In most of these multi-enzyme sequences, the lactone is formed in the final step through oxidation of a cyclic ketone by a Baeyer–Villiger monooxygenase (BVMO). However, systems that derive the lactone from an acyclic precursor have also been reported.
4.1. Formation of Lactones by Baeyer–Villiger Monooxygenases in Cascade Systems
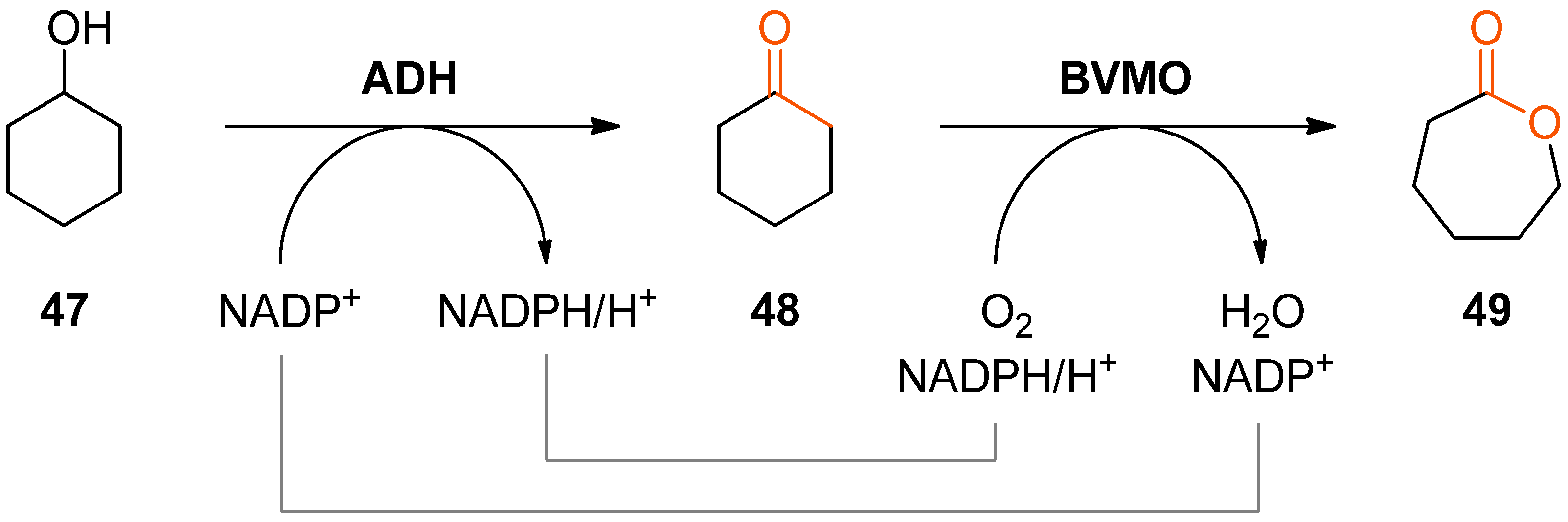
The combination of alcohol oxidation by an alcohol dehydrogenase (ADH) and Baeyer–Villiger oxidation of the resulting ketone by a BVMO represents perhaps the simplest biocatalytic cascade system for lactone production. Although this bi-enzymatic reaction represents a double oxidation, it is redox-neutral with respect to the required nicotinamide cofactor, since the Baeyer–Villiger monooxygenase requires NAD(P)H for reducing one oxygen atom of O2 to water while the other one is incorporated into the substrate. Hence, molecular oxygen is the only stoichiometric co-substrate in the overall process.
The conversion of cyclohexanol (
47) into ɛ-caprolactone (
49) using this cascade concept has been independently reported by two research groups in 2013 (
Scheme 14) [
74,
75].
Both studies show that an efficient transformation of cyclohexanol into ɛ-caprolactone is feasible (94% conversion of 60 mM
47, 80% conversion of 10 mM
47); however, both also observed reduced conversions at elevated substrate concentrations which was attributed to inhibition and deactivation of the Baeyer–Villiger monooxygenase. This issue was addressed in subsequent studies carried out as a joint effort of the two involved research groups: [
76,
77] The ɛ-caprolactone formed underwent in situ ring-opening oligomerisation enabled by lipase A from
Candida antarctica (CAL-A), which alleviated product inhibition and produced oligo-ϵ-caprolactone displaying an average molecular weight of 375 g/mol. It is worth noting that no hydrolysis of
49 to the hydroxycarboxylic acid was observed. In an additional optimisation approach, the authors chose to use a stabilised variant of cyclohexanone monooxygenase (CHMO variant C376L/M400I) [
78] and to employ the ADH and the monooxygenase as separate
E. coli whole-cell preparations. Moreover, despite the theoretical redox-neutral nature of the sequence, acetone and
d-glucose (1 equivalent each) were added to improve cofactor regeneration. These reaction conditions enabled the conversion of 200 mM cyclohexanol (
47) within 48 h going to completion thereby affording a mixture of 75% oligo-ɛ-caprolactone and 25% of monomeric
49. Recently, the co-expression of CHMO and ADH in
E. coli using a Duet™ vector allowed higher conversion values of the substrate
47 in whole-cell biocatalysis compared to an expression of both enzymes from two separate plasmids [
79]. Alternatively, fusion of the alcohol dehydrogenase and the BMVO also allowed to engineer the reaction [
80]. The same cascade could also be achieved using whole cells of
Geotrichum candidum CCT 1205 [
81].
The cascade mentioned above has also been extended by a rhodium-catalyzed hydrogenation step that converts phenol into cyclohexanol (
47), which is then employed in the biocatalytic cascade without purification [
82].
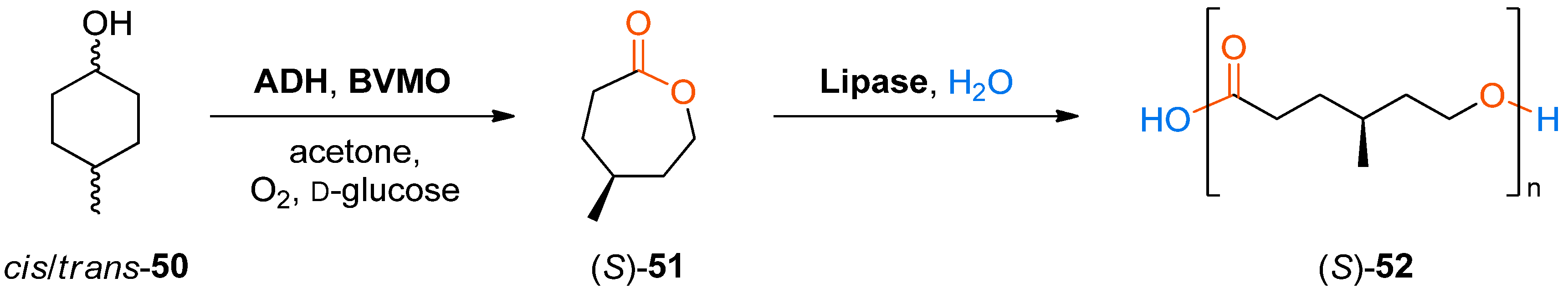
The formation and in situ oligomerisation of a chiral lactone, (
S)-4-methyl-ɛ-caprolactone (
51), from 4-methylcyclohexanol (used as a mixture of
cis and
trans isomers) via the same biocatalytic cascade was also demonstrated (
Scheme 15) [
83].
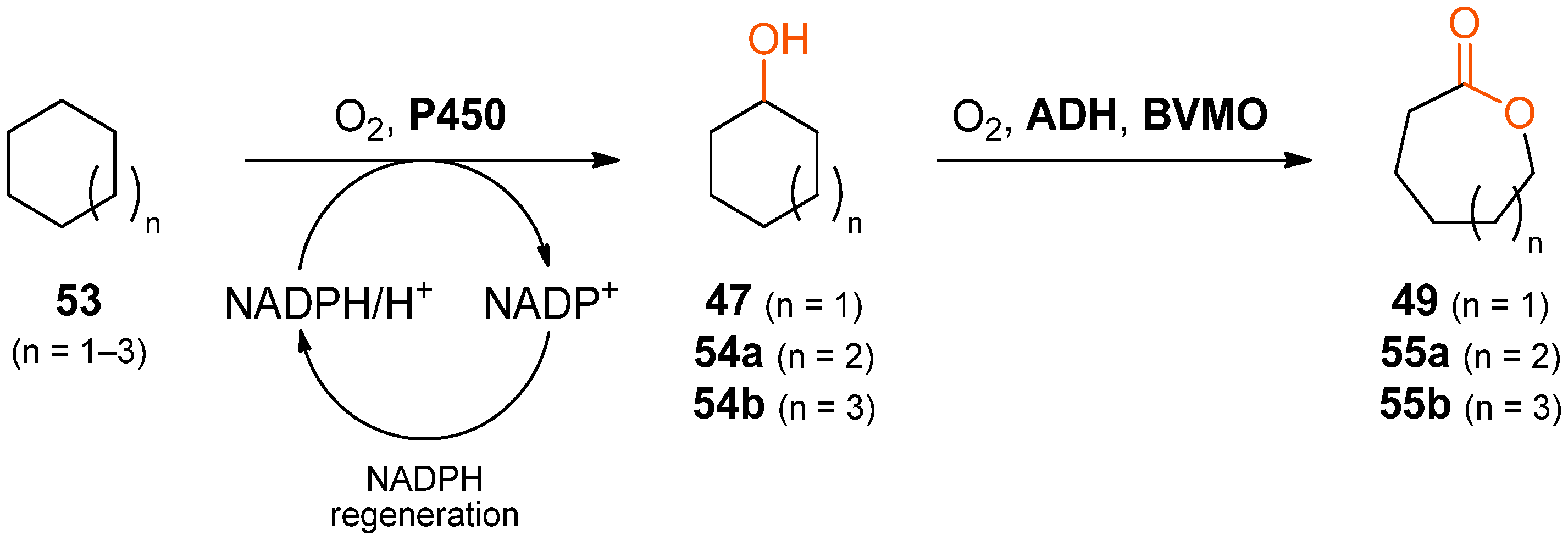
When the alcohol-to-lactone cascade system is extended upstream by a hydroxylation step, lactones can be obtained directly from cycloalkanes. The formation of ɛ-caprolactone (
49), ζ-enantholactone (
55a, n = 2) and η-caprylolactone (
55b, n = 3) from cyclohexane, cycloheptane, and cyclooctane, respectively, according to this concept has been demonstrated using a variant of the P450 monooxygenase BM3 from
Bacillus megaterium for introducing the alcohol functionality in the first step (
Scheme 16) [
84]. The subsequent oxidation to the ketone was performed by an ADH from either a
Thermus sp. or from
Thermoanaerobacter ethanolicus, whereby the latter gave the better results in terms of conversion. The final oxidation to the desired lactone was achieved using a stabilised variant of cyclohexanone monooxygenase from
Acinetobacter sp. NCIMB 9871.
Although the use of a “designer cell” co-expressing all three enzymes did allow production of the desired lactones, yields were generally low (less than 1 mM from 149–185 mM cycloalkane). Therefore, a cascade was tested instead in which the biocatalysts were added as separate cell-free extracts and in which cofactor regeneration was realised by endogenous
E. coli enzymes and the simple addition of
d-glucose and glycerol. Using this reaction setup, 3.2 mM of ζ-enantholactone (
55b) were formed from cycloheptane within 20 h. The addition of an external cofactor recycling system, consisting of sodium formate and a formate dehydrogenase (FDH), increased the final concentration of
55b to 10.6 mM, and raising the biocatalyst amounts by a factor of 3.75 resulted in the formation of 23.3 mM
55b.
Table 7 summarizes the results obtained with all three investigated substrates.
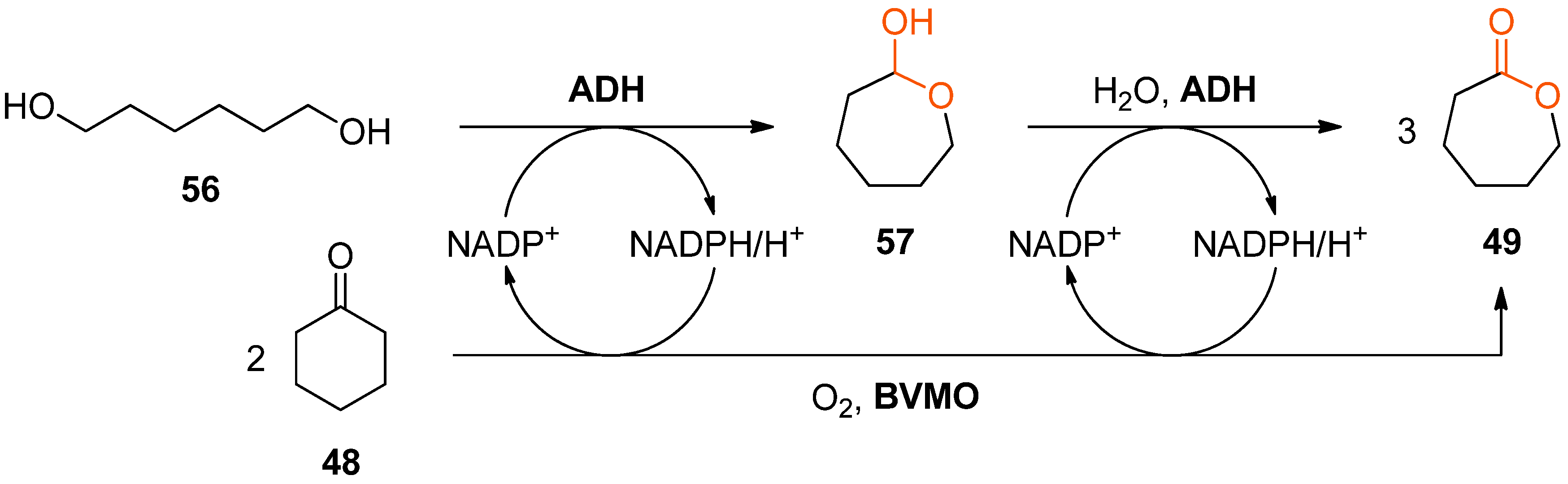
Interestingly, alcohol dehydrogenases and BVMOs have been combined for lactone synthesis also in a second, entirely different way: The Baeyer–Villiger oxidation of cyclohexanone (
48) to ɛ-caprolactone (
49) catalysed by cyclohexanone monooxygenase has been coupled with the double oxidation of hexane-1,6-diol (
56) to
49 (via the intermediate lactol
57) catalysed by the ADH from
Thermoanaerobacter ethanolicus [
85]. The resulting convergent cascade system is redox-neutral with respect to the nicotinamide cofactor and theoretically produces three molecules of ɛ-caprolactone (
49) from two molecules of
48 and one molecule of
56 (
Scheme 17).
However, in practice the formation of three equivalents of ɛ-caprolactone (49) was never observed despite complete depletion of the starting materials due to undesired hydrolysis and oligomerisation of 49 under the reaction conditions. Nevertheless, up to 18.3 mM of 49 were formed from 20 mM of 48 and 10 mM of 56 within a reaction time of 72 h, corresponding to an analytical yield of 61%.
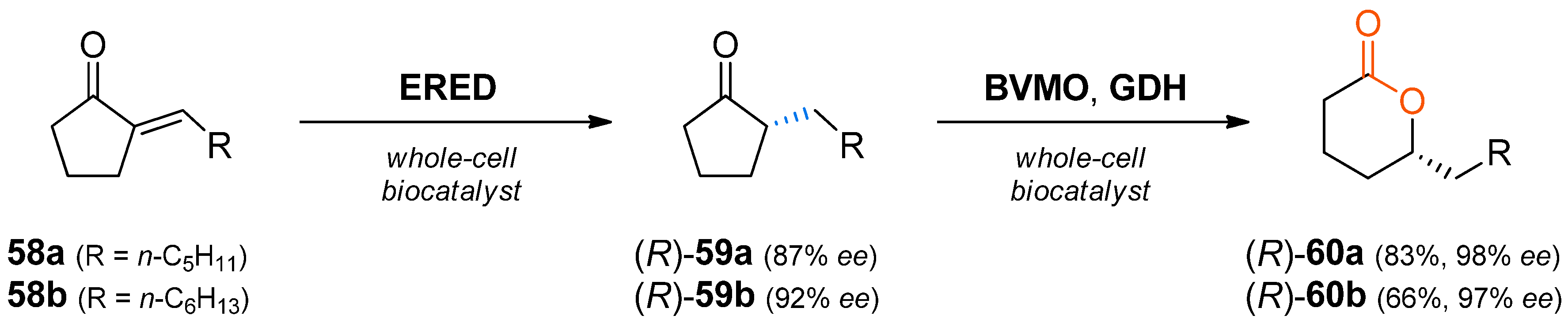
When the asymmetric reduction of prochiral α,β-unsaturated ketones catalysed by ene-reductases is combined with the oxidation by a Baeyer–Villiger monooxygenase catalysed by BVMOs, chiral lactones are obtained. This concept has first been put into practice in a biocatalytic synthesis of 5-alkyl-substituted δ-valerolactones
60 (
Scheme 18), which are flavour and fragrance compounds [
86].
In this study, Acinetobacter sp. RS1 was shown to be the best organism for the C=C-reduction step regarding activity, growth and enantioselectivity by screening 200 alkane-, toluene- or benzene- degrading microbial strains. Already after 2 h, the reaction reached >99% conversion, producing (R)-59a and (R)-59b with 87% and 92% ee, respectively. For the following Baeyer–Villiger oxidation, resting cells of Escherichia coli containing a cyclohexanone monooxygenase (CHMO) and a glucose dehydrogenase (GDH) were employed. In initial experiments, an undesired oxidation activity on substrates 58 was observed; consequently, the one-pot cascade was carried out in a sequential fashion, producing (R)-60a and (R)-60b with 83% and 66% conversion and 98% and 97% ee, respectively. The improved enantiomeric excess of the products 60 compared to the intermediates 59 was shown to be a consequence of the hydrolysis of the lactone catalysed by the Acinetobacter cells. Thereby the (S)-enantiomers of lactones 60 were preferentially converted (E = 8–11). Hence, the optical purity of the desired products increased over time due to removal of the minor enantiomer. As a final proof of concept, preparative-scale transformations with 58a and 58b (40 mg) were performed and the lactones 60a and 60b were isolated in 56% and 41% yield (98% and 97% ee), respectively.
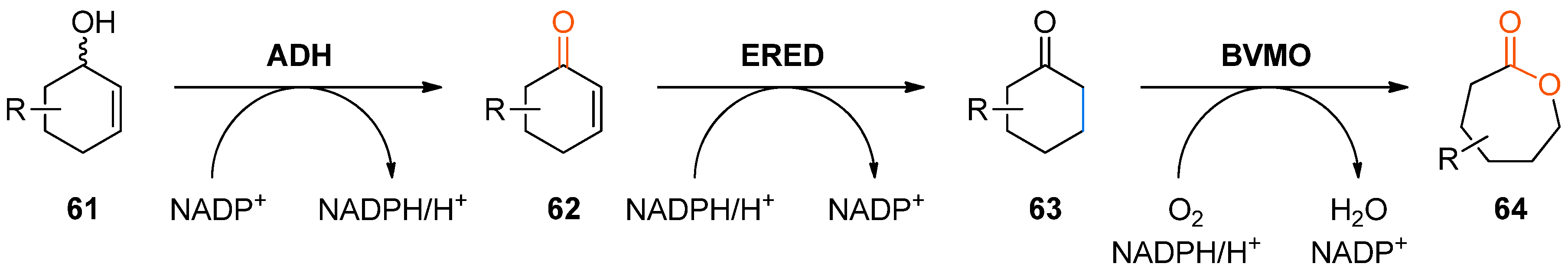
A similar combination of ene-reductases and Baeyer–Villiger monooxygenases, extended by an ADH-catalysed step, has been investigated for the preparation of ε-caprolactone derivatives
64 from cyclohexenol derivatives
61 (
Scheme 19) [
87,
88]. This particular cascade was termed an “artificial metabolic mini pathway” by the authors, meaning that all the required enzymes were co-expressed in
E. coli, while in nature they are not connected in a single organism in a metabolic context.
Suitable enzyme candidates for each individual step of the cascade were identified whereby the ADH from
Lactobacillus kefir, the ene-reductase XenB from
Pseudomonas sp., and the well-known cyclohexanone monooxygenase from
Acinetobacter sp. performed best for most of the tested substrates. Cofactor regeneration was performed via the metabolism of the
E. coli host.
Table 8 summarises the results of the substrates tested.
Interestingly, substrate rac-61d was oxidized by ADH from Lactobacillus kefir with 100% conversion, but the ene-reductase from Pseudomonas sp. did not accept intermediate 62d, although it was active on the regioisomeric 62c. Therefore, an additional ERED was investigated, namely old yellow enzyme 1 (OYE1) from Saccharomyces carlsbergensis, which yielded optically pure (S)-63d, which was then further oxidized to lactone (S)-64d.
Preparative-scale biotransformations with substrates rac-61d and (1S,5S)-61e (100 mg) afforded the products (S)-64d and (3R,6R)-64e in 55% and 60% isolated yield, respectively, and in >99% ee/de.
In a subsequent study, three ene-reductases from
Pseudomonas putida were used in a very similar cascade, in this case implemented using isolated enzymes (purified enzymes and crude cell-free extracts) [
89]. Cyclohexenol, cyclohexanone, and cyclopentenone were investigated, leading to δ-valerolactone or ɛ-caprolactone as products. Conversions between 19% and 99% were obtained at a substrate concentration of 3 mM within 1 h.
4.2. Formation of Lactones from Acyclic Precursors
The formation of lactones form acyclic precursors is not only a common strategy in organic synthesis, but also takes place in the biosynthesis of many naturally occurring lactones, for instance macrolides. These molecules are produced by polyketide synthases (PKSs)—large multi-enzyme complexes—from acyclic precursors that are assembled by consecutive Claisen condensations while bound to an acyl-carrier protein and are finally cyclised by a thioesterase domain of the PKS [
90,
91].
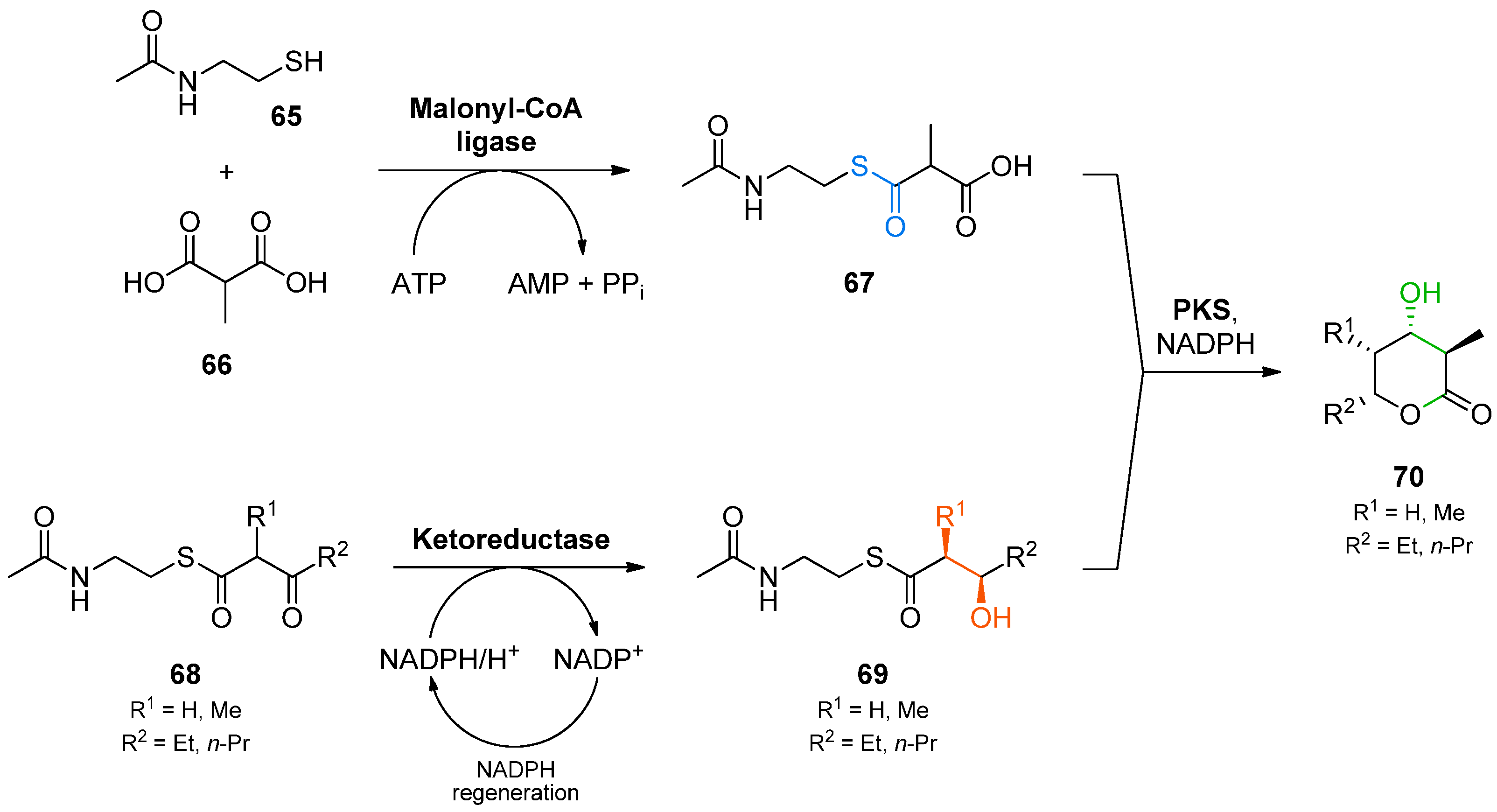
The exploitation of this biocatalytic machinery for polyketide synthesis in vitro has proven challenging, but recently researchers have succeeded in producing triketide lactones via multi-enzyme reactions that combine functional domains from several polyketide synthases [
92]. The authors used the malonyl-CoA ligase from
Streptomyces coelicolor for producing the extender unit
67 from methylmalonic acid (
66) and
N-acetylcysteamine (
65) at the expense of stoichiometric amounts of ATP (
Scheme 20). In parallel, an asymmetric reduction of β-keto-thioesters
68 by isolated ketoreductase domains of bacterial PKSs was performed to furnish the starter units for the PKS-catalysed Claisen condensation. The crude reaction mixtures obtained from the two biotransformations were then combined and supplemented with the terminal PKS module (consisting of acyl carrier protein, acyl transferase, ketoreductase, and ketosynthase domains) fused to the thioesterase of the erythromycin PKS from
Saccharopolyspora erythraea. This “minimal PKS” catalysed the Claisen condensation of
67 and
69 as well as the reduction of the resulting ketone and the final cyclization to the product lactone
70. Although the isolated yields of the overall sequence did not exceed 13%, this method allowed the preparation of substantial amounts of the triketide lactones
70 (3.4–77 mg) as single stereoisomers. The corresponding 3-ketolactones could also be accessed by isolating intermediate
69 and performing the final biotransformation in the absence of an NADPH regeneration system.
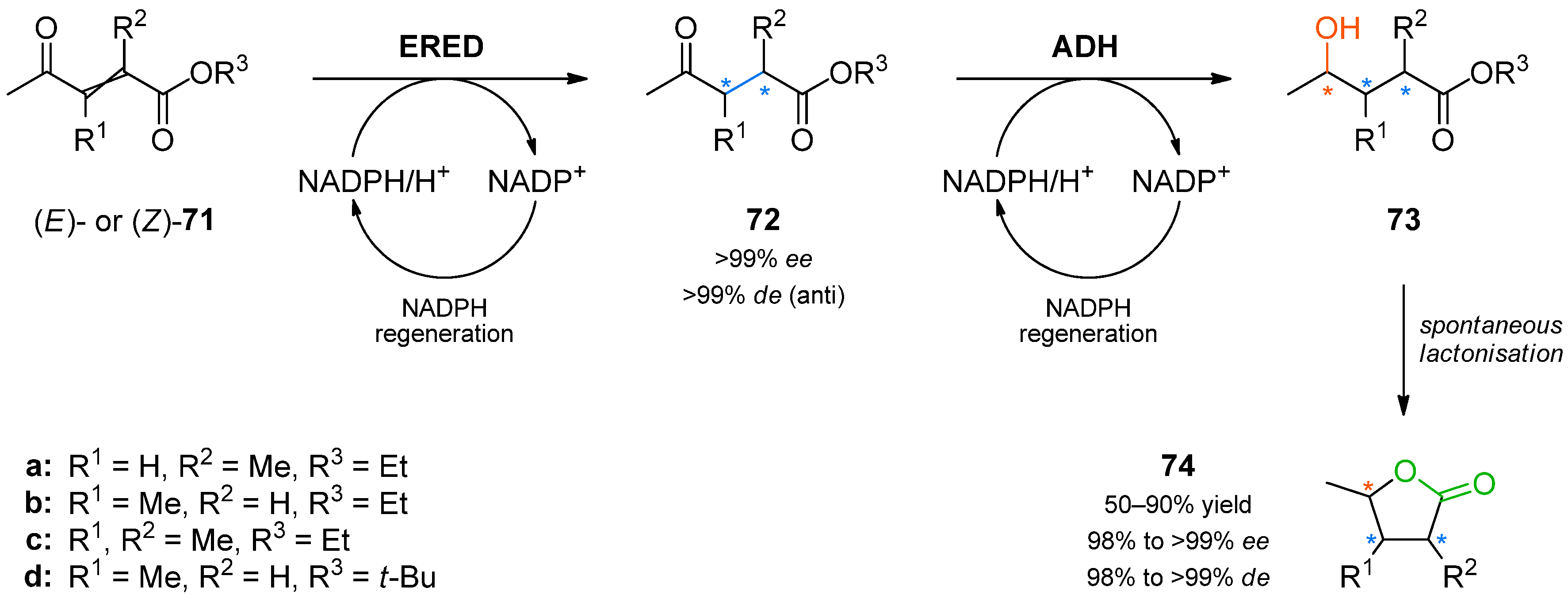
Another study aimed at the synthesis of chiral lactones from acyclic precursors relied on spontaneous ring closure and used ene-reductase YqjM from
Bacillus subtilis and alcohol dehydrogenases from different microbial sources for controlling the formation of up to three contiguous stereogenic centres (
Scheme 21) [
93].
In the first step of the synthetic sequence, (
E)- or (
Z)-oxo-esters
71 were reduced by YqjM—a reaction that proceeded with varying stereochemical outcomes: While both double bond isomers of substrate
71a (R
1 = H, R
2 = Me, R
3 = Et) afforded the same product (
R)-
72a and only the (
E)-isomer of substrate
71c (R
1, R
2 = Me, R
3 = Et) was converted, substrate
71b (R
1 = Me, R
2 = H, R
3 = Et) displayed an
E/
Z-dependent switch in selectivity. The (
E)-isomer was reduced to (
S)-
72b, while the (
Z)-isomer gave the (
R)-enantiomer, and in both cases optically pure products (
ee > 99%) were obtained. The subsequent reduction of intermediates
72 catalysed by alcohol dehydrogenases from
Lactobacillus kefir (
Lk-ADH),
Lactobacillus brevis (
Lb-ADH),
Thermoanaerobacter sp. (ADH-T), or from a commercial supplier (evocatal, evo-1.1.030) was more predictable, proceeding with the expected selectivity in all cases. The ethyl esters
73a–
c underwent spontaneous cyclisation to the desired lactones
74a–
c, while the reaction employing a
tert-butyl ester stopped at the hydroxyacid stage (
73d). All products were easily isolated by extraction and purified by column chromatography. The isolated yields and optical purities are summarised in
Table 9.
In a later study, the cascade was extended, starting from allylic cyclic alcohols using fusion proteins; however, conversions did not exceed 34% [
94].
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

































