Exploring Basic Components Effect on the Catalytic Efficiency of Chevron-Phillips Catalyst in Ethylene Trimerization

 , ,
, , 
Abstract
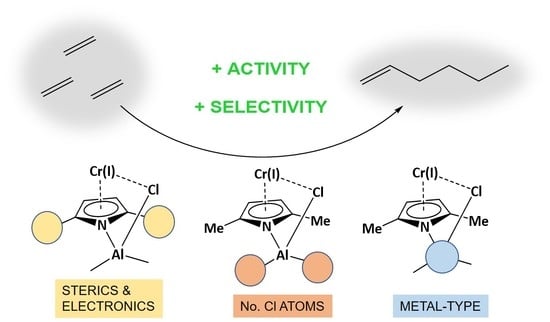
1. Introduction
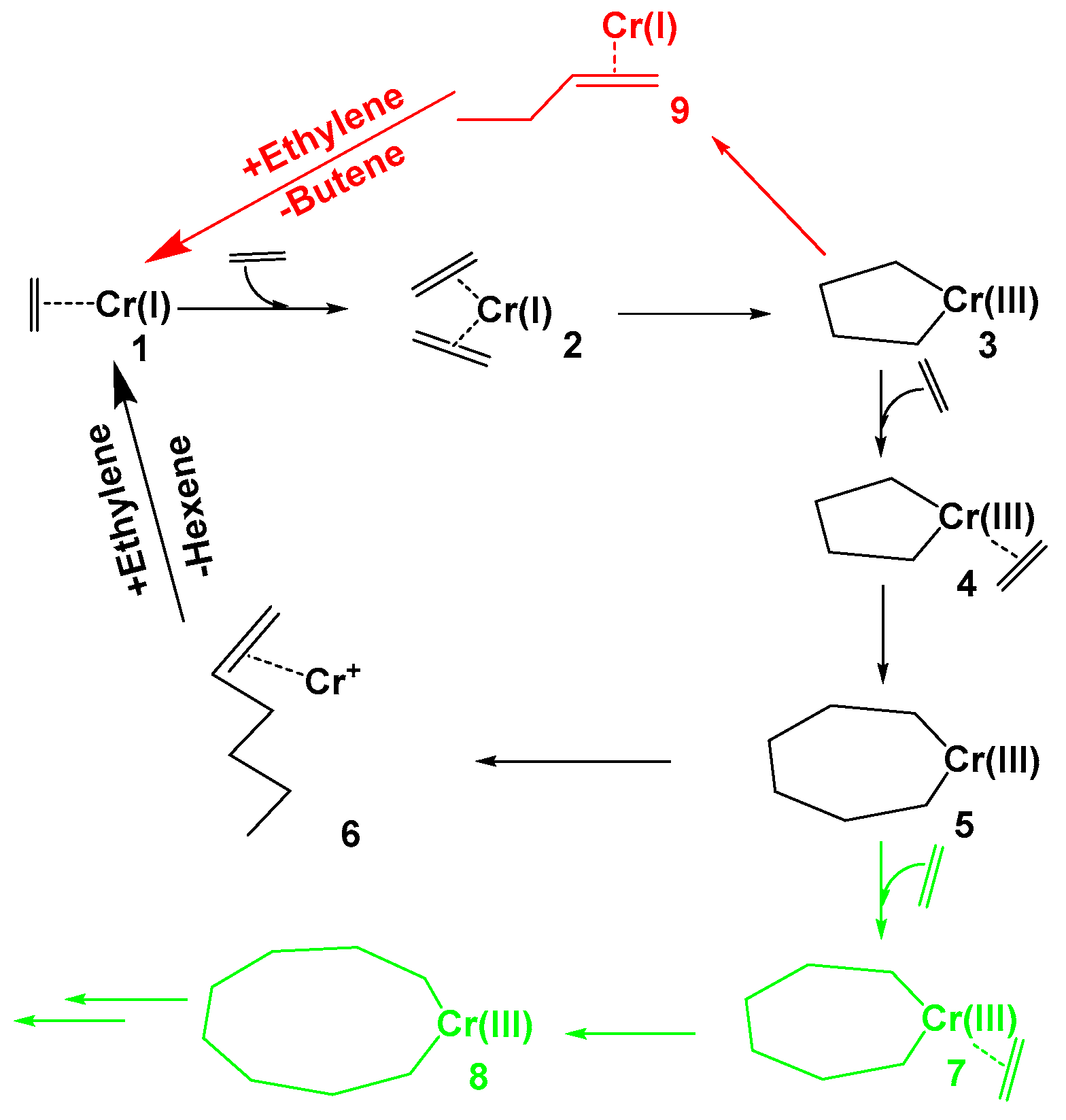
2. Results and Discussion
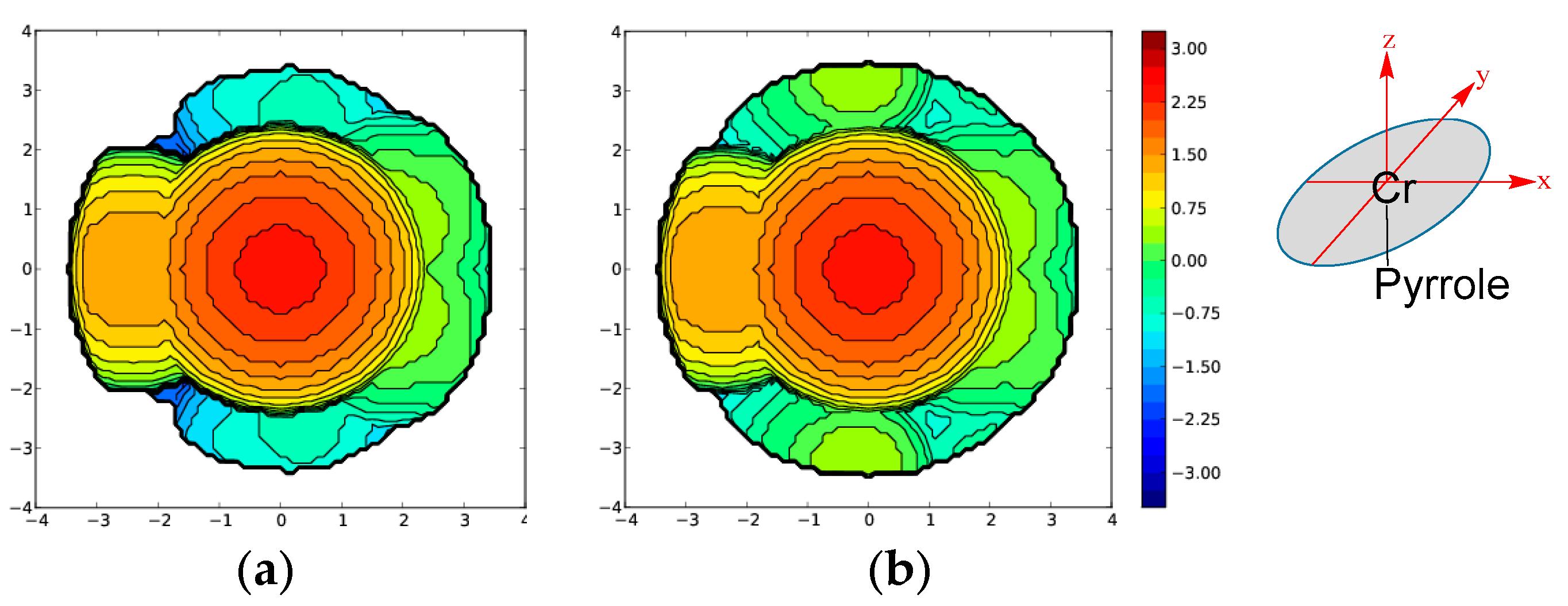
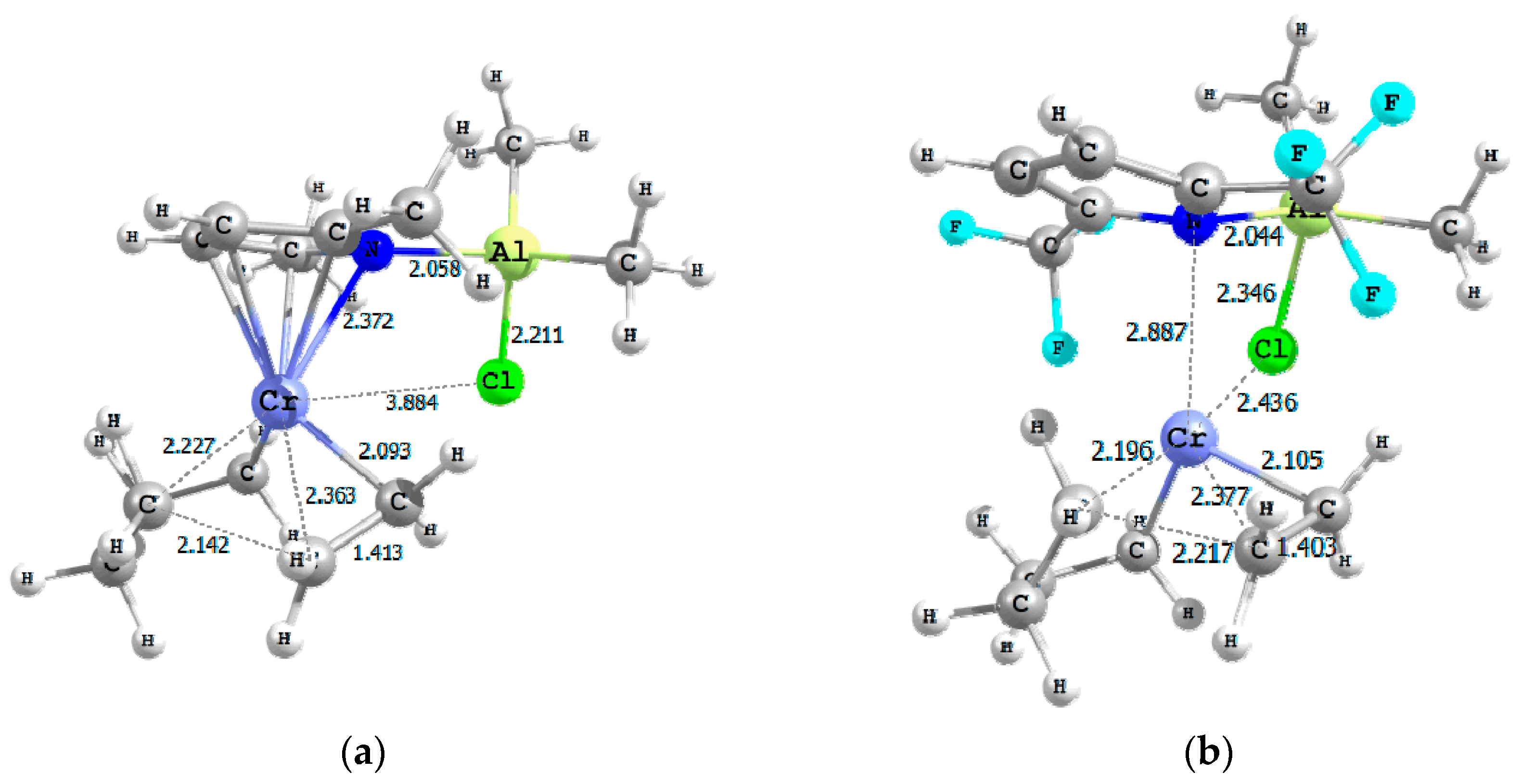
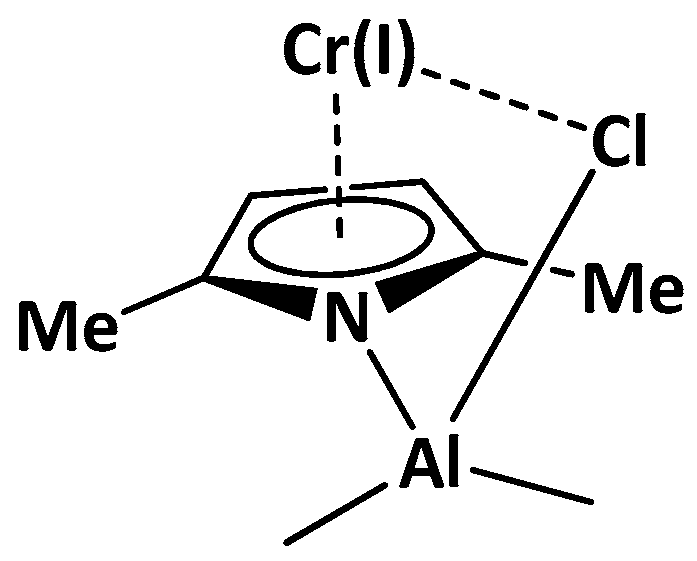
2.1. Effect of Alkyl Substituents on the Pyrrole Ligand
2.2. Effect of the Number of Cl Atoms in Al Compound
2.3. Effect of Metal Type in the Cocatalyst Structure
2.4. Effect of Solvent
3. Conclusions
4. Computational Details
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bazvand, R.; Bahri-Laleh, N.; Nekoomanesh-Haghighi, M.; Abedini, H. Highly efficient FeCl3 doped Mg(OEt)2/TiCl4-based Ziegler–Natta catalysts for ethylene polymerization. Des. Monomers Polym. 2015, 18, 599–610. [Google Scholar] [CrossRef]
- Alferov, K.A.; Babenko, I.A.; Belov, G.P. New catalytic systems on the basis of chromium compounds for selective synthesis of 1-hexene and 1-octene. Petrol. Chem. 2017, 57, 1–30. [Google Scholar] [CrossRef]
- Haghverdi, M.; Tadjarodi, A.; Bahri-laleh, N.; Nekoomanesh Haghighi, M. Synthesis and characterization of Ni(II) complexes bearing of 2-(1H-benzimidazol-2-yl)-phenol derivatives as highly active catalysts for ethylene oligomerization. Appl. Organomet. Chem. 2018, 32, e4015. [Google Scholar] [CrossRef]
- Haghverdi, M.; Tadjarodi, A.; Bahri-laleh, N.; Nekoomanesh Haghighi, M. Cobalt complexes based on 2-(1H-benzimidazol-2-yl)-phenol derivatives: Preparation, spectral studies, DFT calculations and catalytic behavior toward ethylene oligomerization. J. Coord. Chem. 2017, 70, 1800–1814. [Google Scholar] [CrossRef]
- Agapie, T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- Alferov, K.A.; Belov, G.P.; Meng, Y. Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: Recent results. Appl. Catal. A Gen. 2017, 542, 71–124. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in selective ethylene trimerisation—A critical overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- Bahri-Laleh, N.; Karimi, M.; Kalantari, Z.; Fallah, M.; Hanifpour, A.; Nekoomanesh-Haghighi, M. H2 effect in Chevron–Phillips ethylene trimerization catalytic system: An experimental and theoretical investigation. Polym. Bull. 2017. [Google Scholar] [CrossRef]
- Makhaev, V.D.; Petrova, L.A.; Alferov, K.A.; Belov, G.P. A Failed Late-Stage Epimerization Thwarts an Approach to Ineleganolide. Russ. J. Org. Chem. 2013, 86, 1819–1824. [Google Scholar]
- Jiang, T.; Ji, R.; Chen, H.; Cao, C.; Mao, G.; Ning, Y. Effect of Alkylaluminum Activators on Ethylene Trimerization Based on 2,5-DMP/Cr(III)/TCE Catalyst System. Chin. J. Chem. 2011, 29, 1149–1153. [Google Scholar] [CrossRef]
- Gong, M.; Liu, Z.; Li, Y.; Ma, Y.; Sun, Q.; Zhang, J.; Liu, B. Selective Co-Oligomerization of Ethylene and 1-Hexene by Chromium-PNP Catalysts: A DFT Study. Organometallics 2016, 35, 972–981. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Mechanistic DFT Study on Ethylene Trimerization of Chromium Catalysts Supported by a Versatile Pyrrole Ligand System. Organometallics 2014, 33, 2599–2607. [Google Scholar] [CrossRef]
- Skobelev, I.Y.; Panchenko, V.N.; Lyakin, O.Y.; Bryliakov, K.P.; Zakharov, V.A.; Talsi, E.P. In Situ EPR Monitoring of Chromium Species Formed during Cr−Pyrrolyl Ethylene Trimerization Catalyst Formation. Organometallics 2010, 29, 2943–2950. [Google Scholar] [CrossRef]
- Qi, Y.; Dong, Q.; Zhong, L.; Liu, Z.; Qiu, P.; Cheng, R.; He, X.; Vanderbilt, J.; Liu, B. Role of 1, 2-dimethoxyethane in the transformation from ethylene polymerization to trimerization using chromium tris (2-ethylhexanoate)-based catalyst system: A DFT study. Organometallics 2010, 29, 1588–1602. [Google Scholar] [CrossRef]
- Riache, N.; Dery, A.; Callens, E.; Poater, A.; Samantaray, M.; Dey, R.; Hong, J.H.; Lo, K.; Cavallo, L.; Basset, J.M. Silica-supported tungsten carbynes (=SiO)(x)W(CH)(Me)(y) (x = 1, y = 2; x = 2, y = 1): New efficient catalysts for alkyne cyclotrimerization. Organometallics 2015, 34, 690–695. [Google Scholar] [CrossRef]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of Buried Volumes of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Tobisch, S.; Ziegler, T. Catalytic Oligomerization of Ethylene to Higher Linear α-Olefins Promoted by the Cationic Group 4 [(η5-Cp-(CMe2-bridge)-Ph)MII(ethylene)2]+ (M = Ti, Zr, Hf) Active Catalysts: A Density Functional Investigation of the Influence of the Metal on the Catalytic Activity and Selectivity. J. Am. Chem. Soc. 2004, 126, 9059–9071. [Google Scholar] [PubMed]
- Briggs, J.R. The selective trimerization of ethylene to hex-1-ene. J. Chem. Soc. Chem. Commun. 1989, 674–675. [Google Scholar] [CrossRef]
- Manzini, S.; Urbina-Blanco, C.A.; Nelson, D.J.; Poater, A.; Lebl, T.; Meiries, S.; Slawin, A.M.Z.; Falivene, L.; Cavallo, L.; Nolan, S.P. Evaluation of an olefin metathesis pre-catalyst with a bulky and electron-rich N-heterocyclic carbene. J. Organomet. Chem. 2015, 780, 43–48. [Google Scholar] [CrossRef]
- Licciulli, S.; Albahily, K.; Fomitcheva, V.; Korobkov, I.; Gambarotta, S.; Duchateau, R. A Chromium Ethylidene Complex as a Potent Catalyst for Selective Ethylene Trimerization. Angew. Chem. Int. Ed. 2011, 50, 2346–2349. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Nakamura, H.; Nanba, Y.; Okano, T. Process for Producing α-Olefin Oligomer. U.S. Patent 5856612A, 5 January 1999. [Google Scholar]
- Khasbiullin, I.I.; Belov, G.P.; Kharlampidi, K.E.; Vil’ms, A.I. Ethylene oligomerization on the chromium ethylhexanoate-triethylaluminum-2,5-dimethylpyrrol catalytic system in the presence of carbon tetrachloride. Petrol. Chem. 2011, 51, 450–455. [Google Scholar] [CrossRef]
- Tang, S.; Liu, Z.; Yan, X.; Li, N.; Cheng, R.; He, X.; Liu, B. Kinetic studies on the pyrrole–Cr-based Chevron-Phillips ethylene trimerization catalyst system. Appl. Catal. A Gen. 2014, 481, 39–48. [Google Scholar] [CrossRef]
- Jabri, A.; Mason, C.B.; Sim, Y.; Gambarotta, S.; Burchell, T.J.; Duchateau, R. Isolation of Single-Component Trimerization and Polymerization Chromium Catalysts: The Role of the Metal Oxidation. State. Angew. Chem. Int. Ed. 2008, 47, 9717–9721. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.Y.; Park, D.S.; Lee, D.H.; Eo, S.C.; Park, S.Y.; Jeong, M.S.; Kang, Y.Y.; Lee, J.; Lee, B.Y. A chromium precursor for the Phillips ethylene trimerization catalyst: (2-ethylhexanoate)2CrOH. Dalton Trans. 2015, 44, 11004–11012. [Google Scholar] [CrossRef] [PubMed]
- Zilbershtein, T.M.; Kardash, V.A.; Suvorova, V.V.; Golovko, A.K. Decene formation in ethylene trimerization reaction catalyzed by Cr–pyrrole system. Appl. Catal. A Gen. 2014, 475, 371–378. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford CT, USA, 2009. [Google Scholar]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Bahri-Laleh, N.; Nekoomanesh-Haghighi, M.; Mirmohammadi, S.A. A DFT study on the effect of hydrogen in ethylene and propylene polymerization using a Ti-based heterogeneous Ziegler–Natta catalyst. J. Organomet. Chem. 2012, 719, 74–79. [Google Scholar] [CrossRef]
- Bahri-Laleh, N.; Poater, A.; Cavallo, L.; Mirmohammadi, S.A. Exploring the Mechanism of Grignard Methathesis Polymerization of 3-alkylthiophenes. Dalton Trans. 2014, 43, 15143–15150. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Bahri-Laleh, N.; Cavallo, L. How well can DFT reproduce key interactions in Ziegler–Natta systems? Macromol. Chem. Phys. 2013, 214, 1980–1989. [Google Scholar] [CrossRef]
- Schäfer, S.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Costas, M.; Ribas, X.; Poater, A.; López Balvuena, J.M.; Xifra, R.; Company, A.; Duran, M.; Solà, M.; Llobet, A.; Corbella, M.; et al. Copper(II) Hexaaza Macrocyclic Binuclear Complexes Obtained from the Reaction of Their Copper(I) Derivates and Molecular Dioxygen. Inorg. Chem. 2006, 45, 3569–3581. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Ribas, X.; Llobet, A.; Cavallo, L.; Solà, M. Complete Mechanism of σ* Intramolecular Aromatic Hydroxylation through O2 Activation by a Macrocyclic Dicopper(I) Complex. J. Am. Chem. Soc. 2008, 130, 17710–17717. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Chem. Phys. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, H.; Correa, C.; Poater, A.; Costabile, C.; Cavallo, L. Understanding the M-(NHC) (NHC = N-Heterocyclic Carbene) Bond. Coord. Chem. Rev. 2009, 253, 687–703. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Poater, A.; Cavallo, L. Comparing families of olefin polymerization precatalysts using the percentage of buried volume. Dalton Trans. 2009, 2009, 8878–8883. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Falivene, L.; Urbina-Blanco, C.A.; Manzini, S.; Nolan, S.P.; Cavallo, L. How does the Addition of Steric Hindrance to a Typical N-heterocyclic Carbene Ligand Affect Catalytic Activity in Olefin Metathesis? Dalton Trans. 2013, 42, 7433–7439. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-heterocyclic carbene dimerization: The balance of sterics and electronics. Organometallics 2008, 27, 2679–2681. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
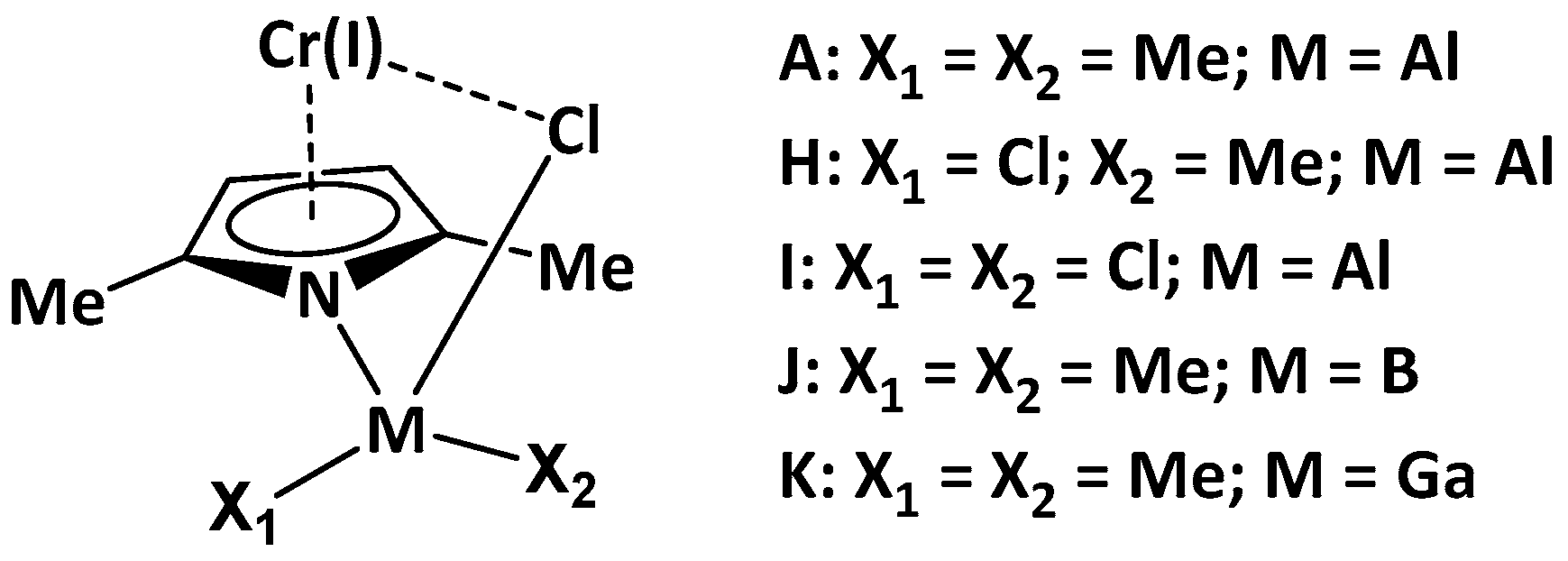{kind=link}
| Model Catalyst | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|
| Active species | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 1 | −3.0 | −2.0 | −3.2 | −3.6 | −0.6 | 2.1 | 2.1 |
| 2 | −1.6 | 0.0 | 2.2 | 4.6 | 7.3 | 0.4 | 5.3 |
| TS-2-3 | 8.4 | 10.4 | 9.6 | 12.1 | 18.3 | 10.4 | 13.9 |
| 3 | −9.4 | −7.7 | −9.4 | −7.8 | −6.9 | −3.8 | −3.1 |
| 4 | −1.6 | 0.2 | 3.6 | 1.2 | 2.8 | 0.8 | 8.3 |
| TS-4-5 | 15.5 | 17.3 | 17.4 | 14.1 | 20.0 | 16.9 | 9.2 |
| 5 | −16.3 | −13.9 | −15.5 | −14.2 | −13.0 | −12.7 | −11.8 |
| TS-5-6 | 4.3 | 6.7 | 5.5 | 4.6 | 10.2 | 3.6 | 6.9 |
| 6 | −20.3 | −18.8 | −19.8 | −20.2 | −17.9 | −14.6 | −14.8 |
| 7 | −7.2 | −3.2 | −5.0 | −0.2 | −2.7 | −5.9 | −1.6 |
| TS-7-8 | 13.1 | 15.0 | 13.4 | 16.8 | 15.3 | 14.0 | 18.0 |
| 8 | −21.0 | −18.9 | −19.5 | −18.3 | −17.9 | −18.2 | −16.1 |
| TS-3-9 | 17.1 | 20.8 | 18.4 | 16.1 | 26.0 | 15.7 | 16.7 |
| 9 | −13.3 | −12.5 | −13.8 | −13.4 | −11.5 | −8.1 | −7.5 |
| Model Catalyst | ETS-4-5-E3 a | ETS-5-6-E5 | ETS-7-8-E5 b | ETS-3-9-E3 |
|---|---|---|---|---|
| A | 24.9 | 20.6 | 29.4 | 26.5 |
| B | 25.0 | 20.6 | 28.9 | 28.5 |
| C | 26.8 | 21.0 | 28.9 | 27.8 |
| D | 21.9 | 18.8 | 31.0 | 23.9 |
| E | 26.9 | 23.2 | 28.3 | 32.9 |
| F | 20.7 | 16.3 | 26.7 | 19.5 |
| G | 12.3 | 18.7 | 29.8 | 19.8 |
| H | 21.6 | 15.8 | 25.3 | 24.7 |
| I | 18.8 | 16.0 | 24.1 | 22.1 |
| J | 28.7 | 22.4 | 38.7 | 25.6 |
| K | 15.1 | 22.5 | 31.2 | 25.8 |
| Model Catalyst | A | A * | H | I | J | K |
|---|---|---|---|---|---|---|
| Active species | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 1 | −3.0 | −3.4 | −1.9 | −0.6 | 5.2 | −3.1 |
| 2 | −1.6 | −2.0 | −2.8 | −3.2 | 16.6 | −0.1 |
| TS-2-3 | 8.4 | 8.0 | 8.2 | 7.2 | 29.1 | 9.9 |
| 3 | −9.4 | −10.0 | −7.7 | −6.4 | −1.9 | −9.4 |
| 4 | −1.6 | −1.9 | −2.8 | −4.2 | 11.1 | 0.8 |
| TS-4-5 | 15.5 | 15.2 | 13.9 | 12.4 | 26.8 | 14.3 |
| 5 | −16.3 | −16.9 | −13.8 | −14.1 | −9.4 | −16.3 |
| TS-5-6 | 4.3 | 3.9 | 2.0 | 1.9 | 13.0 | 6.2 |
| 6 | −20.3 | −20.9 | −18.9 | −18.2 | −11.8 | −20.4 |
| 7 | −7.2 | −7.6 | −9.6 | −10.2 | 11.3 | −5.7 |
| TS-7-8 | 13.1 | 12.7 | 11.5 | 10.0 | 29.3 | 14.9 |
| 8 | −21.0 | −21.6 | −19.4 | −18.8 | −13.7 | −21.2 |
| TS-3-9 | 17.1 | 16.8 | 17.0 | 15.7 | 23.7 | 16.4 |
| 9 | −13.3 | −13.8 | −12.2 | −11.1 | −5.8 | −13.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naji-Rad, E.; Gimferrer, M.; Bahri-Laleh, N.; Nekoomanesh-Haghighi, M.; Jamjah, R.; Poater, A. Exploring Basic Components Effect on the Catalytic Efficiency of Chevron-Phillips Catalyst in Ethylene Trimerization. Catalysts 2018, 8, 224. https://doi.org/10.3390/catal8060224
Naji-Rad E, Gimferrer M, Bahri-Laleh N, Nekoomanesh-Haghighi M, Jamjah R, Poater A. Exploring Basic Components Effect on the Catalytic Efficiency of Chevron-Phillips Catalyst in Ethylene Trimerization. Catalysts. 2018; 8(6):224. https://doi.org/10.3390/catal8060224
Chicago/Turabian StyleNaji-Rad, Ebtehal, Martí Gimferrer, Naeimeh Bahri-Laleh, Mehdi Nekoomanesh-Haghighi, Roghieh Jamjah, and Albert Poater. 2018. "Exploring Basic Components Effect on the Catalytic Efficiency of Chevron-Phillips Catalyst in Ethylene Trimerization" Catalysts 8, no. 6: 224. https://doi.org/10.3390/catal8060224
APA StyleNaji-Rad, E., Gimferrer, M., Bahri-Laleh, N., Nekoomanesh-Haghighi, M., Jamjah, R., & Poater, A. (2018). Exploring Basic Components Effect on the Catalytic Efficiency of Chevron-Phillips Catalyst in Ethylene Trimerization. Catalysts, 8(6), 224. https://doi.org/10.3390/catal8060224