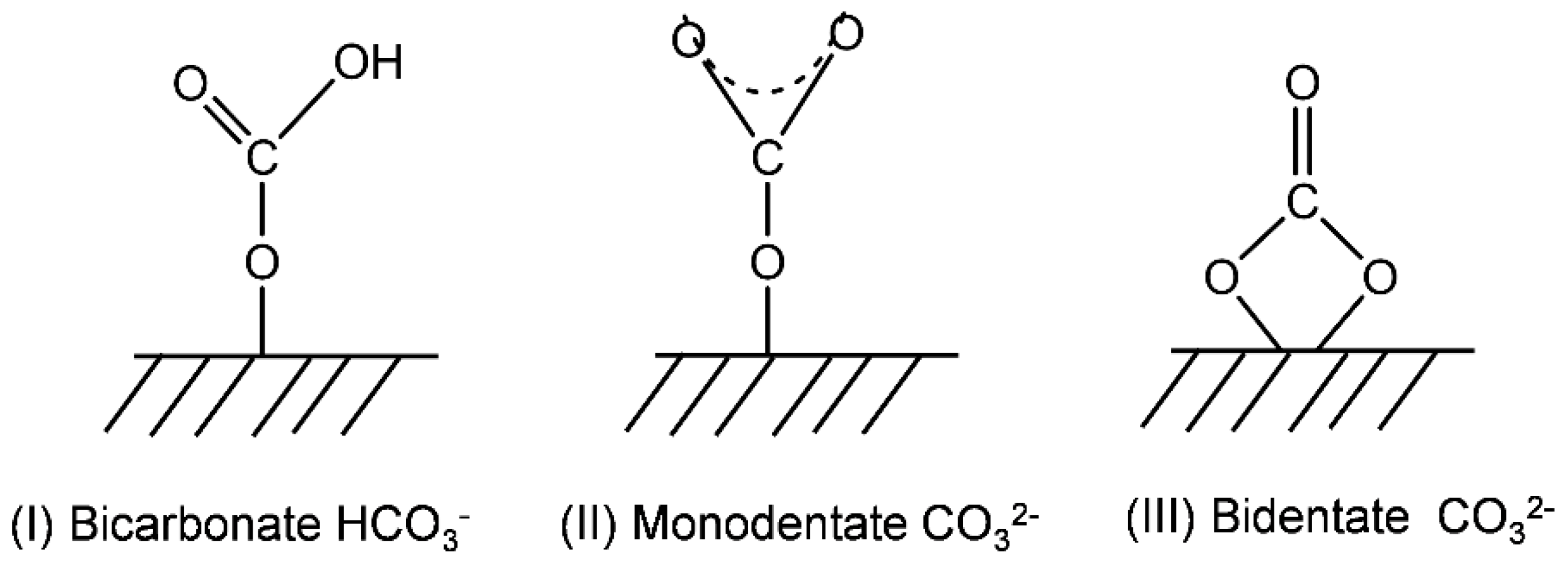
2.2. Catalyst Characteristics
Structure and Composition
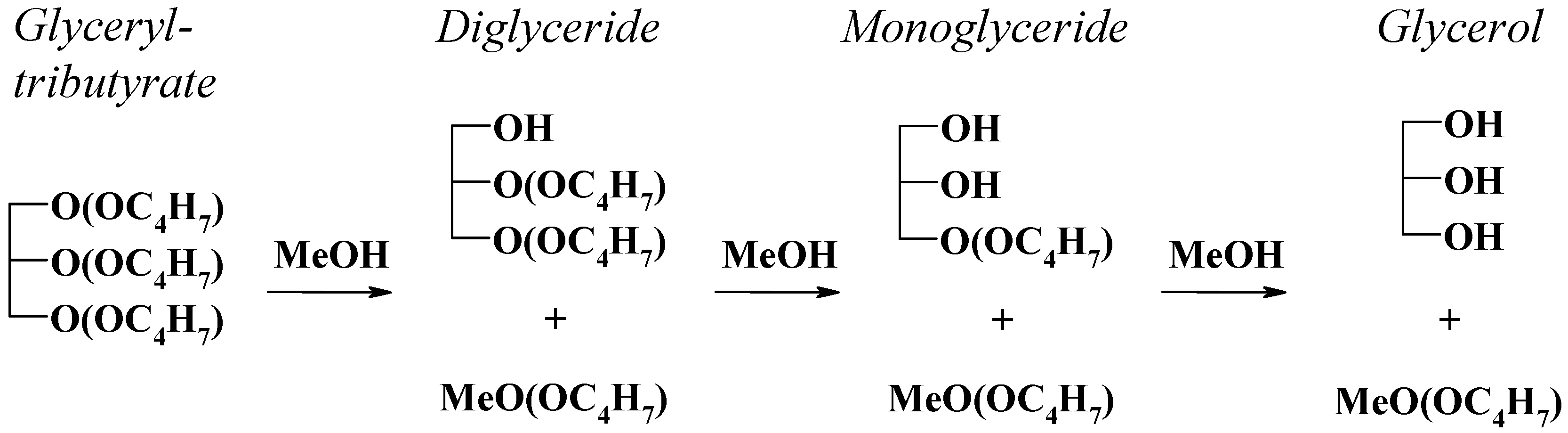
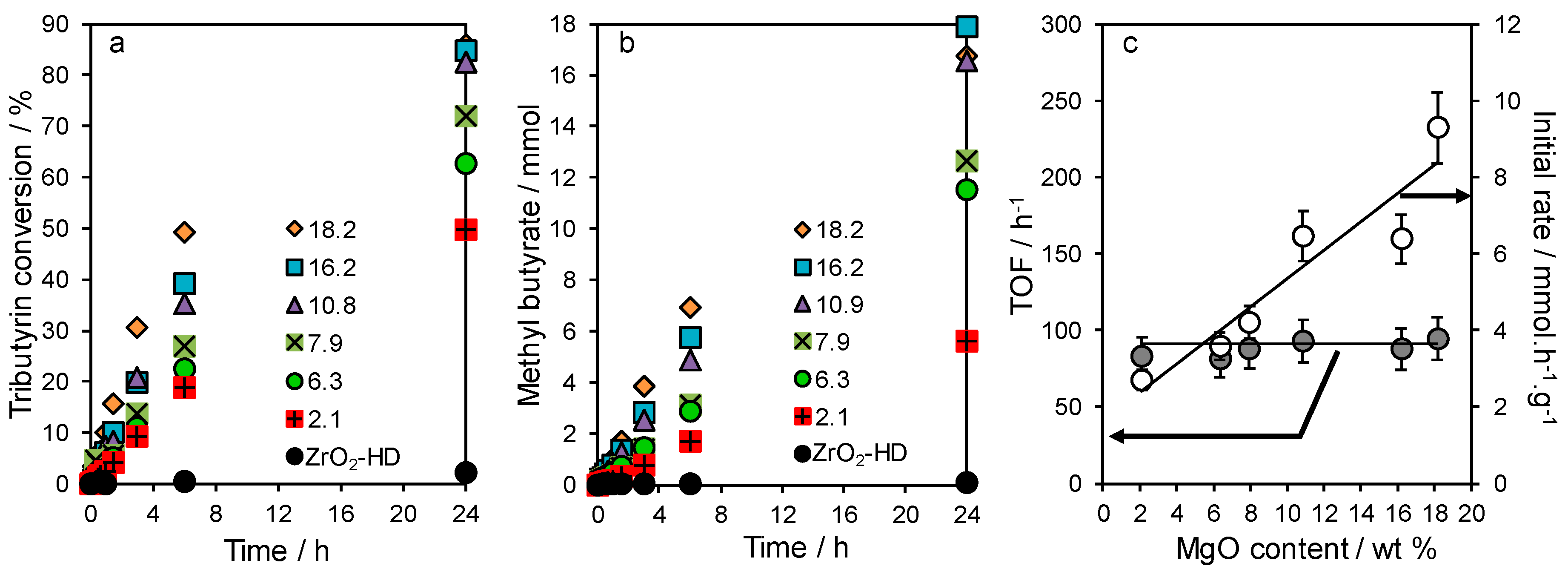
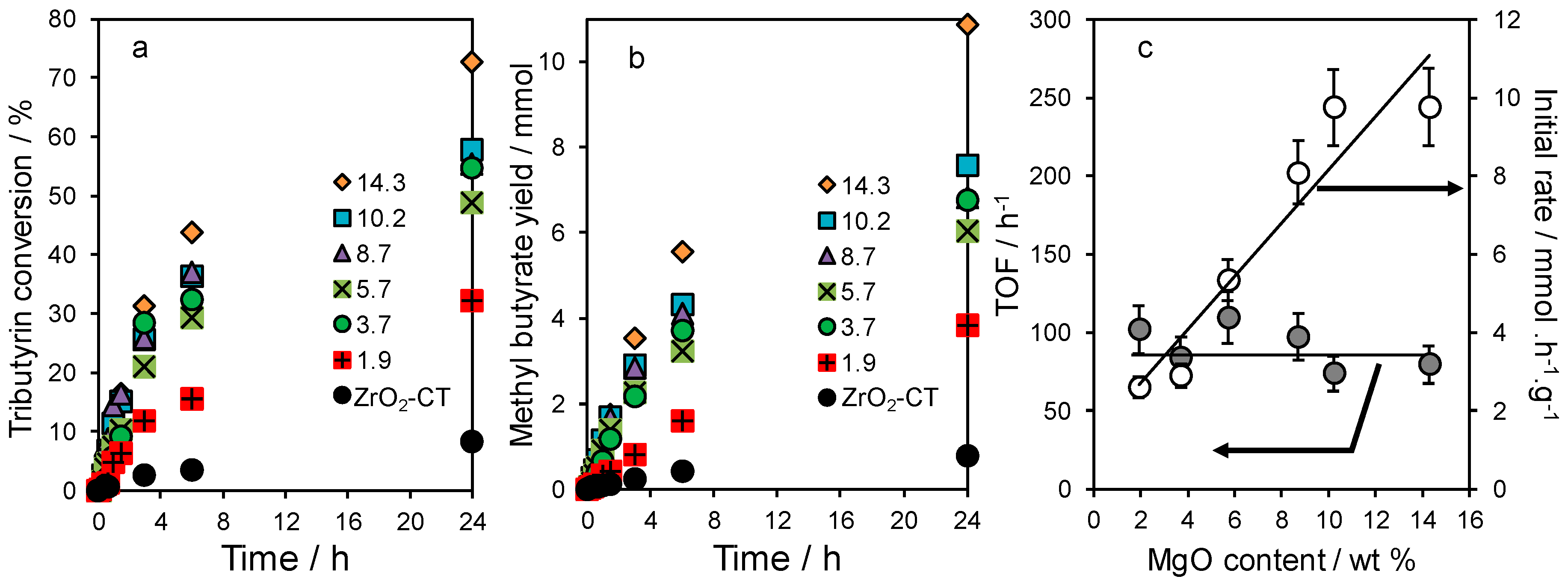
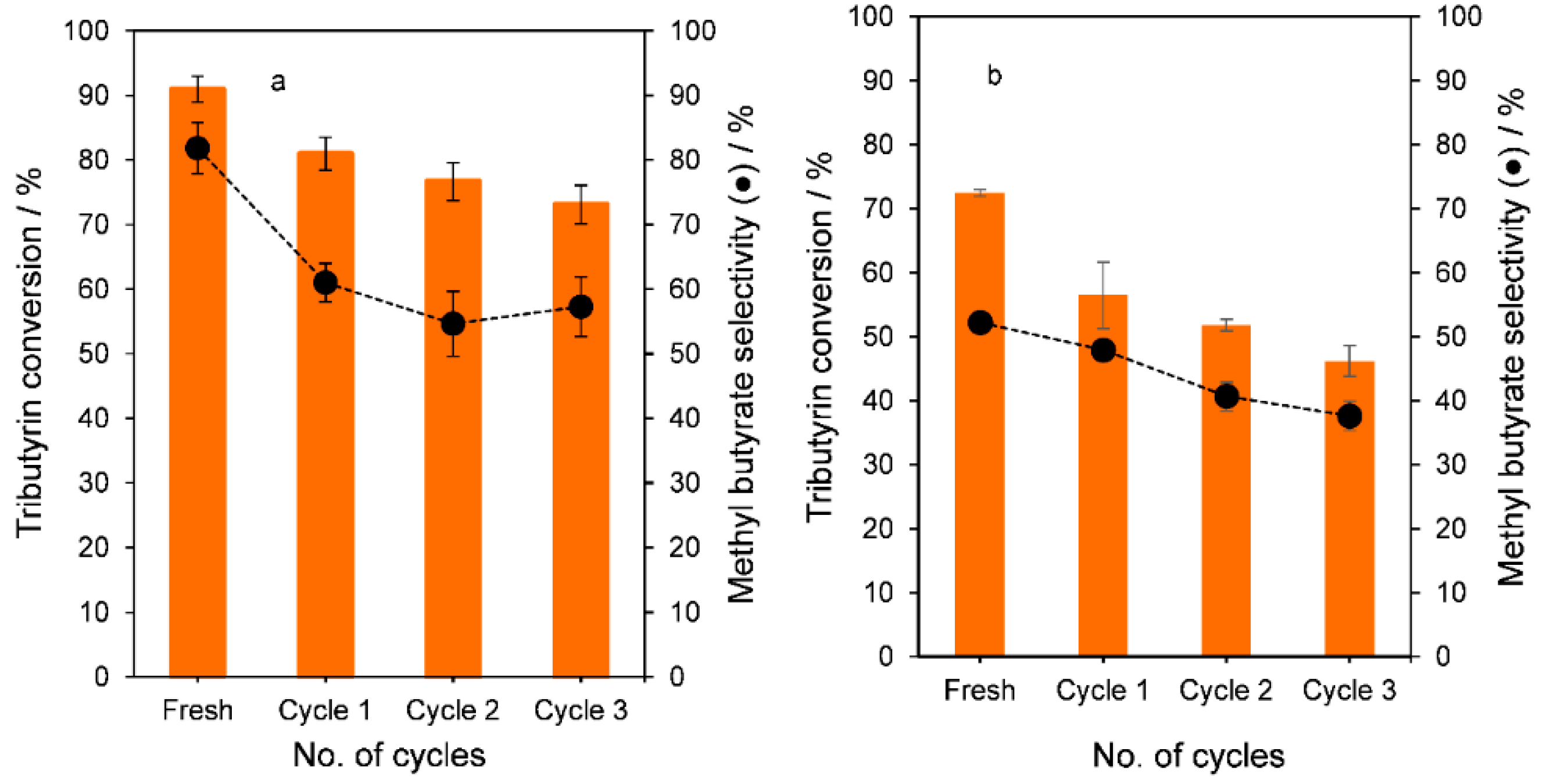
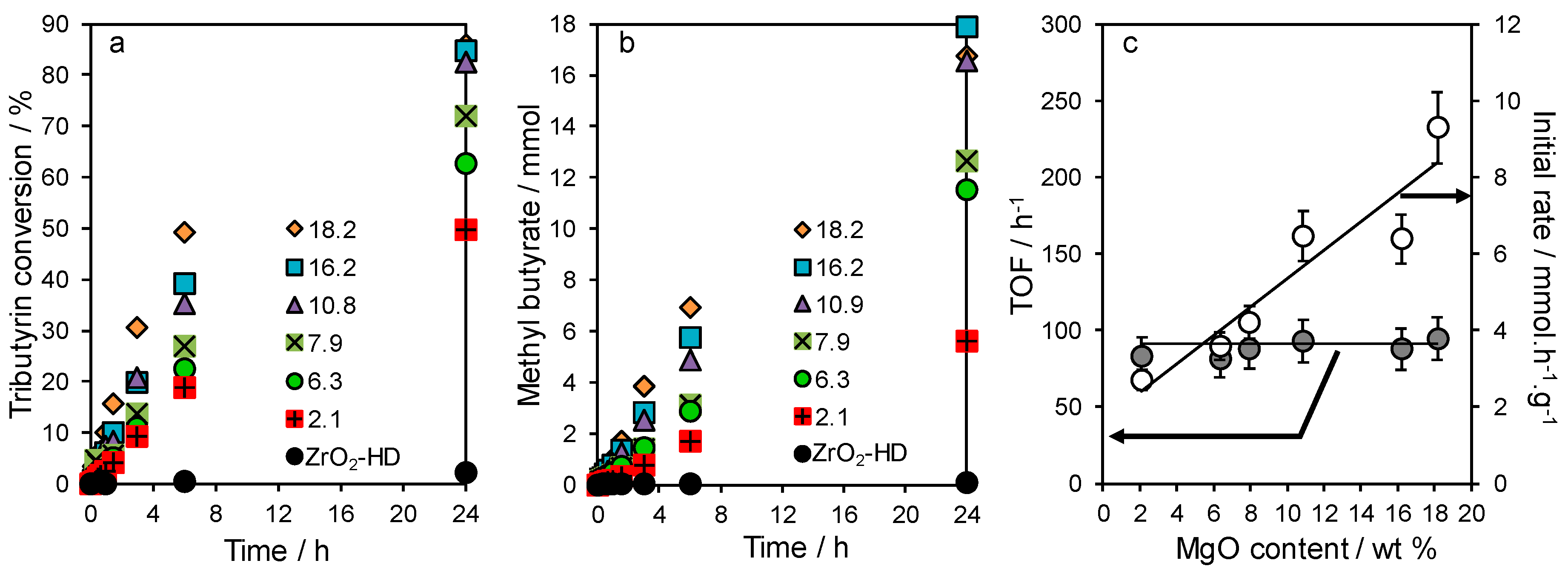
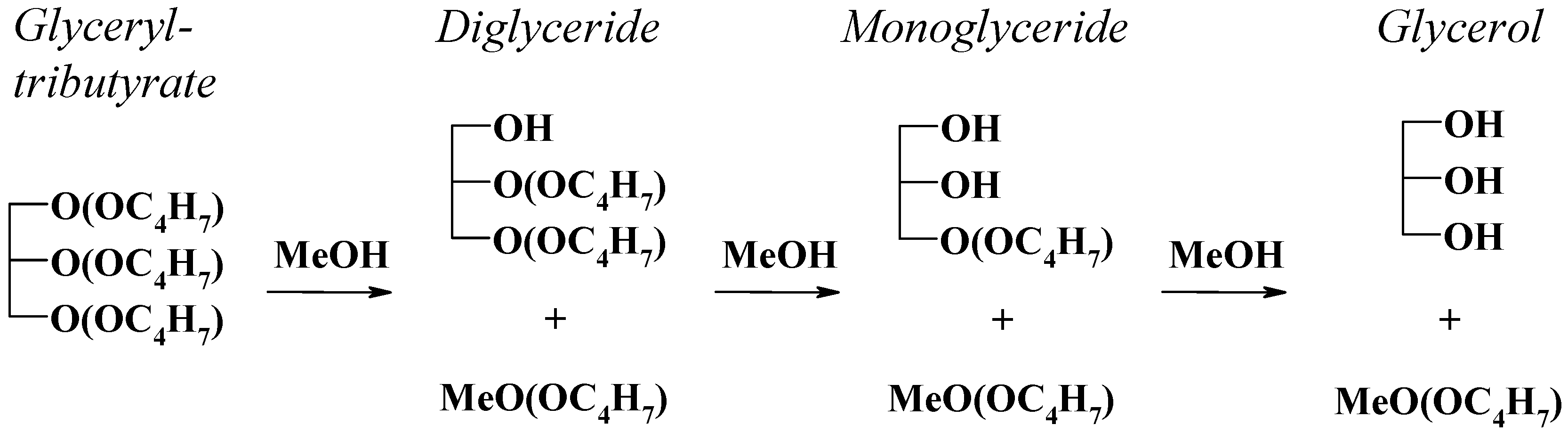
2.3. Tributyrin Transesterification
3. Experimental Details
3.1. Catalyst Synthesis
3.1.1. Non-Aqueous Impregnation
3.1.2. Citrate-Mediated Sol–Gel
3.2. Catalyst Characterization
3.3. Transesterification Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gawande, M.B.; Pandey, R.K.; Jayaram, R.V. Role of mixed metal oxides in catalysis science—Versatile applications in organic synthesis. Catal. Sci. Technol. 2012, 2, 1113–1125. [Google Scholar] [CrossRef]
- Aramendía, M.A.; Boráu, V.; Jiménez, C.; Marinas, J.M.; Marinas, A.; Porras, A.; Urbano, F.J. Synthesis and characterization of ZrO2as acid–basic catalysts: Reactivity of 2-methyl-3-butyn-2-ol. J. Catal. 1999, 183, 240–250. [Google Scholar] [CrossRef]
- Tanabe, K.; Yamaguchi, T. Acid-base bifunctional catalysis by ZrO2 and its mixed oxides. Catal. Today 1994, 20, 185–197. [Google Scholar] [CrossRef]
- Komanoya, T.; Nakajima, K.; Kitano, M.; Hara, M. Synergistic Catalysis by Lewis Acid and Base Sites on ZrO2 for Meerwein–Ponndorf–Verley Reduction. J. Phys. Chem. C 2015, 119, 26540–26546. [Google Scholar] [CrossRef]
- Kozlowski, J.T.; Aronson, M.T.; Davis, R.J. Transesterification of tributyrin with methanol over basic Mg:Zr mixed oxide catalysts. Appl. Catal. B Environ. 2010, 96, 508–515. [Google Scholar] [CrossRef]
- Liu, S.; Ma, J.; Guan, L.; Li, J.; Wei, W.; Sun, Y. Mesoporous CaO–ZrO2 nano-oxides: A novel solid base with high activity and stability. Microporous Mesoporous Mater. 2009, 117, 466–471. [Google Scholar] [CrossRef]
- Koirala, R.; Gunugunuri, K.R.; Pratsinis, S.E.; Smirniotis, P.G. Effect of zirconia doping on the structure and stability of CaO-based sorbents for CO2 capture during extended operating cycles. J. Phys. Chem. C 2011, 115, 24804–24812. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Zhao, N.; Wei, W.; Sun, Y. CaO–ZrO2 solid solution: A highly stable catalyst for the synthesis of dimethyl carbonate from propylene carbonate and methanol. Catal. Lett. 2005, 105, 253–257. [Google Scholar] [CrossRef]
- Gawande, M.B.; Branco, P.S.; Parghi, K.; Shrikhande, J.J.; Pandey, R.K.; Ghumman, C.; Bundaleski, N.; Teodoro, O.; Jayaram, R.V. Synthesis and characterization of versatile MgO–ZrO2 mixed metal oxide nanoparticles and their applications. Catal. Sci. Technol. 2011, 1, 1653–1664. [Google Scholar] [CrossRef]
- Faba, L.; Díaz, E.; Ordóñez, S. Gas phase acetone self-condensation over unsupported and supported Mg–Zr mixed-oxides catalysts. Appl. Catal. B Environ. 2013, 142, 387–395. [Google Scholar] [CrossRef]
- Kozlowski, J.T.; Behrens, M.; Schlögl, R.; Davis, R.J. Influence of the Precipitation Method on Acid–Base-Catalyzed Reactions over Mg–Zr Mixed Oxides. ChemCatChem 2013, 5, 1989–1997. [Google Scholar] [CrossRef]
- Sádaba, I.; Ojeda, M.; Mariscal, R.; Fierro, J.; Granados, M.L. Catalytic and structural properties of co-precipitated Mg–Zr mixed oxides for furfural valorization via aqueous aldol condensation with acetone. Appl. Catal. B Environ. 2011, 101, 638–648. [Google Scholar] [CrossRef]
- Gawande, M.B.; Jayaram, R.V. A novel catalyst for the Knoevenagel condensation of aldehydes with malononitrile and ethyl cyanoacetate under solvent free conditions. Catal. Commun. 2006, 7, 931–935. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Li, J.; Zhao, N.; Wei, W.; Sun, Y. Preparation and application of stabilized mesoporous MgO–ZrO2 solid base. Catal. Commun. 2008, 9, 1527–1532. [Google Scholar] [CrossRef]
- Sree, R.; Babu, N.S.; Prasad, P.S.; Lingaiah, N. Transesterification of edible and non-edible oils over basic solid Mg/Zr catalysts. Fuel Process. Technol. 2009, 90, 152–157. [Google Scholar] [CrossRef]
- Gawande, M.B.; Rathi, A.K.; Branco, P.S.; Potewar, T.M.; Velhinho, A.; Nogueira, I.D.; Tolstogouzov, A.; Ghumman, C.A.; Teodoro, O.M. Nano-MgO–ZrO2 mixed metal oxides: Characterization by SIMS and application in the reduction of carbonyl compounds and in multicomponent reactions. RSC Adv. 2013, 3, 3611–3617. [Google Scholar] [CrossRef]
- Wilson, K.; Hardacre, C.; Lee, A.F.; Montero, J.M.; Shellard, L. The application of calcined natural dolomitic rock as a solid base catalyst in triglyceride transesterification for biodiesel synthesis. Green Chem. 2008, 10, 654–659. [Google Scholar] [CrossRef]
- Ma, F.; Hanna, M.A. Biodiesel production: A review1 Journal Series #12109, Agricultural Research Division, Institute of Agriculture and Natural Resources, University of Nebraska–Lincoln. 1. Bioresour. Technol. 1999, 70, 1–15. [Google Scholar] [CrossRef]
- Gosch, B.J.; Magnusson, M.; Paul, N.A.; Nys, R. Total lipid and fatty acid composition of seaweeds for the selection of species for oil-based biofuel and bioproducts. GCB Bioenergy 2012, 4, 919–930. [Google Scholar] [CrossRef]
- Carpenter, D.L.; Lehmann, J.; Mason, B.S.; Slover, H.T. Lipid composition of selected vegetable oils. J. Am. Oil Chem. Soc. 1976, 53, 713–718. [Google Scholar] [CrossRef]
- Pinzi, S.; Garcia, I.L.; Lopez-Gimenez, F.J.; de Castro, M.D.L.; Dorado, G.; Dorado, M.P. The Ideal Vegetable Oil-based Biodiesel Composition: A Review of Social, Economical and Technical Implications. Energy Fuels 2009, 23, 2325–2341. [Google Scholar] [CrossRef]
- Demirbas, A. Biodiesel from Vegetable Oils with MgO Catalytic Transesterification in Supercritical Methanol. Energy Sources Part A Recover. Util. Environ. Eff. 2008, 30, 1645–1651. [Google Scholar] [CrossRef]
- Verziu, M.; Cojocaru, B.; Hu, J.; Richards, R.; Ciuculescu, C.; Filip, P.; Parvulescu, V.I. Sunflower and rapeseed oil transesterification to biodiesel over different nanocrystalline MgO catalysts. Green Chem. 2008, 10, 373–381. [Google Scholar] [CrossRef]
- Gai, P.L.; Montero, J.M.; Lee, A.F.; Wilson, K.; Boyes, E.D. In situ Aberration Corrected-Transmission Electron Microscopy of Magnesium Oxide Nanocatalysts for Biodiesels. Catal. Lett. 2009, 132, 182–188. [Google Scholar] [CrossRef]
- Montero, J.M.; Brown, D.R.; Gai, P.L.; Lee, A.F.; Wilson, K. In situ studies of structure–reactivity relations in biodiesel synthesis over nanocrystalline MgO. Chem. Eng. J. 2010, 161, 332–339. [Google Scholar] [CrossRef]
- Montero, J.M.; Gai, P.; Wilson, K.; Lee, A.F. Structure-sensitive biodiesel synthesis over MgO nanocrystals. Green Chem. 2009, 11, 265–268. [Google Scholar] [CrossRef]
- Man, I.-C.; Soriga, S.G.; Parvulescu, V. Theoretical aspects of methyl acetate and methanol activation on MgO (100) and (501) catalyst surfaces with application in FAME production. Appl. Surf. Sci. 2017, 392, 920–928. [Google Scholar] [CrossRef]
- Lee, A.F.; Wilson, K. Recent developments in heterogeneous catalysis for the sustainable production of biodiesel. Catal. Today 2015, 242, 3–18. [Google Scholar] [CrossRef]
- Jiang, D.-E.; Pan, G.; Zhao, B.; Ran, G.; Xie, Y.; Min, E. Preparation of ZrO2-supported MgO with high surface area and its use in mercaptan oxidation of jet fuel. Appl. Catal. A Gen. 2000, 201, 169–176. [Google Scholar] [CrossRef]
- Roehr, M.; Kubicek, C.P.; Komínek, J. Citric acid. In Biotechnology Set, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 307–345. [Google Scholar]
- Danks, A.; Hall, S.; Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef]
- Lajavardi, M.; Kenney, D.J.; Lin, S.H. Time-Resolved Phase Transitions of the Nanocrystalline Cubic to Submicron Monoclinic Phase in Zirconia. J. Chin. Chem. Soc. 2000, 47, 1043–1053. [Google Scholar] [CrossRef]
- Rabee, A.I.; Durndell, L.J.; Fouad, N.E.; Frattini, L.; Isaacs, M.A.; Lee, A.F.; Mekhemer, G.A.; dos Santos, V.C.; Wilson, K.; Zaki, M.I. Citrate-mediated sol–gel synthesis of Al-substituted sulfated zirconia catalysts for α-pinene isomerization. Mol. Catal. 2017. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared via a route of solid-state reaction. Catal. Commun. 2011, 12, 1095–1098. [Google Scholar] [CrossRef]
- Nefedov, V.; Firsov, M.; Shaplygin, I. Electronic structures of MRhO2, MRh2O4, RhMO4 and Rh2MO6 on the basis of X-ray spectroscopy and ESCA data. J. Electron Spectrosc. Relat. Phenom. 1982, 26, 65–78. [Google Scholar] [CrossRef]
- Barr, T.L. Recent advances in x-ray photoelectron spectroscopy studies of oxides. J. Vac. Sci. Technol. A Vac. Surf. Films 1991, 9, 1793–1805. [Google Scholar] [CrossRef]
- Mekhemer, G.; Halawy, S.; Mohamed, M.; Zaki, M. Qualitative and quantitative assessments of acid and base sites exposed on polycrystalline MgO surfaces: Thermogravimetric, calorimetric, and in-situ FTIR spectroscopic study combination. J. Phys. Chem. B 2004, 108, 13379–13386. [Google Scholar] [CrossRef]
- Lavalley, J. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Davydov, A. The Nature of Oxide Surface Centers. In Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003; pp. 27–179. [Google Scholar]
- Busca, G.; Lorenzelli, V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces. Mater. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Bennici, S.; Vega, A.; Ordónez, S.; Auroux, A. Adsorption of CO2 on hydrotalcite-derived mixed oxides: Sorption mechanisms and consequences for adsorption irreversibility. Ind. Eng. Chem. Res. 2010, 49, 3663–3671. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, tuning, and exploiting the basicity of hydrotalcites for applications in heterogeneous catalysis. Chem. A Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef] [PubMed]
- Prinetto, F.; Ghiotti, G.; Durand, R.; Tichit, D. Investigation of acid–base properties of catalysts obtained from layered double hydroxides. J. Phys. Chem. B 2000, 104, 11117–11126. [Google Scholar] [CrossRef]
- Martyanov, I.N.; Sayari, A. Comparative study of triglyceride transesterification in the presence of catalytic amounts of sodium, magnesium, and calcium methoxides. Appl. Catal. A Gen. 2008, 339, 45–52. [Google Scholar] [CrossRef]
- Alifanti, M.; Baps, B.; Blangenois, N.; Naud, J.; Grange, P.; Delmon, B. Characterization of CeO2−ZrO2 mixed oxides. Comparison of the citrate and sol−gel preparation methods. Chem. Mater. 2003, 15, 395–403. [Google Scholar] [CrossRef]
- Che, M.; Védrine, J.C. Characterization of Solid Materials and Heterogeneous Catalysts: From Structure to Surface Reactivity; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET a/m2 g−1 | Crystallite Size b/nm | Basicity c/μmol·g−1 |
|---|---|---|---|
| ZrO2-HD | 59 | 15.4 | 11.7 |
| 2.1MZ–HD | 152 | 7.2 | 32.7 |
| 6.3MZ–HD | 190 | 7.3 | 44.0 |
| 7.9MZ–HD | 129 | 7.6 | 47.8 |
| 10.9MZ–HD | 137 | 7.5 | 69.8 |
| 16.2MZ–HD | 149 | 7.4 | 72.9 |
| 18.2MZ–HD | 140 | 7.1 | 94.8 |
| ZrO2-CT | 50 | 8.8 | 4.9 |
| 1.9MZ–CT | 87 | 6.5 | 7.3 |
| 3.7MZ–CT | 114 | 4.8 | 9.8 |
| 5.7MZ–CT | 125 | 4.4 | 13.9 |
| 8.7MZ–CT | 107 | 4.4 | 23.8 |
| 10.2MZ–CT | 105 | 4.4 | 37.6 |
| 14.3MZ–CT | 117 | 4.6 | 35.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabee, A.I.M.; Manayil, J.C.; Isaacs, M.A.; Parlett, C.M.A.; Durndell, L.J.; Zaki, M.I.; Lee, A.F.; Wilson, K. On the Impact of the Preparation Method on the Surface Basicity of Mg–Zr Mixed Oxide Catalysts for Tributyrin Transesterification. Catalysts 2018, 8, 228. https://doi.org/10.3390/catal8060228
Rabee AIM, Manayil JC, Isaacs MA, Parlett CMA, Durndell LJ, Zaki MI, Lee AF, Wilson K. On the Impact of the Preparation Method on the Surface Basicity of Mg–Zr Mixed Oxide Catalysts for Tributyrin Transesterification. Catalysts. 2018; 8(6):228. https://doi.org/10.3390/catal8060228
Chicago/Turabian StyleRabee, Abdallah I. M., Jinesh C. Manayil, Mark A. Isaacs, Christopher M. A. Parlett, Lee J. Durndell, Mohamed I. Zaki, Adam F. Lee, and Karen Wilson. 2018. "On the Impact of the Preparation Method on the Surface Basicity of Mg–Zr Mixed Oxide Catalysts for Tributyrin Transesterification" Catalysts 8, no. 6: 228. https://doi.org/10.3390/catal8060228
APA StyleRabee, A. I. M., Manayil, J. C., Isaacs, M. A., Parlett, C. M. A., Durndell, L. J., Zaki, M. I., Lee, A. F., & Wilson, K. (2018). On the Impact of the Preparation Method on the Surface Basicity of Mg–Zr Mixed Oxide Catalysts for Tributyrin Transesterification. Catalysts, 8(6), 228. https://doi.org/10.3390/catal8060228