Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing


Abstract
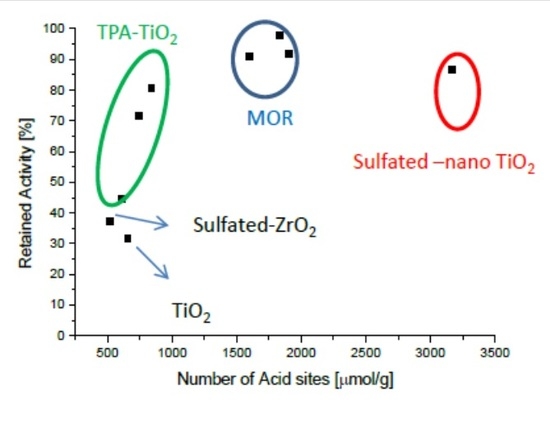
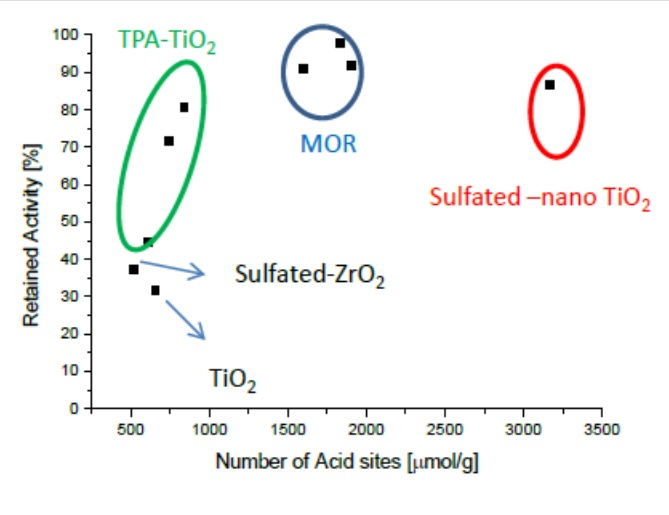{kind=link}
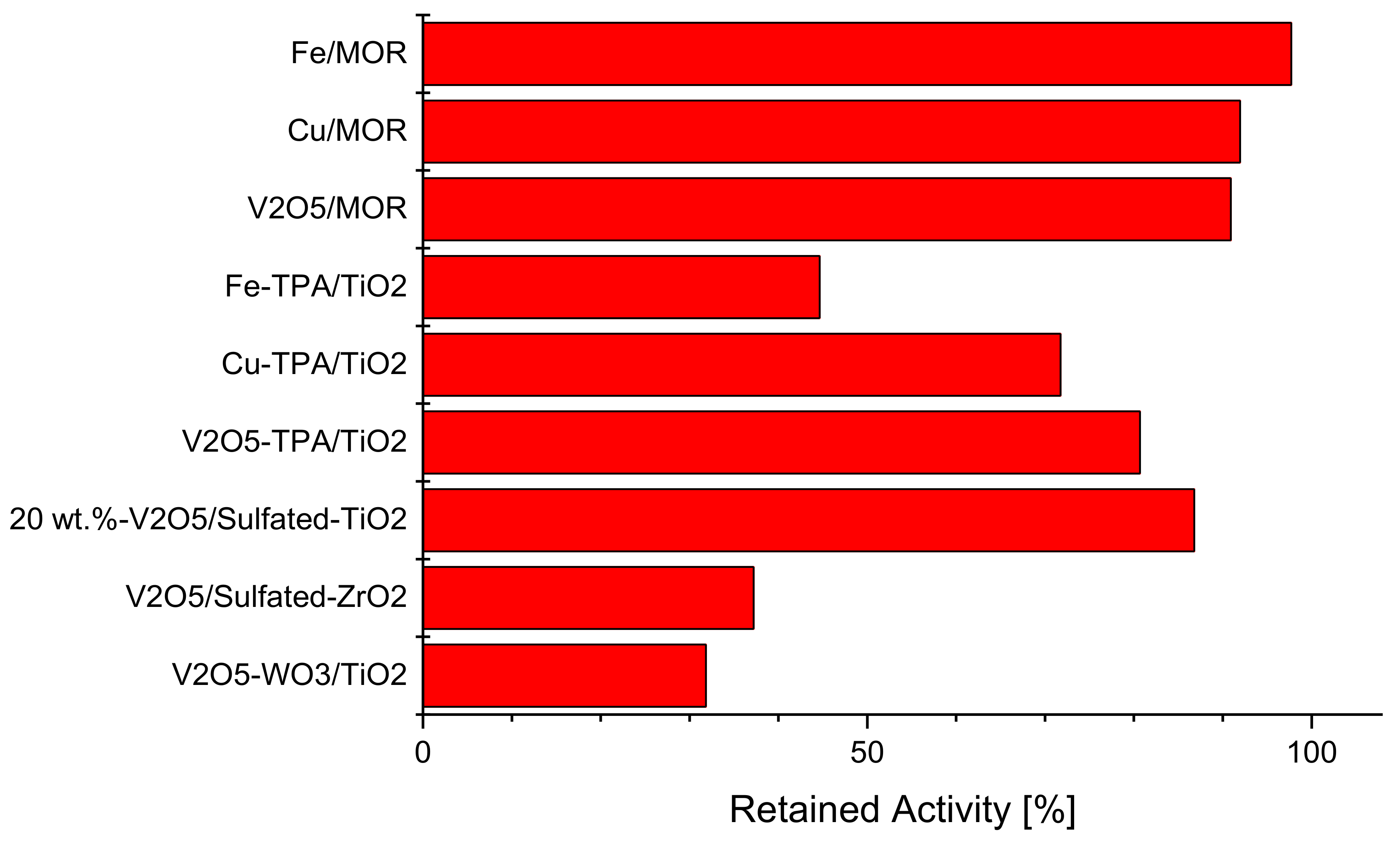{kind=link}
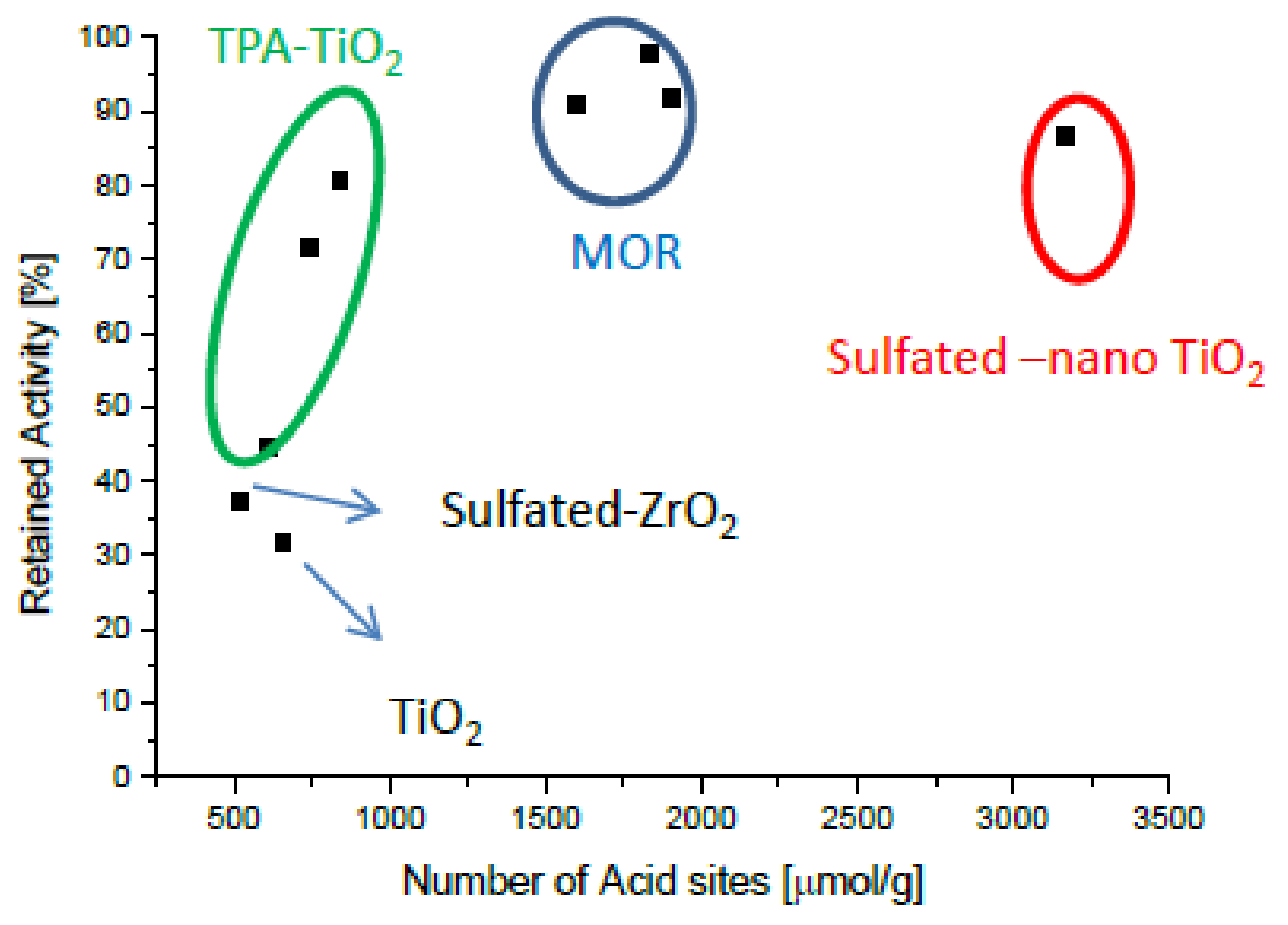{kind=link}
1. Introduction
2. Strategies Coping with Potassium Rich Fuels
2.1. Potassium Removal by Adsorption
2.2. Tail-End Placement of the SCR Unit
2.3. Coating Monoliths with Basic Substances
2.4. Intrinsically Potassium Resistant Catalysts
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bjerg, J.; Aden, R.; Ogand, J.A.; Arrieta, J.A.; Hahlbrock, A.; Holmquist, L.; Kellberg, C.; KIp, W.N.; Koch, J.; Langnickel, U.; et al. Biomass 2020: Opportunities, Challenges and Solutions. Eurelectric 2001, 72, 18. [Google Scholar]
- Agbor, E.; Zhang, X.; Kumar, A. A review of biomass co-firing in North America. Renew. Sustain. Energy Rev. 2014, 40, 930–943. [Google Scholar] [CrossRef]
- Livingston, W.R. The Status of Large Scale Biomass Firing: The Milling and Combustion of Biomass Materials in Large Pulverised Coal Boilers; IEA Bioenergy Report 2016; IEA Bioenergy: Paris, France, 2006; ISBN 9781910154267. Available online: http://www.ieabcc.nl/publications/IEA_Bioenergy_T32_cofiring_2016.pdf (accessed on 27 February 2018).
- Khorshidi, Z.; Ho, M.T.; Wiley, D.E. Techno-economic study of biomass co-firing with and without CO2 capture in an Australian black coal-fired power plant. Energy Procedia 2013, 37, 6035–6042. [Google Scholar] [CrossRef]
- Nuamah, A.; Malmgren, A.; Riley, G.; Lester, E. Biomass co-firing. Compr. Renew. Energy 2012, 5, 55–73. [Google Scholar] [CrossRef]
- Mann, M.; Spath, P. A life cycle assessment of biomass cofiring in a coal-fired power plant. Clean Prod. Process. 2001, 3, 81–91. [Google Scholar] [CrossRef]
- Kadiyala, A.; Kommalapati, R.; Huque, Z. Evaluation of the Life Cycle Greenhouse Gas Emissions from Different Biomass Feedstock Electricity Generation Systems. Sustainability 2016, 8, 1181. [Google Scholar] [CrossRef]
- Renewable Energy Agency. Renewable Power Generation Costs in 2017; International Renewable Energy Agency: Abu Dhabi, UAE, 2018. [Google Scholar]
- British Petroleum. BP Statistical Review of World Energy 2017; British Petroleum: London, UK, 2017; pp. 1–52. Available online: http://www.bp.com/content/dam/bp/en/corporate/pdf/energy-economics/statistical-review-2017/bp-statistical-review-of-world-energy-2017-full-report.pdf (accessed on 26 February 2018).
- Lüdge, S. The value of flexibility for fossil-fired power plants under the conditions of the Strommark 2.0. VGB PowerTech J. 2017, 3, 212–214. [Google Scholar]
- Clery, D.S.; Mason, P.E.; Rayner, C.M.; Jones, J.M. The effects of an additive on the release of potassium in biomass combustion. Fuel 2018, 214, 647–655. [Google Scholar] [CrossRef]
- Klimczak, M.; Kern, P.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts-Part I: V2O5-WO3/TiO2 catalysts. Appl. Catal. B Environ. 2010, 95, 39–47. [Google Scholar] [CrossRef]
- Kern, P.; Klimczak, M.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts. Part II: Fe-zeolite catalysts. Appl. Catal. B Environ. 2010, 95, 48–56. [Google Scholar] [CrossRef]
- Jensen-holm, H.; Lindenhoff, P.; Safronov, S. SCR Design Issues in Reduction of NOx Emissions from Thermal Power Plants; Haldor Topsøe A/S: Kongens Lyngby, Denmark, 2007. [Google Scholar]
- Wieck-Hansen, K.; Overgaard, P.; Larsen, O.H. Cofiring coal and straw in a 150 MWe power boiler experiences. Biomass Bioenergy 2000, 19, 395–409. [Google Scholar] [CrossRef]
- Baxter, L. Biomass Impacts on SCR Catalyst Performance; IEA Task 32 Bioenergy Report; IEA Bioenergy: Paris, France, 2005; Available online: http://task32.ieabioenergy.com/wp-content/uploads/2017/03/Combined_Final_Report_SCR.pdf (accessed on 25 February 2018).
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Kiełtyka, M.A. Influence of Biomass Co-Firing on SCR Catalyst Deactivation 2; Techniques of NOx Emission Reduction; Instituto Superior Técnico, Universidade de Lisboa: Lisbon, Portugal, 2010. [Google Scholar]
- Mladenović, M.; Paprika, M.; Marinković, A. Denitrification techniques for biomass combustion. Renew. Sustain. Energy Rev. 2018, 82, 3350–3364. [Google Scholar] [CrossRef]
- Forzatti, P. Present status and perspectives in de-NOx SCR catalysis. Appl. Catal. A Gen. 2003, 222, 221–236. [Google Scholar] [CrossRef]
- Busca, G.; Lietti, L.; Ramis, G.; Berti, F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review. Appl. Catal. B Environ. 1998, 18. [Google Scholar] [CrossRef]
- Knudsen, J.N.; Jensen, P.A.; Dam-Johansen, K. Transformation and release to the gas phase of Cl, K, and S during combustion of annual biomass. Energy Fuels 2004, 18, 1385–1399. [Google Scholar] [CrossRef]
- Kristensen, S.B. deNOx Catalysts for Biomass Combustion. Ph.D. Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, April 2013. [Google Scholar]
- Wang, L.; Hustad, J.E.; Skreiberg, Ø.; Skjevrak, G.; Grønli, M. A critical review on additives to reduce ash related operation problems in biomass combustion applications. Energy Procedia 2012, 20, 20–29. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, J.; Preto, F.; Zhu, J.; Xu, C. Ash deposition in biomass combustion or co-firing for power/heat generation. Energies 2012, 5, 5171–5189. [Google Scholar] [CrossRef]
- Davidsson, K.O.; Steenari, B.M.; Eskilsson, D. Kaolin addition during biomass combustion in a 35 MW circulating fluidized-bed boiler. Energy Fuels 2007, 21, 1959–1966. [Google Scholar] [CrossRef]
- Dahlin, S.; Nilsson, M.; Bäckström, D.; Bergman, S.L.; Bengtsson, E.; Bernasek, S.L.; Pettersson, L.J. Multivariate analysis of the effect of biodiesel-derived contaminants on V2O5-WO3/TiO2SCR catalysts. Appl. Catal. B Environ. 2016, 183, 377–385. [Google Scholar] [CrossRef]
- Khodayari, R.; Odenbrand, C.U.I. Regeneration of commercial SCR catalysts by washing and sulphation: Effect of sulphate groups on the activity. Appl. Catal. B Environ. 2001, 33, 277–291. [Google Scholar] [CrossRef]
- Forzatti, P.; Nova, I.; Tronconi, E.; Kustov, A.; Thøgersen, J.R. Effect of operating variables on the enhanced SCR reaction over a commercial V2O5-WO3/TiO2 catalyst for stationary applications. Catal. Today 2012, 184, 153–159. [Google Scholar] [CrossRef]
- Lisi, L.; Lasorella, G.; Malloggi, S.; Russo, G. Single and combined deactivating effect of alkali metals and HCl on commercial SCR catalysts. Appl. Catal. B Environ. 2004, 50, 251–258. [Google Scholar] [CrossRef]
- Zheng, Y.; Jensen, A.D.; Johnsson, J.E.; Thøgersen, J.R. Deactivation of V2O5-WO3-TiO2SCR catalyst at biomass fired power plants: Elucidation of mechanisms by lab- and pilot-scale experiments. Appl. Catal. B Environ. 2008, 83, 186–194. [Google Scholar] [CrossRef]
- Zeuthen, J.H.; Jensen, P.A.; Jensen, J.P.; Livbjerg, H. Aerosol Formation during the Combustion of Straw with Addition of Sorbents. Energy Fuels 2007, 860–870. [Google Scholar] [CrossRef]
- Sippula, O.; Lind, T.; Jokiniemi, J. Effects of chlorine and sulphur on particle formation in wood combustion performed in a laboratory scale reactor. Fuel 2008, 87, 2425–2436. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2catalysts on selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2011, 170, 531–537. [Google Scholar] [CrossRef]
- Wu, X.; Yu, W.; Si, Z.; Weng, D. Chemical deactivation of V2O5-WO3/TiO2 SCR catalyst by combined effect of potassium and chloride. Front. Environ. Sci. Eng. 2013, 7, 420–427. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Huang, X.; Li, X.; Su, W.; Sun, X.; Wang, D.; Hao, J. Deactivation mechanism of potassium on the V2O 5/CeO2 catalysts for SCR reaction: Acidity, reducibility and adsorbed-NOx. Environ. Sci. Technol. 2014, 48, 4515–4520. [Google Scholar] [CrossRef] [PubMed]
- Putluru, S.S.R.; Jensen, A.D. Alternative Alkali Resistant deNOx Technologies; Appendix-I: PSO Project 7318; Department of Chemical and Biochemical Engineering, Technical University of Denmark: Kongens Lyngby, Denmark, 2011; Available online: http://orbit.dtu.dk/files/6454472/NEI-DK-5569.pdf (accessed on 25 February 2018).
- Sorvajävi, T.; Maunula, J.; Silvennoinen, J.; Toivone, J. Optical Monitoring of KCl Vapor in 4 MW CFB Boiler during Straw Comubstion and Ferric Sulfate Injection; Optics Laboratory, Department of Physics, Tampere University of Technology: Tampere, Finnland, 2014. [Google Scholar]
- Niwa, M.; Katada, N.; Okumura, K. Characterization and Design of Zeolite Catalysts (Solid Acidity, Shape Selectivity and Loading Properties); Springer: Berlin/Heidelberg, Germany, 2010; p. 25. ISBN 9783642126192. [Google Scholar]
- Wu, H.; Shafique, M.; Arendt, P.; Sander, B.; Glarborg, P. Impact of coal fly ash addition on ash transformation and deposition in a full-scale wood suspension-firing boiler. Fuel 2013, 113, 632–643. [Google Scholar] [CrossRef]
- Steenari, B.M.; Lindqvist, O. High-temperature reactions of straw ash and the anti-sintering additives kaolin and dolomite. Biomass Bioenergy 1998, 14, 67–76. [Google Scholar] [CrossRef]
- Overgaard, P.; Sander, B.; Junker, H.; Friborg, K.; Larsen, O.H. Small-scale CHP Plant based on a 75 kWel Hermetic Eight Cylinder Stirling Engine for Biomass Fuels—Development, Technology and Operating Experiences. In Proceedings of the 2nd World Conference on Biomass for Energy, Industry and Climate Protection, Rome, Italy, 10–14 May 2004; pp. 1261–1264. [Google Scholar]
- Henderson, C. Cofiring of biomass in coal-fired power plants—European experience The role of biomass in Europe. Presented at the FCO/IEA CCC Workshops on Policy and Investment Frameworks to Introduce CCT in Hebei and Shandong Provinces, China, 8–9 and 13–14 January 2015. [Google Scholar]
- Jensen-holm, H.; Castellino, F.; White, T.N. SCR DeNOx catalyst considerations when using biomass in power generation. In Proceedings of the Power Plant Air Pollutant Control “MEGA” Symposium, Baltimore, MD, USA, 20–23 August 2012; Available online: https://www.topsoe.com/sites/default/files/scr_denox_catalyst_considerations_when_using_biomass_in_power_generation_2012.ashx__0.pdf (accessed on 23 February 2018).
- Castellino, F.; Jensen, A.D.; Johnsson, J.E.; Fehrmann, R. Influence of reaction products of K-getter fuel additives on commercial vanadia-based SCR catalysts. Part I. Potassium phosphate. Appl. Catal. B Environ. 2009, 86, 196–205. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Schill, L.; Godiksen, A.; Poreddy, R.; Mossin, S.; Jensen, A.D.; Fehrmann, R. Promoted V2O5/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2016, 183, 282–290. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Kristensen, S.B.; Due-Hansen, J.; Riisager, A.; Fehrmann, R. Alternative alkali resistant deNOx catalysts. Catal. Today 2012, 184, 192–196. [Google Scholar] [CrossRef]
- Olsen, B.K.; Kügler, F.; Castellino, F.; Jensen, A.D. Poisoning of vanadia based SCR catalysts by potassium: Influence of catalyst composition and potassium mobility. Catal. Sci. Technol. 2016, 6, 2249–2260. [Google Scholar] [CrossRef][Green Version]
- Kristensen, S.B.; Kunov-Kruse, A.J.; Riisager, A.; Rasmussen, S.B.; Fehrmann, R. High performance vanadia-anatase nanoparticle catalysts for the Selective Catalytic Reduction of NO by ammonia. J. Catal. 2011, 284, 60–67. [Google Scholar] [CrossRef]
- Fu, M.; Li, C.; Lu, P.; Qu, L.; Zhang, M.; Zhou, Y.; Yu, M.; Fang, Y. A review on selective catalytic reduction of NOx by supported catalysts at 100–300 °C—Catalysts, mechanism, kinetics. Catal. Sci. Technol. 2014, 4, 14–25. [Google Scholar] [CrossRef]
- Schill, L. Alternative Catalysts and Technologies for NOx Removal from Biomass- and Waste-Fired Plants. Ph.D. Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, April 2014. [Google Scholar]
- Casapu, M.; Kröcher, O.; Elsener, M. Screening of doped MnOx-CeO2 catalysts for low-temperature NO-SCR. Appl. Catal. B Environ. 2009, 88, 413–419. [Google Scholar] [CrossRef]
- Fehrmann, R.; Jensen, A.D. Low Temperature deNOx Technologies for Biomass and Waste Fired Power Plants; Forskel Energinet.dk Project No. 12096; Department of Chemistry, Technical University of Denmark: Kongens Lyngby, Denmark, 2017; Available online: https://energiforskning.dk/sites/energiteknologi.dk/files/slutrapporter/12096_slutrapport.pdf (accessed on 20 February 2018).
- Gao, C.; Shi, J.-W.; Fan, Z.; Gao, G.; Niu, C. Sulfur and Water Resistance of Mn-Based Catalysts for Low-Temperature Selective Catalytic Reduction of NOx: A Review. Catalysts 2018, 8, 11. [Google Scholar] [CrossRef]
- Zheng, Y.; Jensen, A.D.; Johnsson, J.E. Deactivation of V2O5-WO3-TiO2SCR catalyst at a biomass-fired combined heat and power plant. Appl. Catal. B Environ. 2005, 60, 253–264. [Google Scholar] [CrossRef]
- Olsen, B.J.; Kügler, F.; Castellino, F.; Schill, L.; Fehrmann, R.; Jensen, A.D. Deactivation of SCR Catalysts by Potassium: A Study of Potential Alkali Barrier Materials. VGB PowerTech J. 2017, 3, 56–64. [Google Scholar]
- Jensen, A.D.; Castellino, F.; Rams, P.D.; Pedersen, J.B.; Putluru, S.S.R. Deactivation Resistant Catalyst for Selective Catalytic Reduction of NOx. U.S. Patent 2012/0315206A1, 13 December 2012. [Google Scholar]
- Putluru, S.S.R.; Riisager, A.; Fehrmann, R. Alkali resistant Cu/zeolite deNOxcatalysts for flue gas cleaning in biomass fired applications. Appl. Catal. B Environ. 2011, 101, 183–188. [Google Scholar] [CrossRef]
- Cheng, Y.; Lambert, C.; Kim, D.H.; Kwak, J.H.; Cho, S.J.; Peden, C.H.F. The different impacts of SO2 and SO3 on Cu/zeolite SCR catalysts. Catal. Today 2010, 151, 266–270. [Google Scholar] [CrossRef]
- Kumar, A.; Smith, M.A.; Kamasamudram, K.; Currier, N.W.; Yezerets, A. Chemical deSOx: An effective way to recover Cu-zeolite SCR catalysts from sulfur poisoning. Catal. Today 2016, 267, 10–16. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Jensen, A.D.; Riisager, A.; Fehrmann, R. Heteropoly acid promoted V2O5/TiO2 catalysts for NO abatement with ammonia in alkali containing flue gases. Catal. Sci. Technol. 2011, 1, 631. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Mossin, S.; Riisager, A.; Fehrmann, R. Heteropoly acid promoted Cu and Fe catalysts for the selective catalytic reduction of NO with ammonia. Catal. Today 2011, 176, 292–297. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Shi, W.; Xu, J.; Hao, J. Design strategies for development of SCR catalyst: Improvement of alkali poisoning resistance and novel regeneration method. Environ. Sci. Technol. 2012, 46, 12623–12629. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, X.; Fu, Y.; Gao, F.; Luo, Z.; Cen, K. The co-effect of Sb and Nb on the SCR performance of the V2O5/TiO2catalyst. J. Colloid Interface Sci. 2012, 368, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, P.; Yu, F.; Wang, H.; Wu, Z. Dual resistance to alkali metals and SO2: Vanadium and cerium supported on sulfated zirconia as an efficient catalyst for NH3-SCR. Catal. Sci. Technol. 2016, 6, 8148–8156. [Google Scholar] [CrossRef]
- Schill, L.; Sankar, S.; Putluru, R.; Funk, C.; Houmann, C.; Fehrmann, R.; Degn, A. Applied Catalysis B: Environmental Ethanol-selective catalytic reduction of NO by Ag/Al2O3 catalysts: Activity and deactivation by alkali salts. Appl. Catal. B Environ. 2012, 127, 323–329. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schill, L.; Fehrmann, R. Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing. Catalysts 2018, 8, 135. https://doi.org/10.3390/catal8040135
Schill L, Fehrmann R. Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing. Catalysts. 2018; 8(4):135. https://doi.org/10.3390/catal8040135
Chicago/Turabian StyleSchill, Leonhard, and Rasmus Fehrmann. 2018. "Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing" Catalysts 8, no. 4: 135. https://doi.org/10.3390/catal8040135
APA StyleSchill, L., & Fehrmann, R. (2018). Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing. Catalysts, 8(4), 135. https://doi.org/10.3390/catal8040135