Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series


Abstract
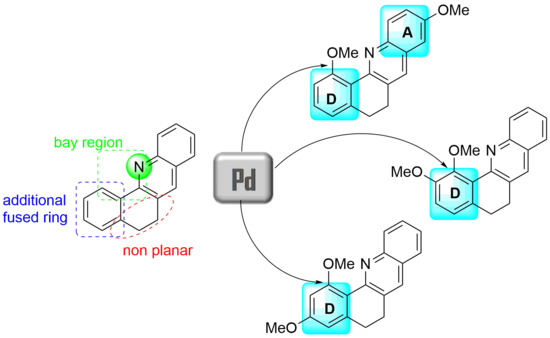
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
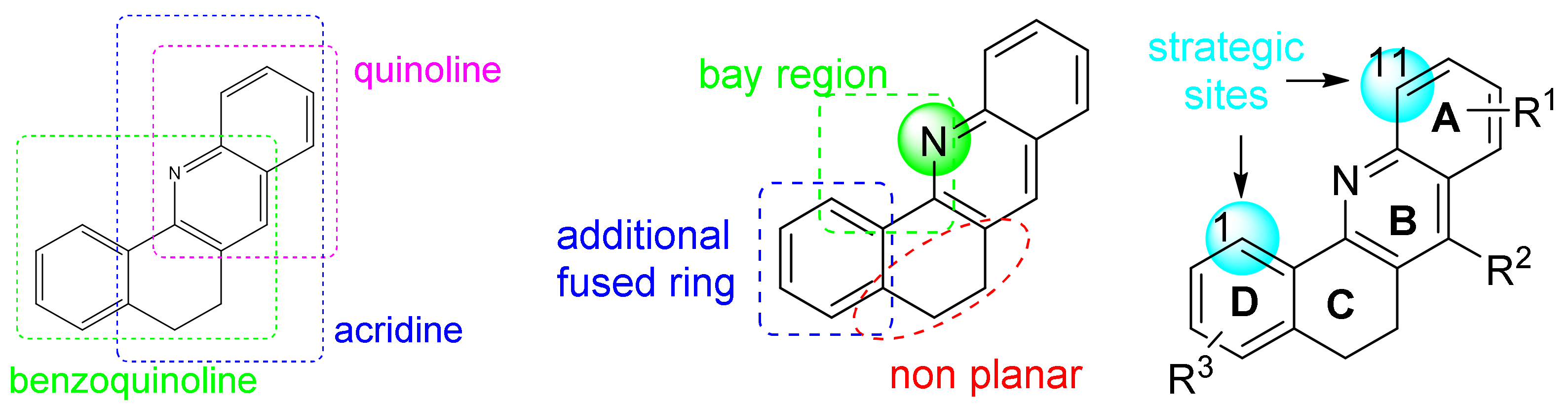
1. Introduction
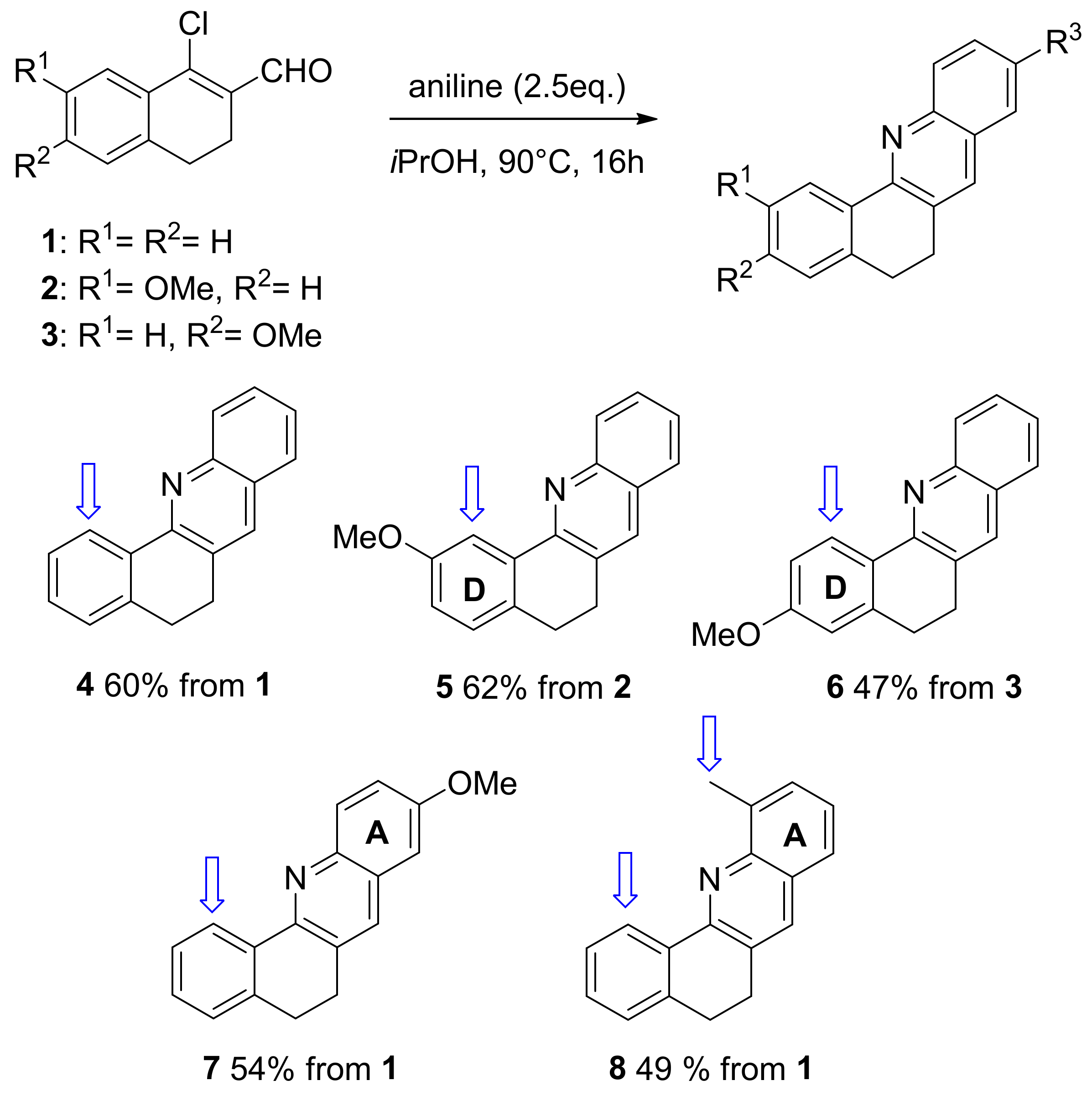
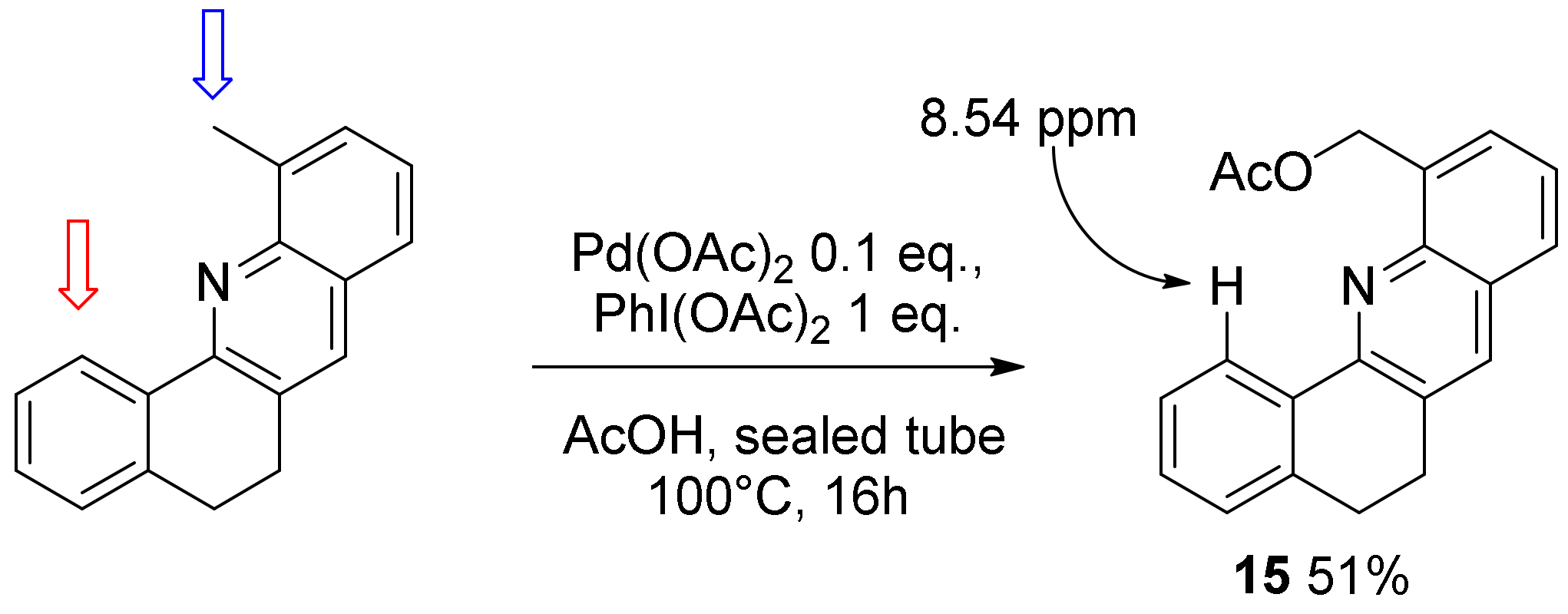
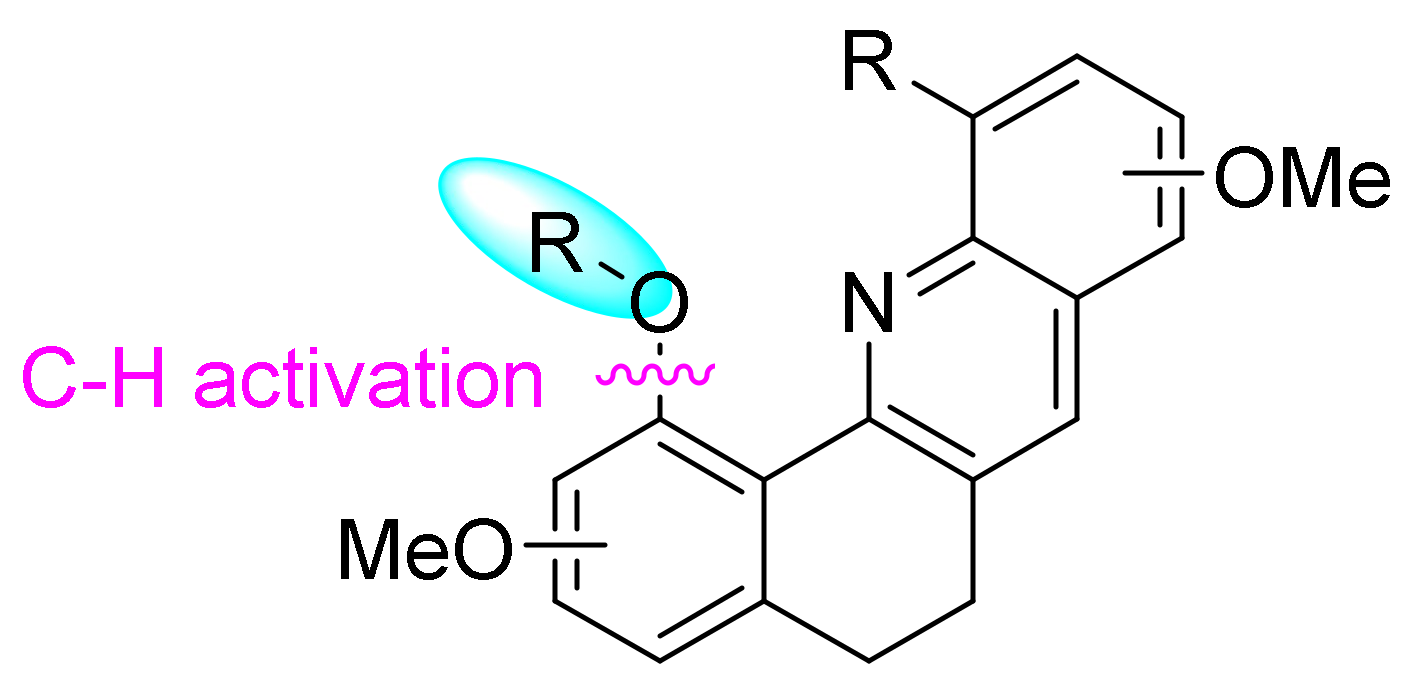
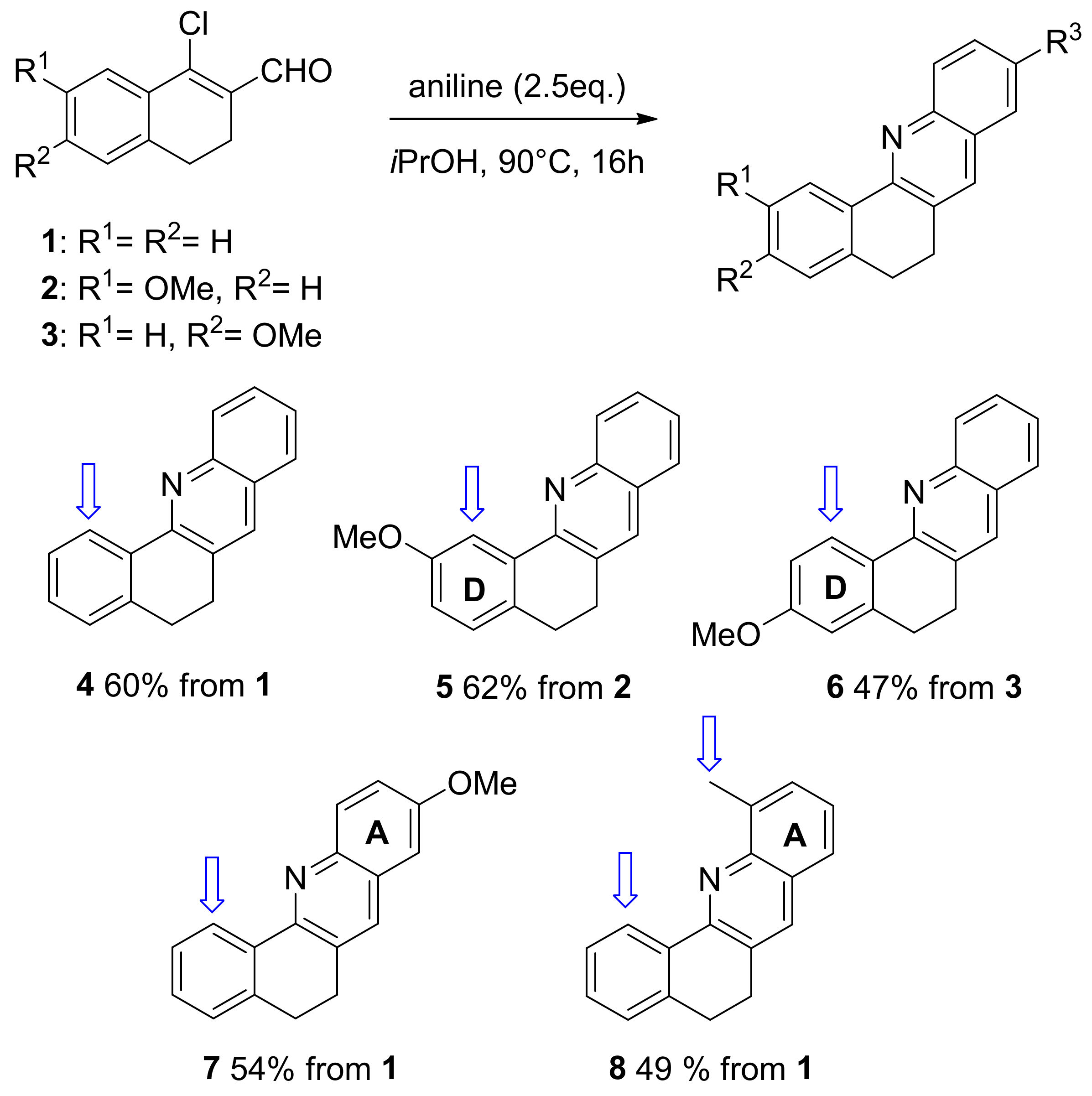
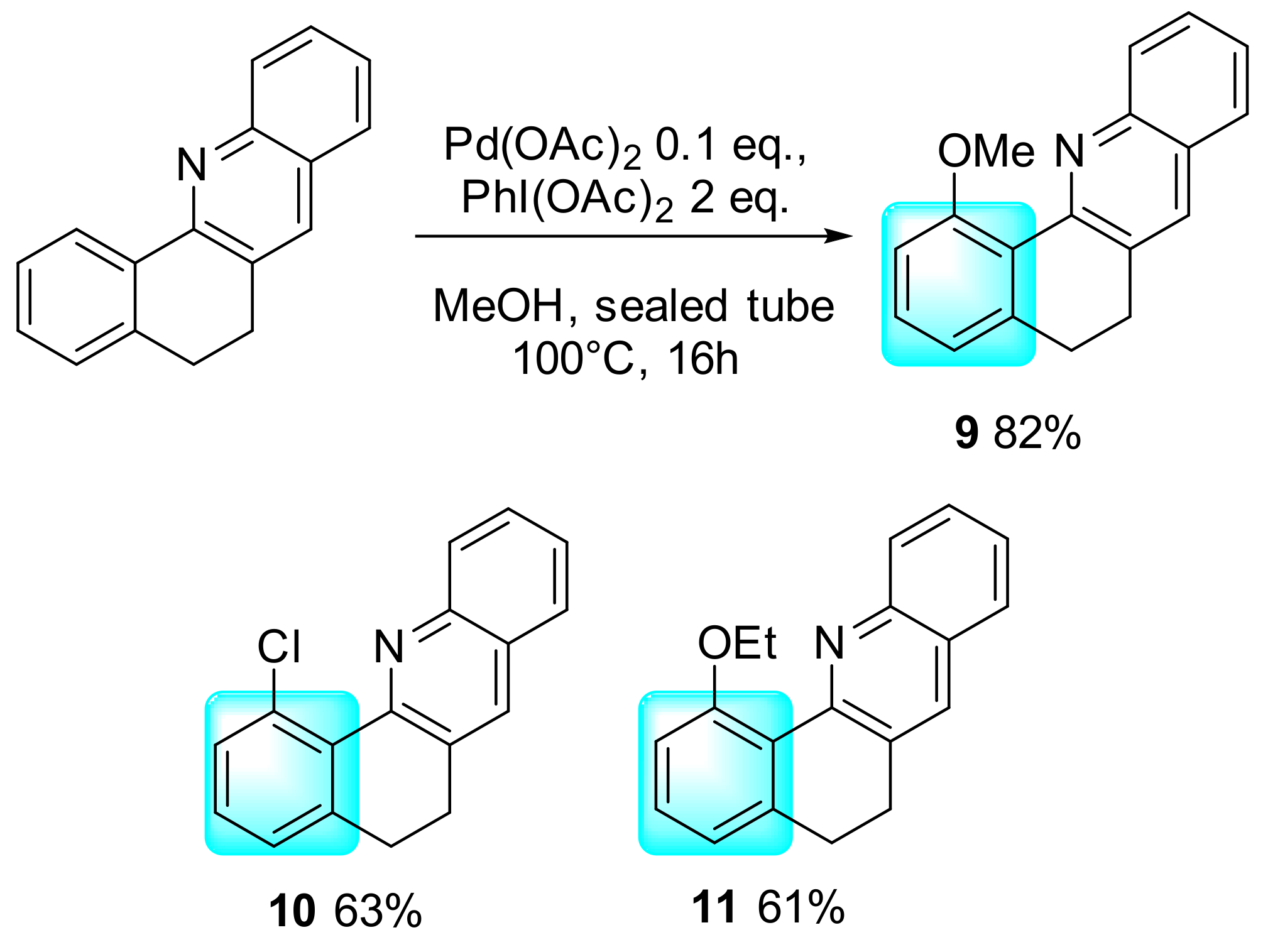
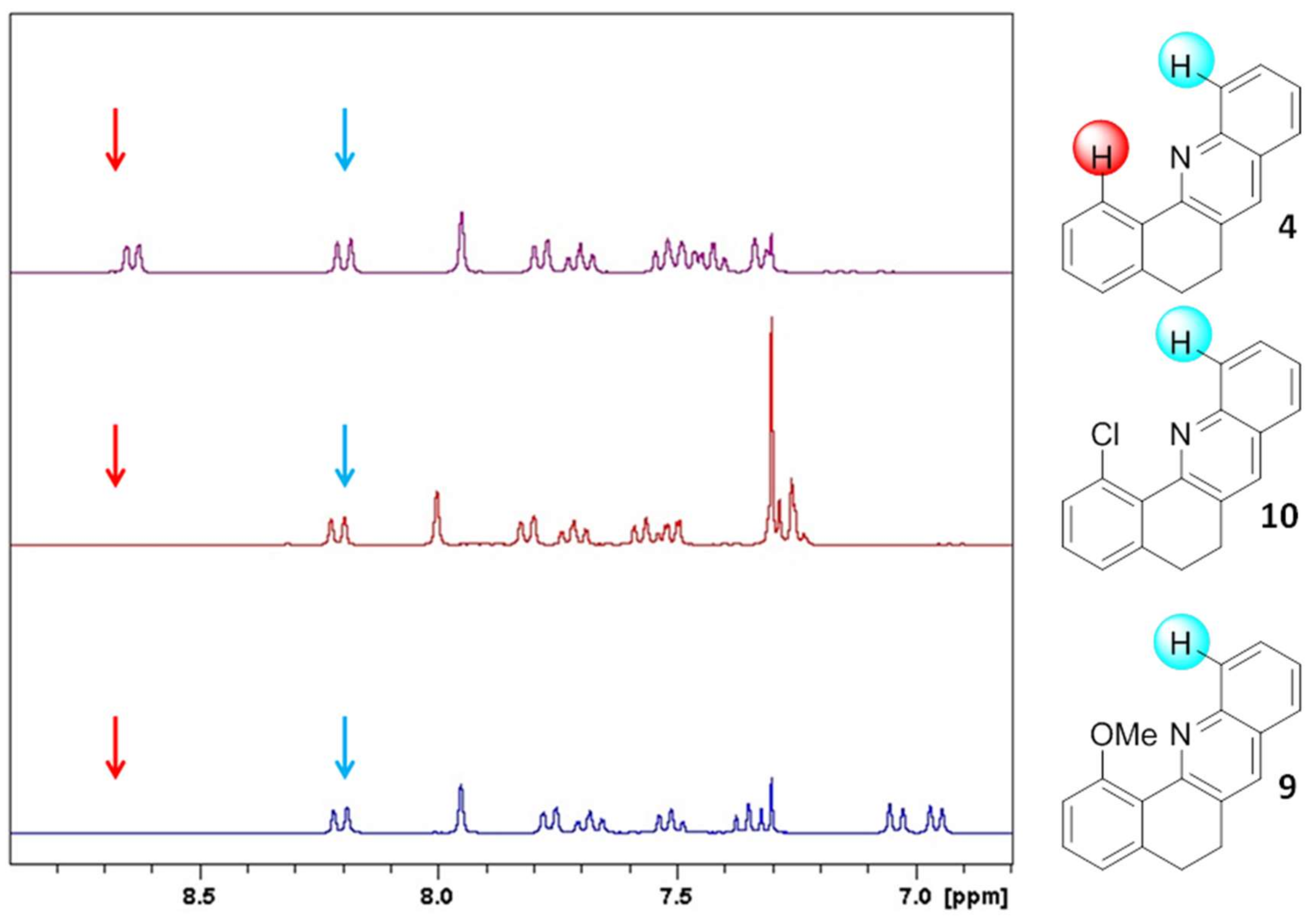
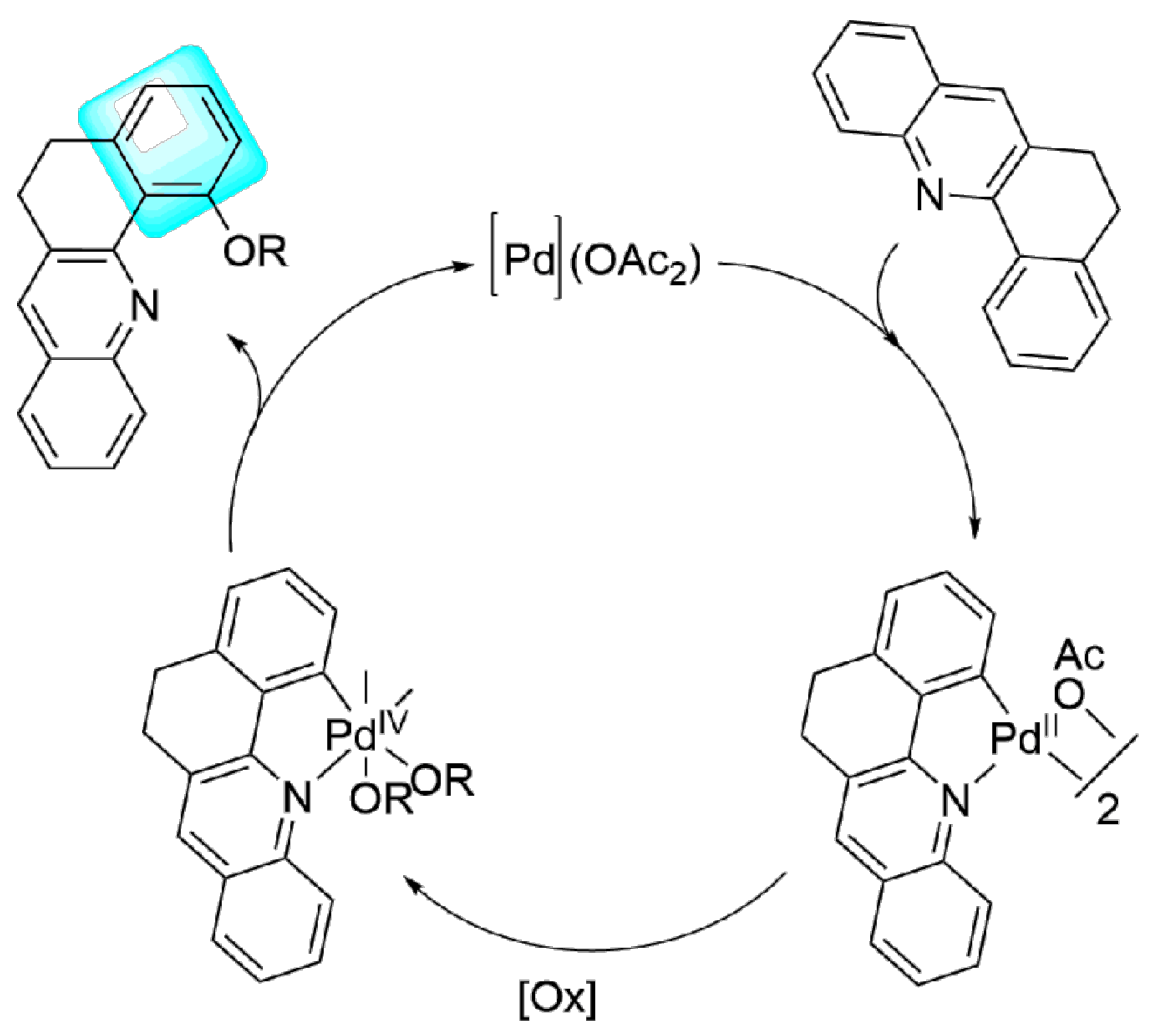
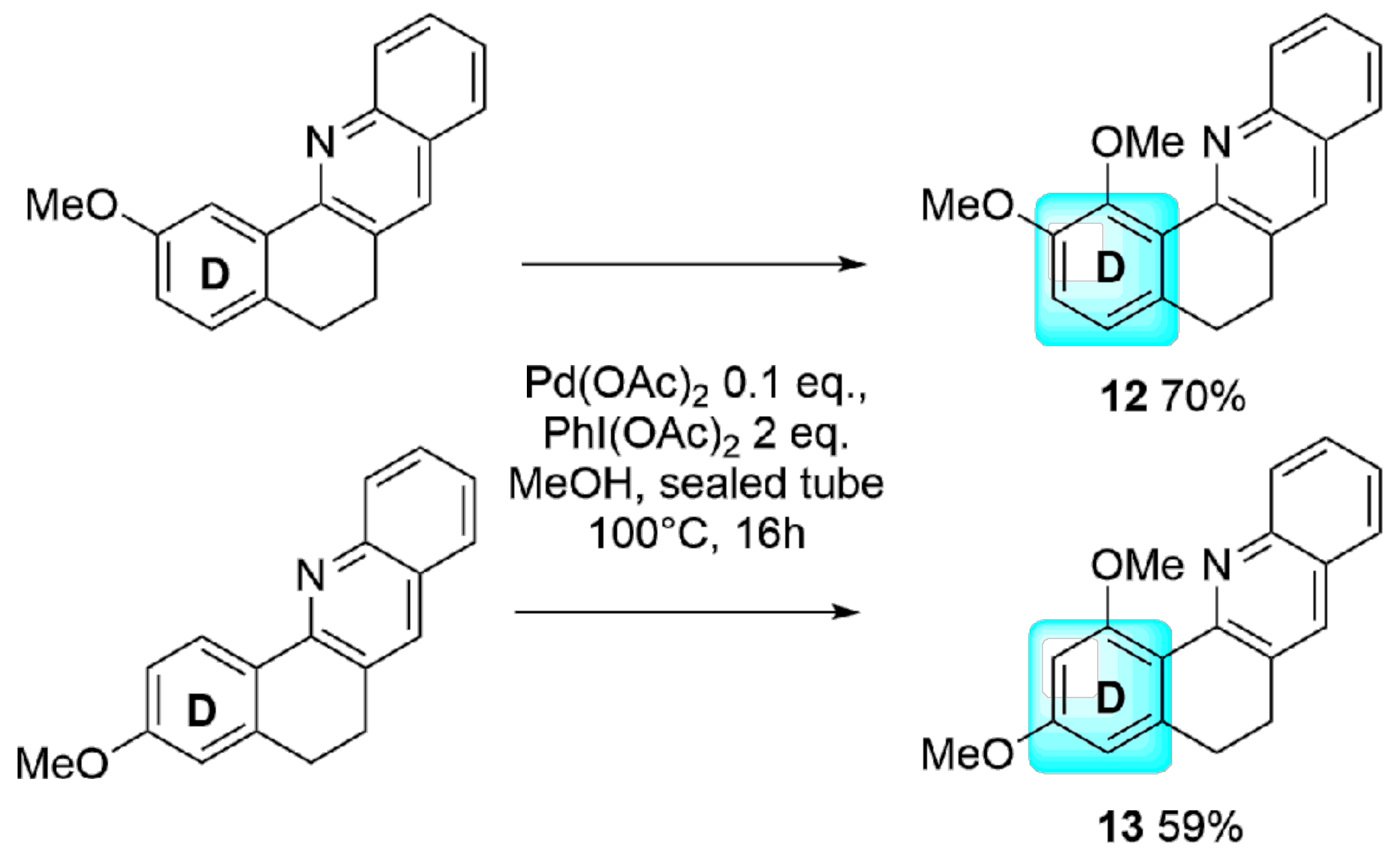
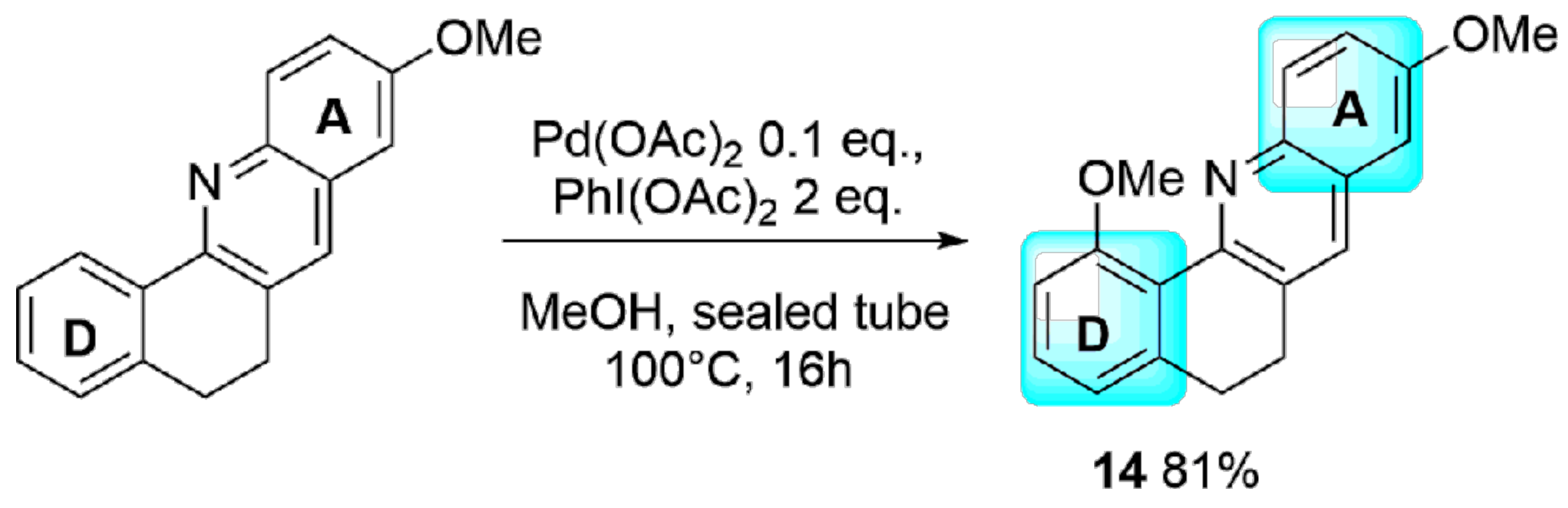
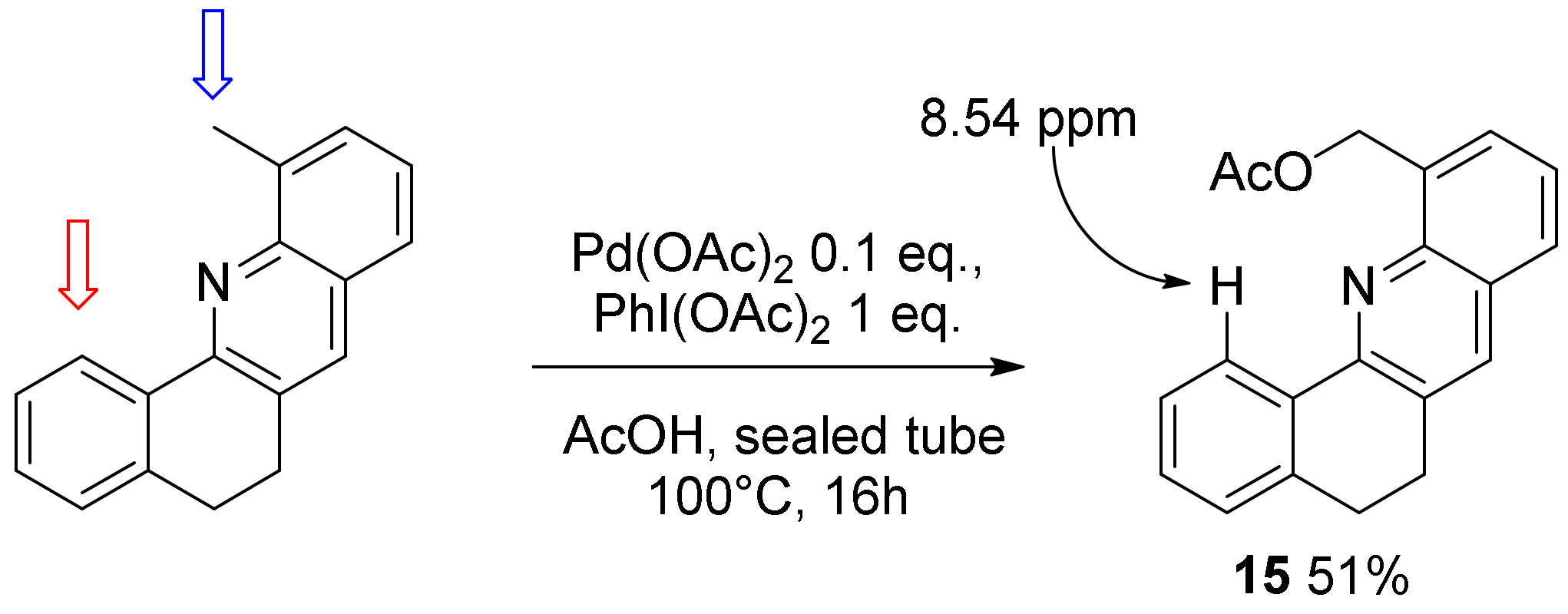
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. General Procedure for Pd-Catalyzed Alkoxylation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, Y.-J.; Chen, D.-S.; Li, Y.-L.; Liu, Y.; Wang, X.-S. Combinatorial synthesis of pyrrolo[3,2-f]quinoline and pyrrolo[3,2-a]acridine derivatives via a three-component reaction under catalyst-free conditions. ACS Comb. Sci. 2013, 15, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-M.; Ramiandrasoa, F.; Guetzoyan, L.; Pradines, B.; Quintino, E.; Gadelle, D.; Forterre, P.; Cresteil, T.; Mahy, J.-M.; Pethe, S. Synthesis and biological evaluation of acridine derivatives as antimalarial agents. ChemMedChem 2012, 7, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Suryadi, J.; Bierbach, U. Cellular recognition and repair of monofunctional-interactive platinum-DNA adducts. Chem. Res. Toxicol. 2015, 28, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Geddes, C.D. Optical thin film polymeric sensors for the determination of aqueous chloride, bromide and iodide ions at high pH, based on the quenching of fluorescence of two acridinium dyes. Dyes Pigment. 2000, 45, 243–251. [Google Scholar] [CrossRef]
- Warther, D.; Bolze, F.; Leonard, J.; Gug, S.; Specht, A.; Puliti, D.; Sun, X.-H.; Kessler, P.; Lutz, Y.; Vonesch, J.-L.; et al. Live-cell one- and two-photon uncaging of a far-red Emitting acridinone fluorophore. J. Am. Chem. Soc. 2009, 132, 2585–2590. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Yoo, C.; Lee, Y. Direct CO2 addition to a Ni(0)–CO species allows the selective generation of a Nickel(II) carboxylate with expulsion of CO. J. Am. Chem. Soc. 2018, 140, 2179–2185. [Google Scholar] [CrossRef]
- Dos Santos, P.L.; Ward, J.S.; Bryce, M.R.; Monkman, A.P. Using guest–host interactions to optimize the efficiency of TADF Oleds. J. Phys. Chem. Lett. 2016, 7, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Kumar, V.; Singh, S.P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R.S. Non-aggregating solvatochromic bipolar benzo[f]quinolines and benzo[a]acridines for organic electronics. J. Mater. Chem. 2012, 22, 14880–14888. [Google Scholar] [CrossRef]
- Martins, A.P.; Frizzo, C.P.; Moreira, D.N.; Buriol, L.; Machado, P. Solvent-free heterocyclic synthesis. Chem. Rev. 2009, 109, 4140–4182. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fan, Q.; Jiang, X. Nitrogen-iodine exchange of dirayliodonium salts: Access to acridine and carbazole. Org. Lett. 2018, 20, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, P.; He, M.; Wu, Q.; Ye, L.; Mu, Y. Facile synthesis of acridine derivatives by ZnCl2-promoted intramolecular cyclization of o-arylaminophenyl Schiff bases. Org. Lett. 2014, 16, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-J.; Chen, W.-W.; Li, Y.; Xu, M.-H. Facile synthesis of acridines via Pd(0)-diphosphine complex-catalyzed tandem coupling/cyclization protocol. Org. Biomol. Chem. 2015, 13, 6580–6586. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Kindelin, P.J.; Klumpp, D.A. Charge migration in dicationic electrophiles and its application to the synthesis of aza-polycyclic aromatic compounds. Org. Lett. 2006, 8, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, P.; Meena Rani, A.; Saiganesh, R.; Balasubramanian, K.K.; Kabilan, S. Synthesis of 5,6-dihydrobenz[c]acridines: A comparative study. Tetrahedron 2009, 65, 811–821. [Google Scholar] [CrossRef]
- Souibgui, A.; Gaucher, A.; Marrot, J.; Bourdreux, F.; Aloui, F.; Ben Hassine, B.; Prim, D. New series of acridines and phenanthrolines: Synthesis and characterization. Tetrahedron 2014, 70, 3042–3048. [Google Scholar] [CrossRef]
- Gogoi, S.; Shekarrao, K.; Duarah, A.; Bora, T.C.; Gogoi, S.; Boruah, R.C. A microwave promoted solvent-free approach to steroidal quinolines and their in vitro evaluation for antimicrobial activities. Steroids 2012, 77, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Mishra, S.; Kakde, B.N.; Dey, D.; Bisai, A. Expeditious approach to pyrrolophenanthridones, phenanthridines, and benzo[c]phenanthridines via organocatalytic direct biaryl-coupling promoted by potassium tert-butoxide. J. Org. Chem. 2013, 78, 7823–7844. [Google Scholar] [CrossRef] [PubMed]
- Jierry, L.; Harthong, S.; Aronica, C.; Mulatier, J.-C.; Guy, L.; Guy, S. Efficient dibenzo[c]acridine helicene-like synthesis and resolution: Scale up, structural control, and high chiroptical properties. Org. Lett. 2012, 14, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Speed, S.; Pointillart, F.; Mulatier, J.-C.; Guy, L.; Golhen, S.; Cador, O.; Le Guennic, B.; Riobé, F.; Maury, O.; Ouahab, L. Photophysical and magnetic properties in complexes containing 3d/4f elements and chiral phenanthroline-based helicate-like ligands. Eur. J. Inorg. Chem. 2017, 14, 2100–2111. [Google Scholar] [CrossRef]
- Souibgui, A.; Gaucher, A.; Marrot, J.; Aloui, F.; Mahuteau-Betzer, F.; Ben Hassine, B.; Prim, D. A Flexible strategy towards thienyl-, oxazolyl- and pyridyl-fused fluorenones. Eur. J. Org. Chem. 2013, 21, 4515–4522. [Google Scholar] [CrossRef]
- Solmont, K.; Boufroura, H.; Souibgui, A.; Fornarelli, P.; Gaucher, A.; Mahuteau-Betzer, F.; Ben Hassine, B.; Prim, D. Divergent strategy in the synthesis of original dihydro benzo- and dihydronaphtho-acridines. Org. Biomol. Chem. 2015, 13, 6269–6277. [Google Scholar] [CrossRef] [PubMed]
- Some, S.; Ray, J.K. Chemoselective arylamination of β-bromovinylaldehydes followed by acid catalyzed cyclization: A general method for polycyclic quinolines. Tetrahedron Lett. 2007, 48, 5013–5016. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A highly selective catalytic method for the oxidative functionalization of C-H bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Seki, B. Arylation using ruthenium catalyst. In Catalytic Transformations via C-H Activation; Yu, J.-Q., Ed.; Georg thieme Verlag KG: Stuttgart, Germany, 2016; Volume 1, pp. 119–153. ISBN 978-3-13-171141-0. [Google Scholar]
- Powers, D.C.; Benitez, D.; Tkatchouk, E.; Goddard, W.A., III; Ritter, T. Bimetallic reductive elimination from dinuclear Pd(III)complexes. J. Am. Chem. Soc. 2010, 132, 14092–14103. [Google Scholar] [CrossRef] [PubMed]
- Aiello, I.; Crispini, A.; Ghedini, M.; La Deda, M.; Barigelletti, F. Synthesis and characterization of a homologous series of mononuclear palladium complexes containing different cyclometalated ligands. Inorg. Chim. Acta 2000, 308, 121–128. [Google Scholar] [CrossRef]
- Selbin, J.; Gutierrez, M.A. Cyclometallation IV. Palladium(II) compounds with benzo[h]quinoline and substituted 2,6-diraylpyridines. J. Organomet. Chem. 1983, 246, 95–104. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Yuan, J.; Guo, S. Palladacycles incorporating a carboxylate-functionalized phosphine ligand: Syntheses, characterization and their catalytic applications toward Suzuki couplings in water. Transit. Met. Chem. 2017, 42, 727–738. [Google Scholar] [CrossRef]
- Li, C.; Sun, P.; Yan, L.; Pan, Y.; Cheng, C.-H. Synthesis and electroluminescent properties of Ir complexes with benzo[c]acridine or 5,6-dihydro-benzo[c]acridine ligands. Thin Solid Films 2008, 516, 6186–6190. [Google Scholar] [CrossRef]
- Sako, M.; Takeuchi, Y.; Tsujihara, T.; Kodera, J.; Kawano, T.; Takizawa, S.; Sasai, H. Efficient enantioselective synthesis of oxahelicenes using redox/acid cooperative catalysts. J. Am. Chem. Soc. 2016, 138, 11481–11484. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Schardt, B.C.; Hill, C.L. Preparation of iodobenzene dimethoxide. A new synthesis of [18O]iodosylbenzene and a reexamination of its infrared spectrum. Inorg. Chem. 1983, 22, 1563–1565. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Large, B.; Bourdreux, F.; Damond, A.; Gaucher, A.; Prim, D. Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series. Catalysts 2018, 8, 139. https://doi.org/10.3390/catal8040139
Large B, Bourdreux F, Damond A, Gaucher A, Prim D. Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series. Catalysts. 2018; 8(4):139. https://doi.org/10.3390/catal8040139
Chicago/Turabian StyleLarge, Benjamin, Flavien Bourdreux, Aurélie Damond, Anne Gaucher, and Damien Prim. 2018. "Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series" Catalysts 8, no. 4: 139. https://doi.org/10.3390/catal8040139
APA StyleLarge, B., Bourdreux, F., Damond, A., Gaucher, A., & Prim, D. (2018). Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series. Catalysts, 8(4), 139. https://doi.org/10.3390/catal8040139