Vanadium Supported on Alumina and/or Zirconia Catalysts for the Selective Transformation of Ethane and Methanol


Abstract
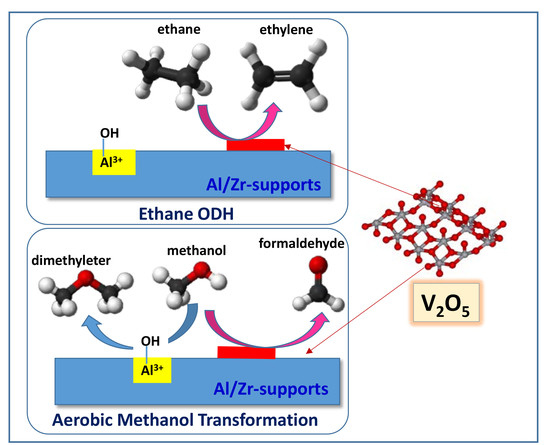
:
1. Introduction
2. Results and Discussion
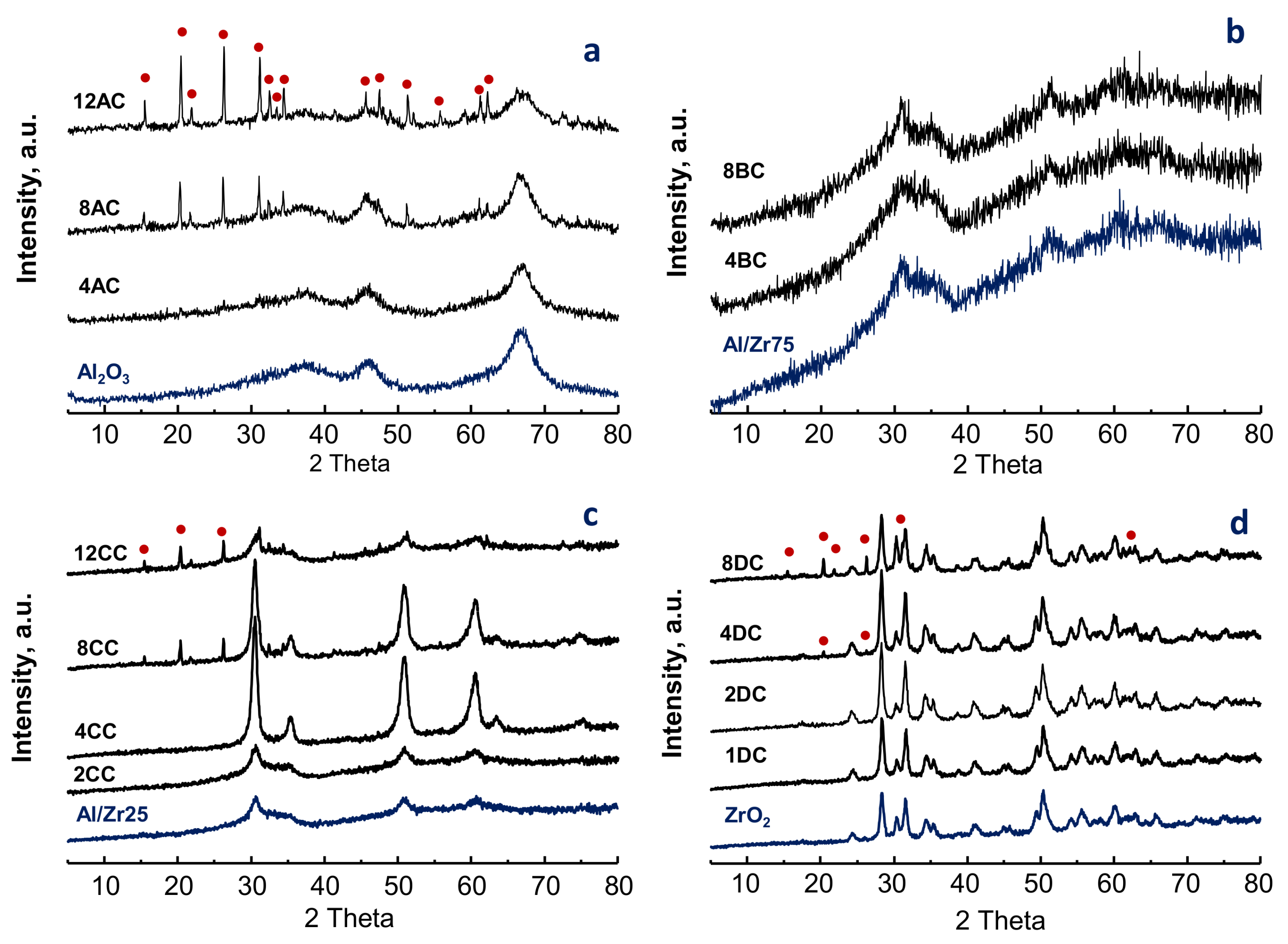
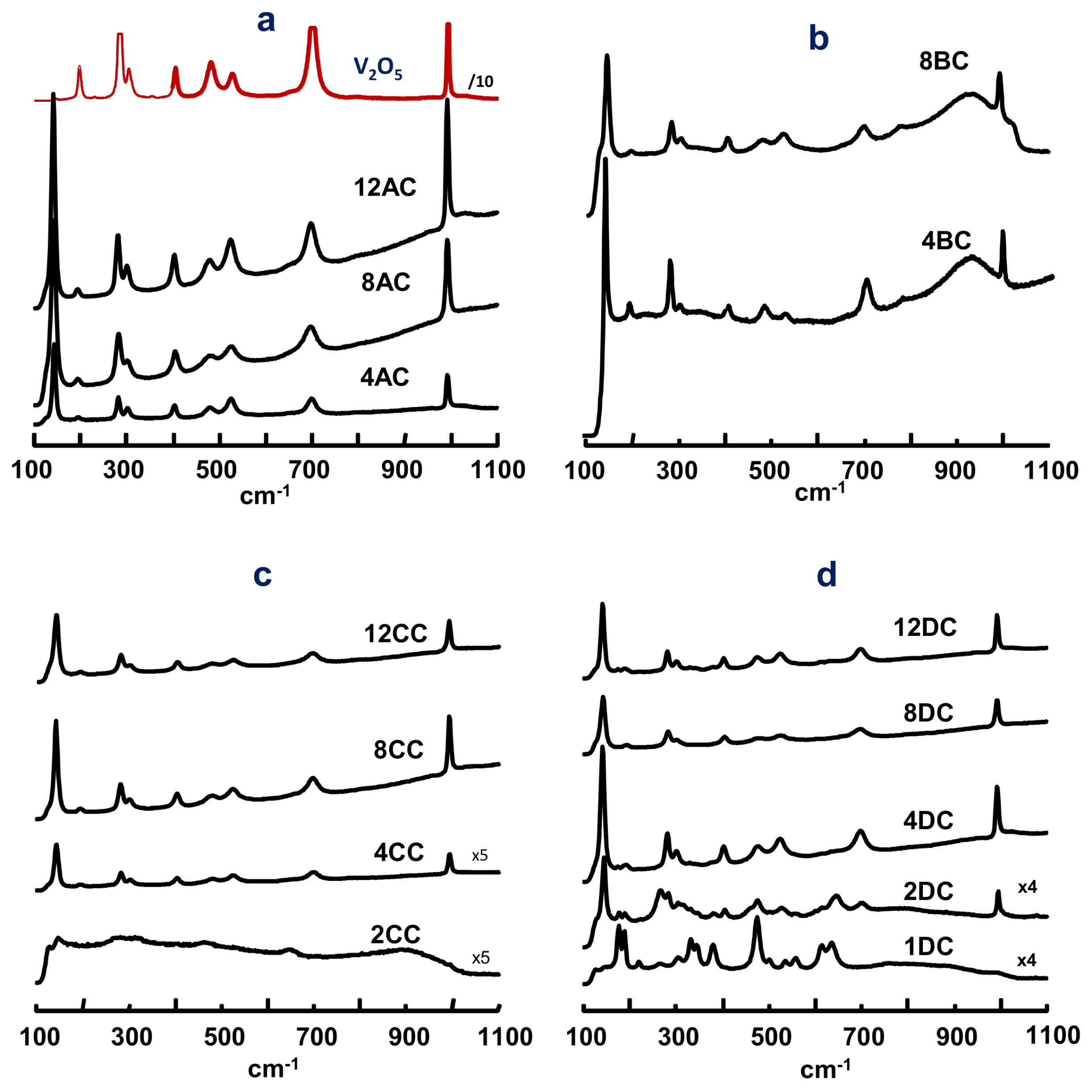
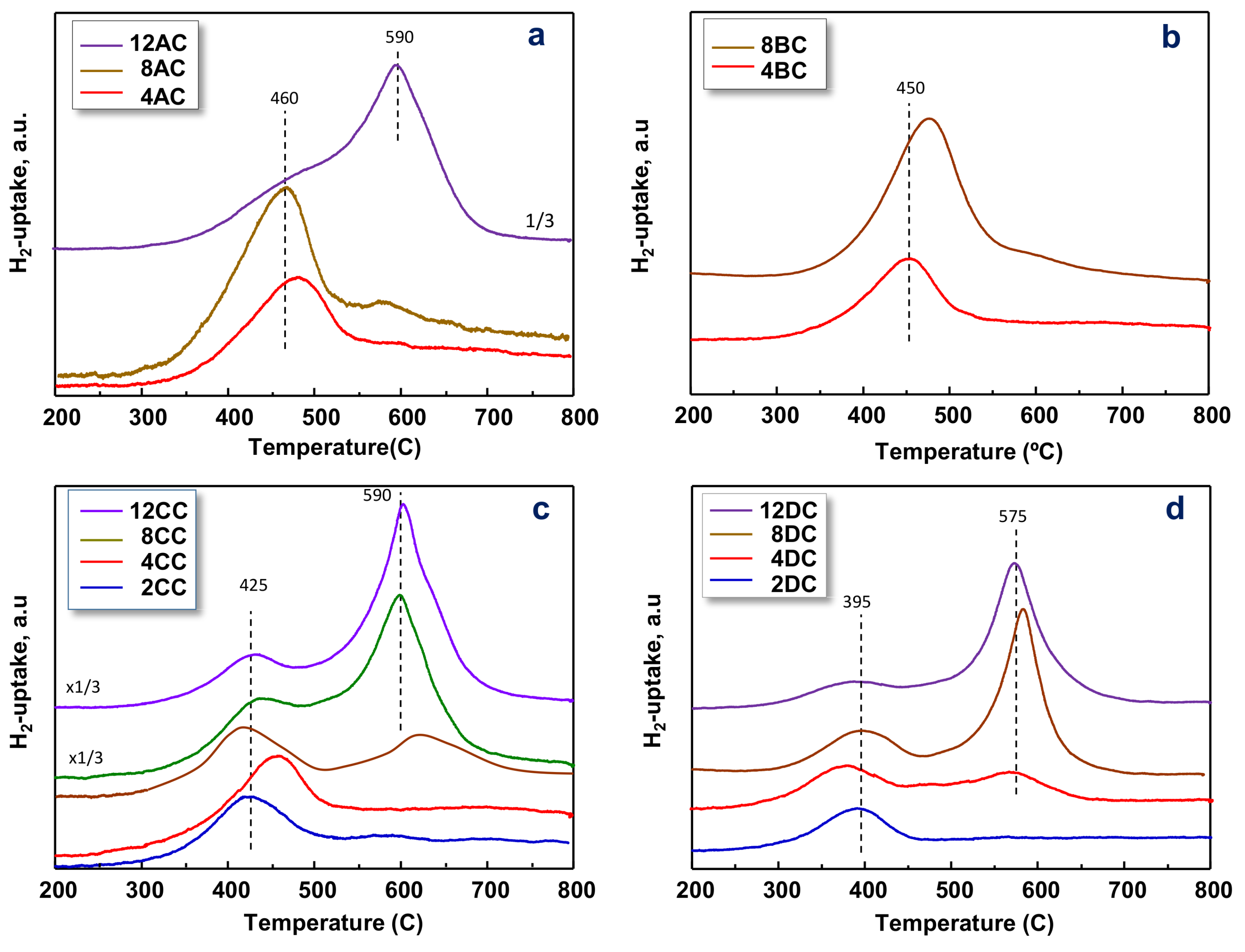
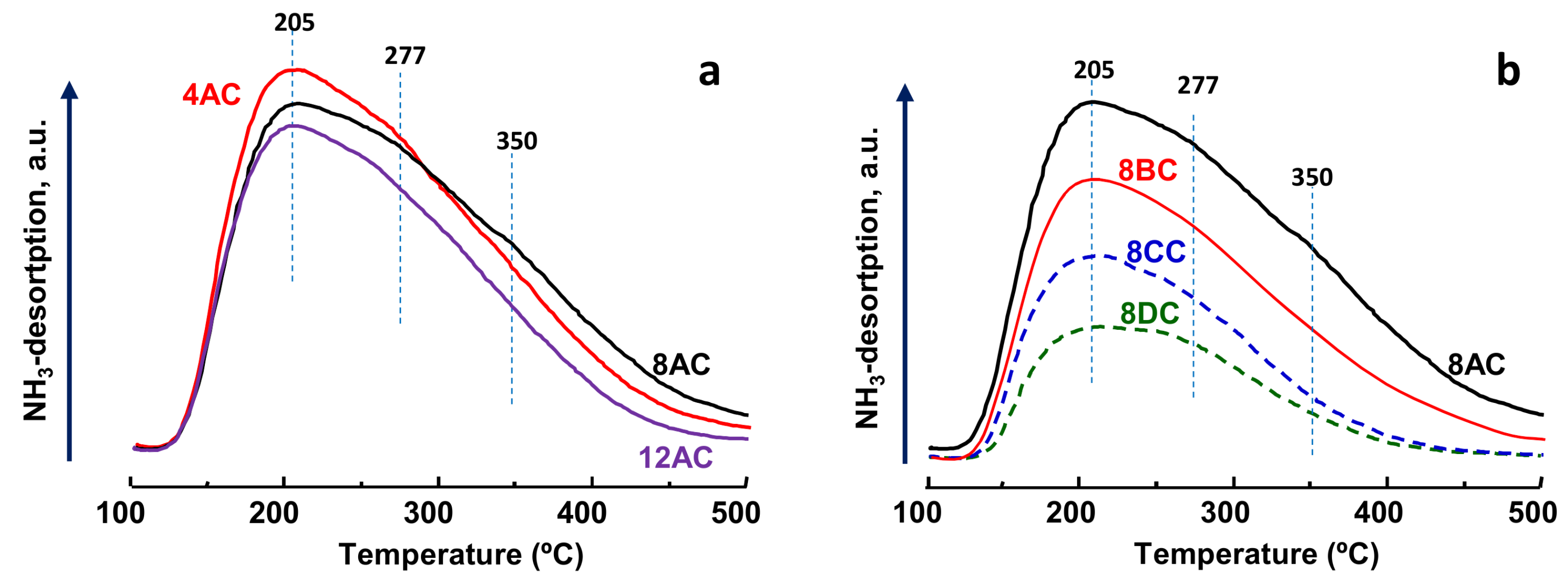
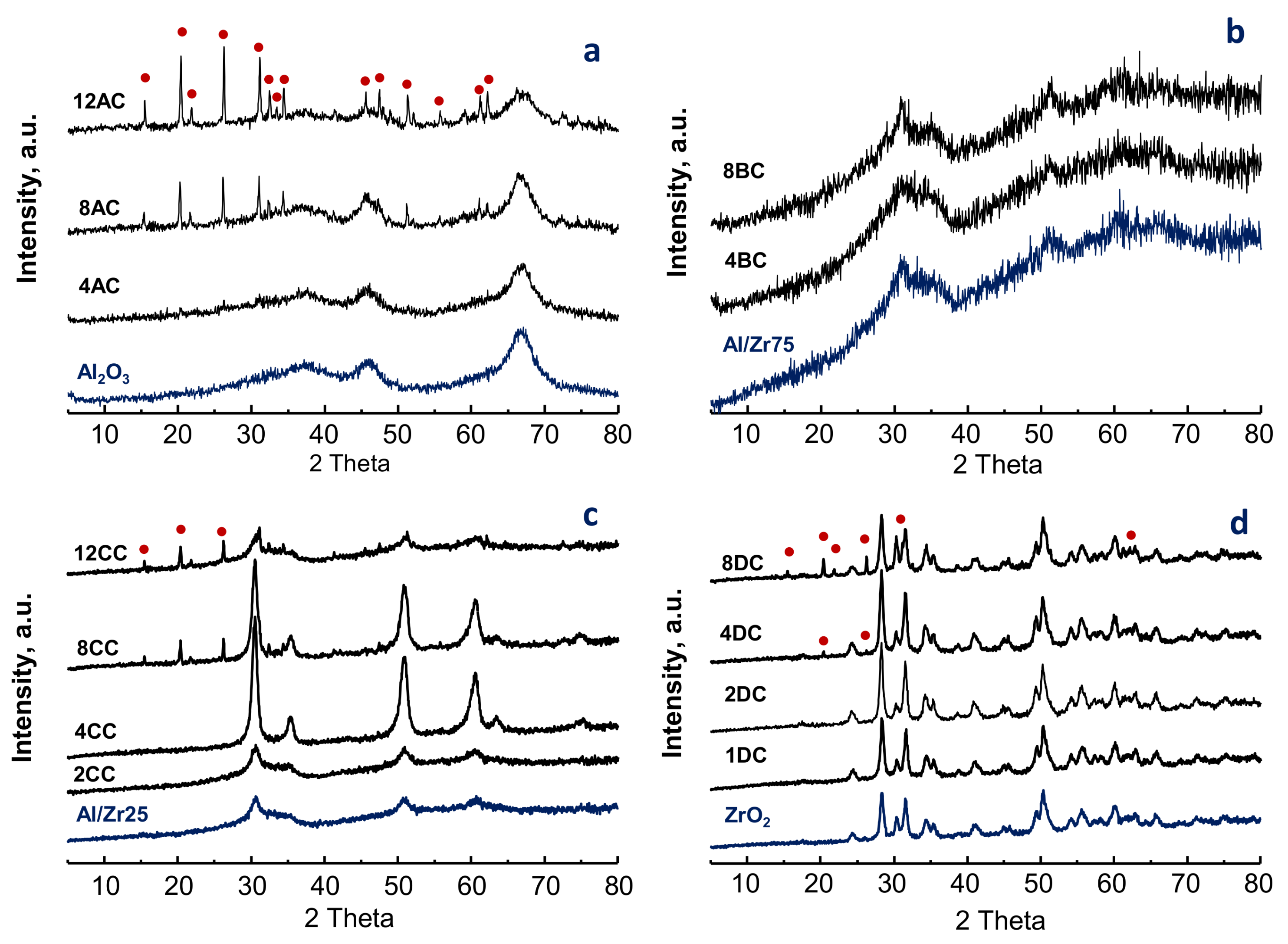
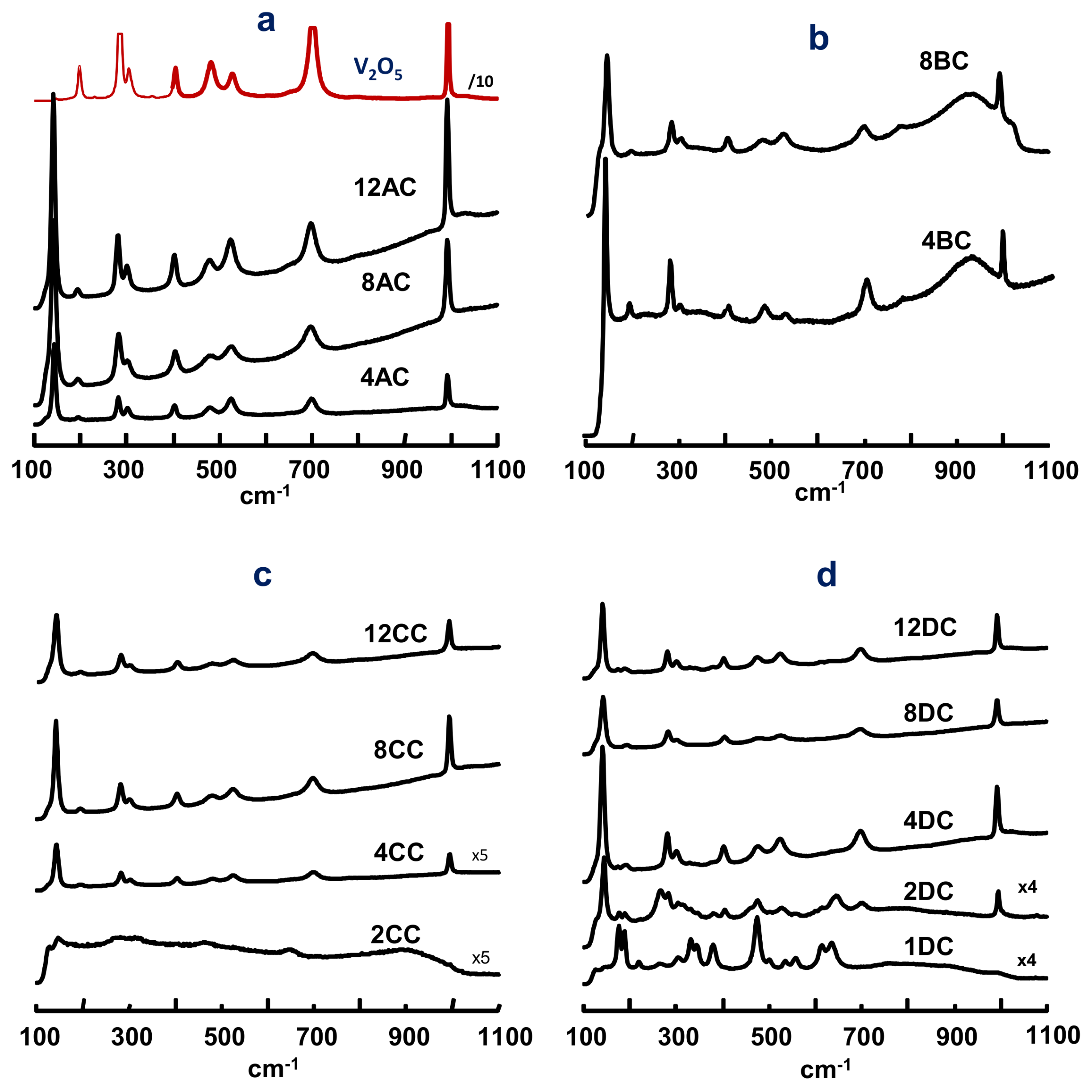
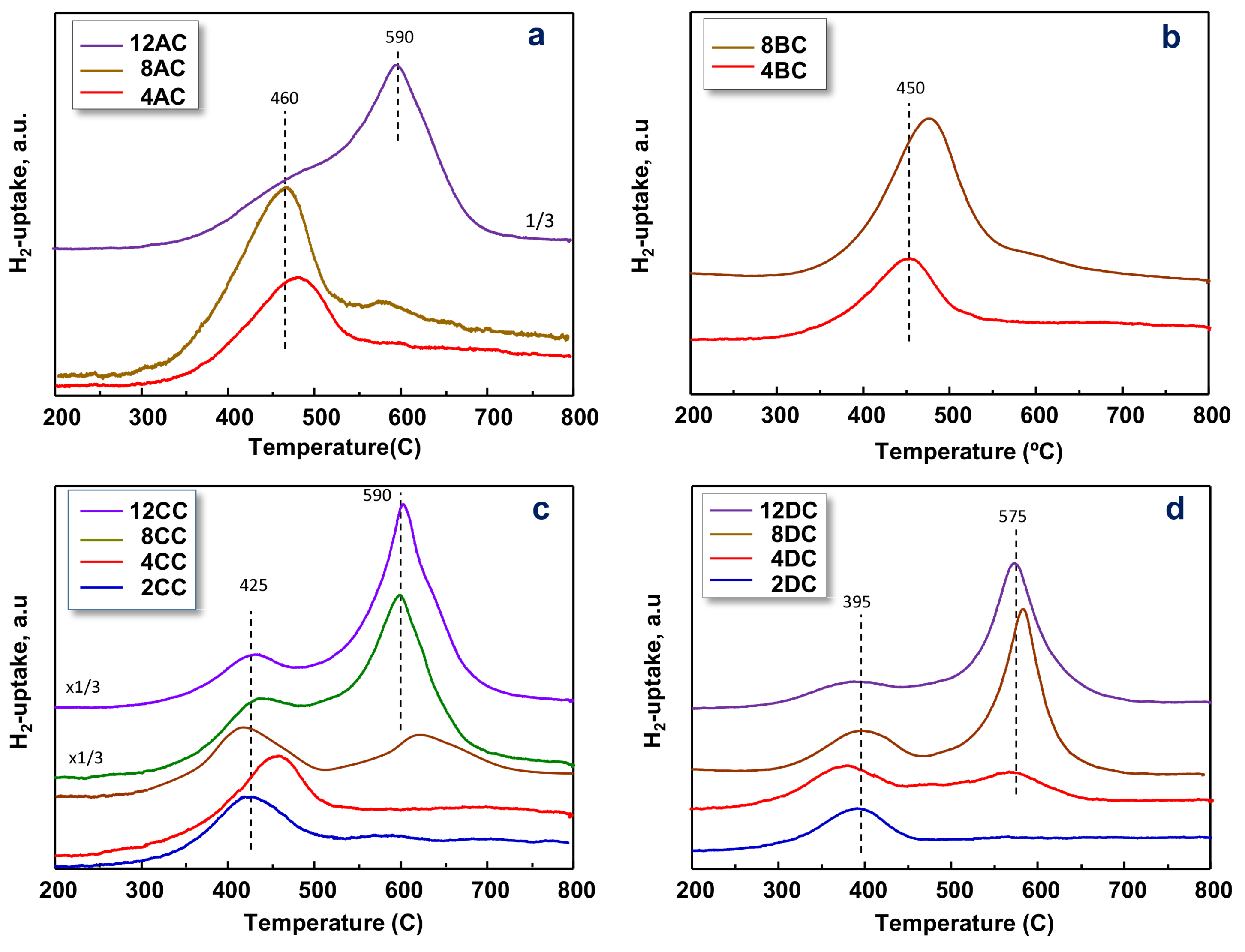
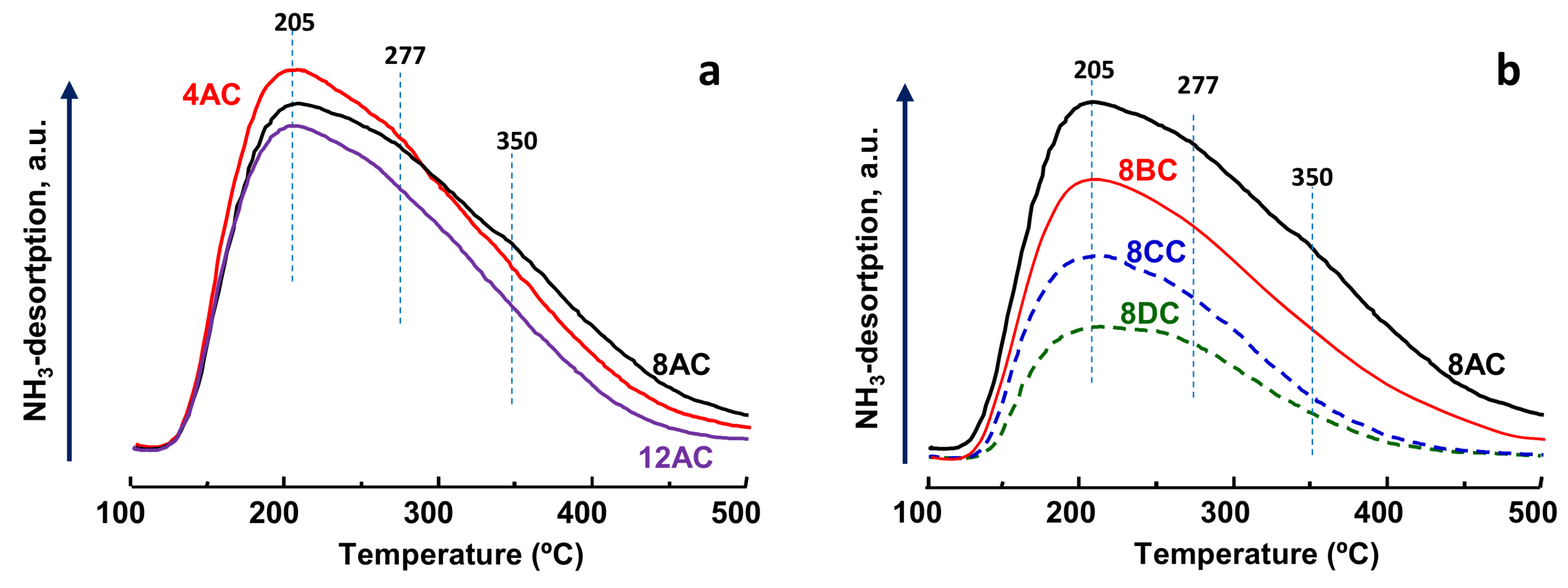
2.1. Characterization of Catalysts
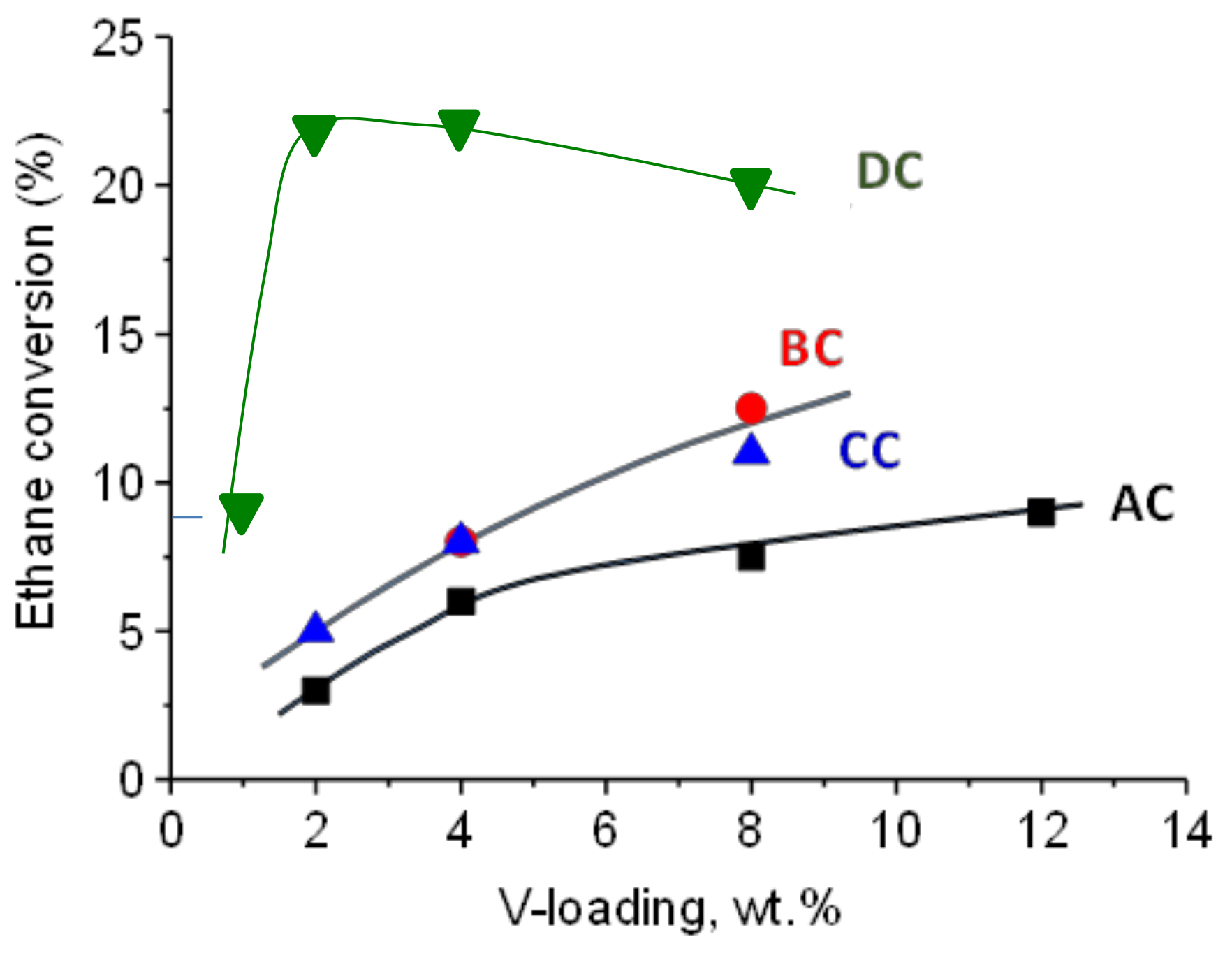
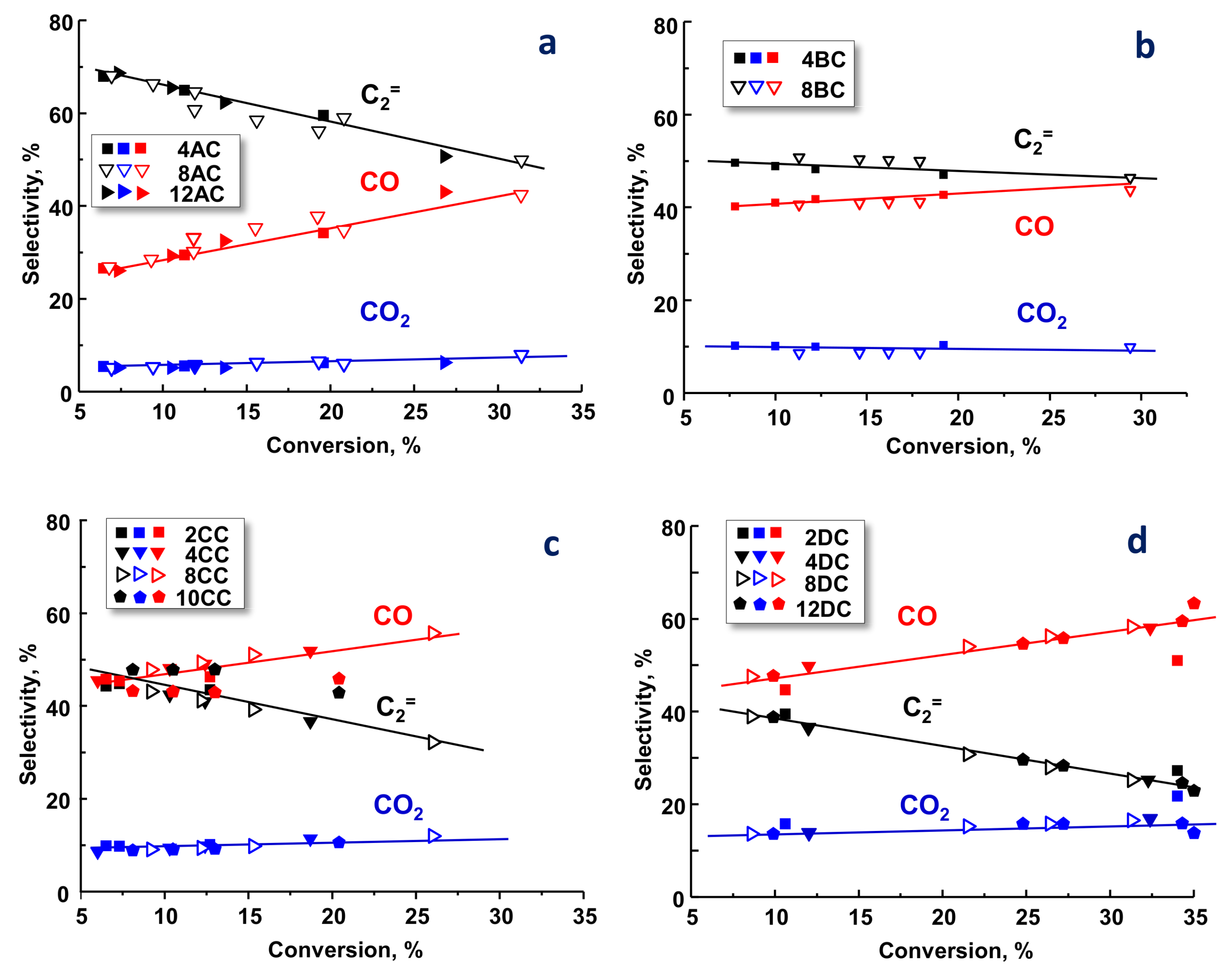
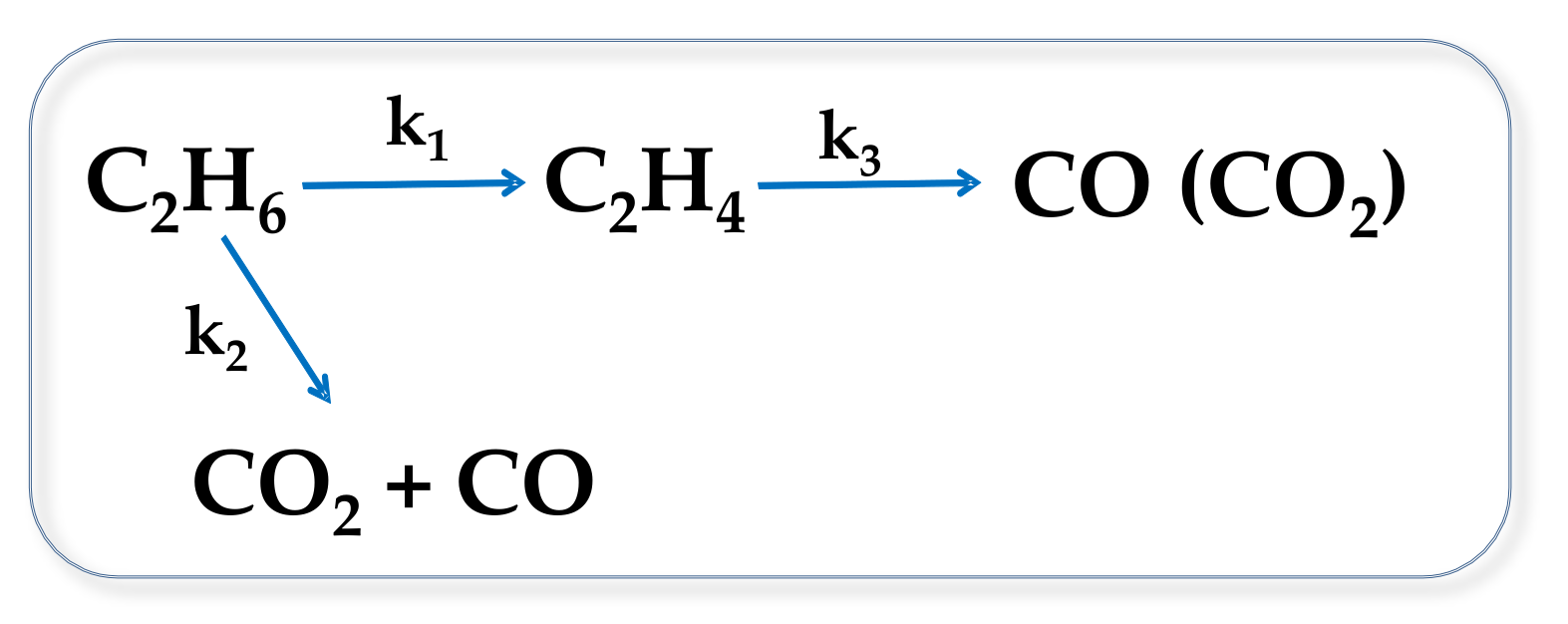
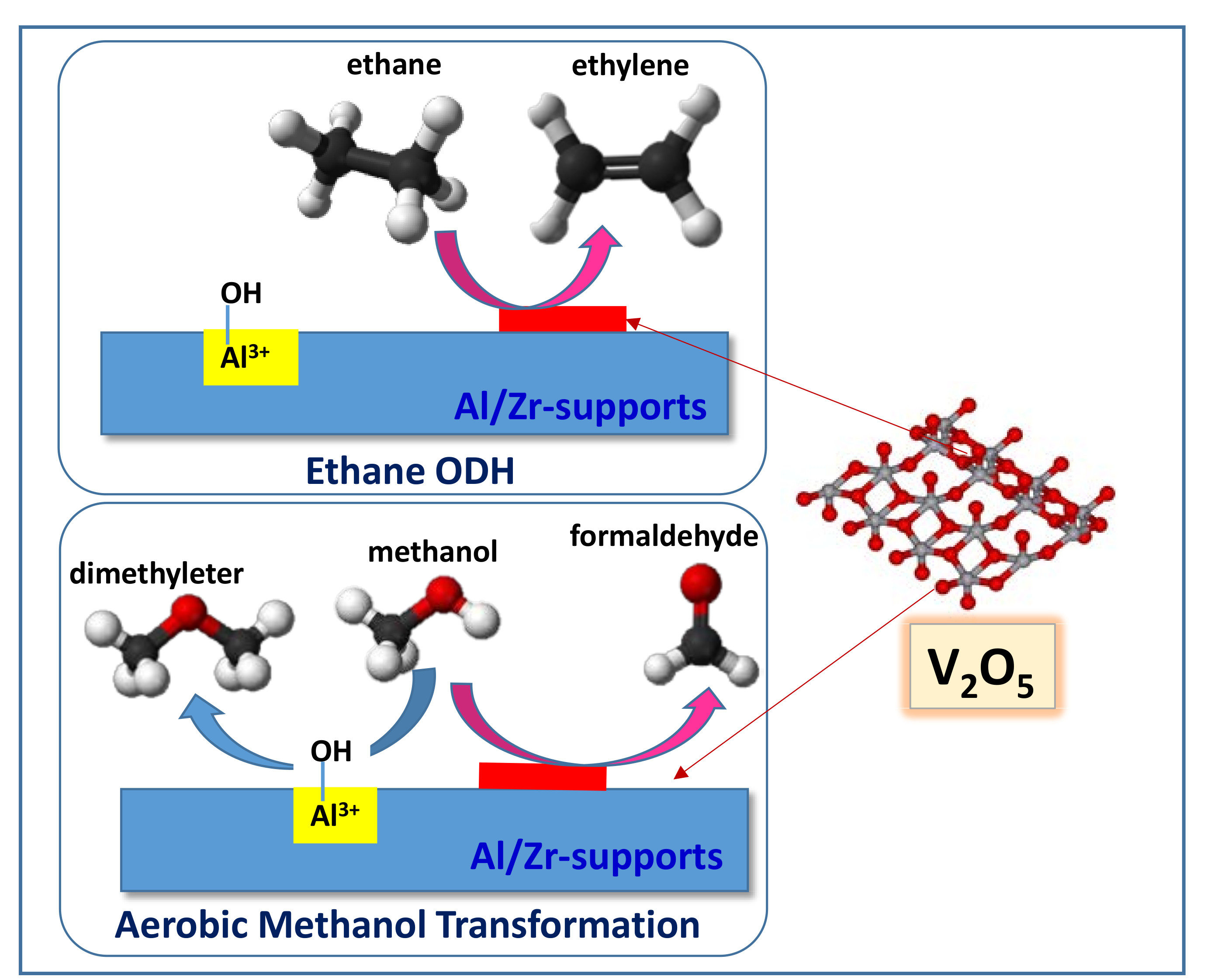
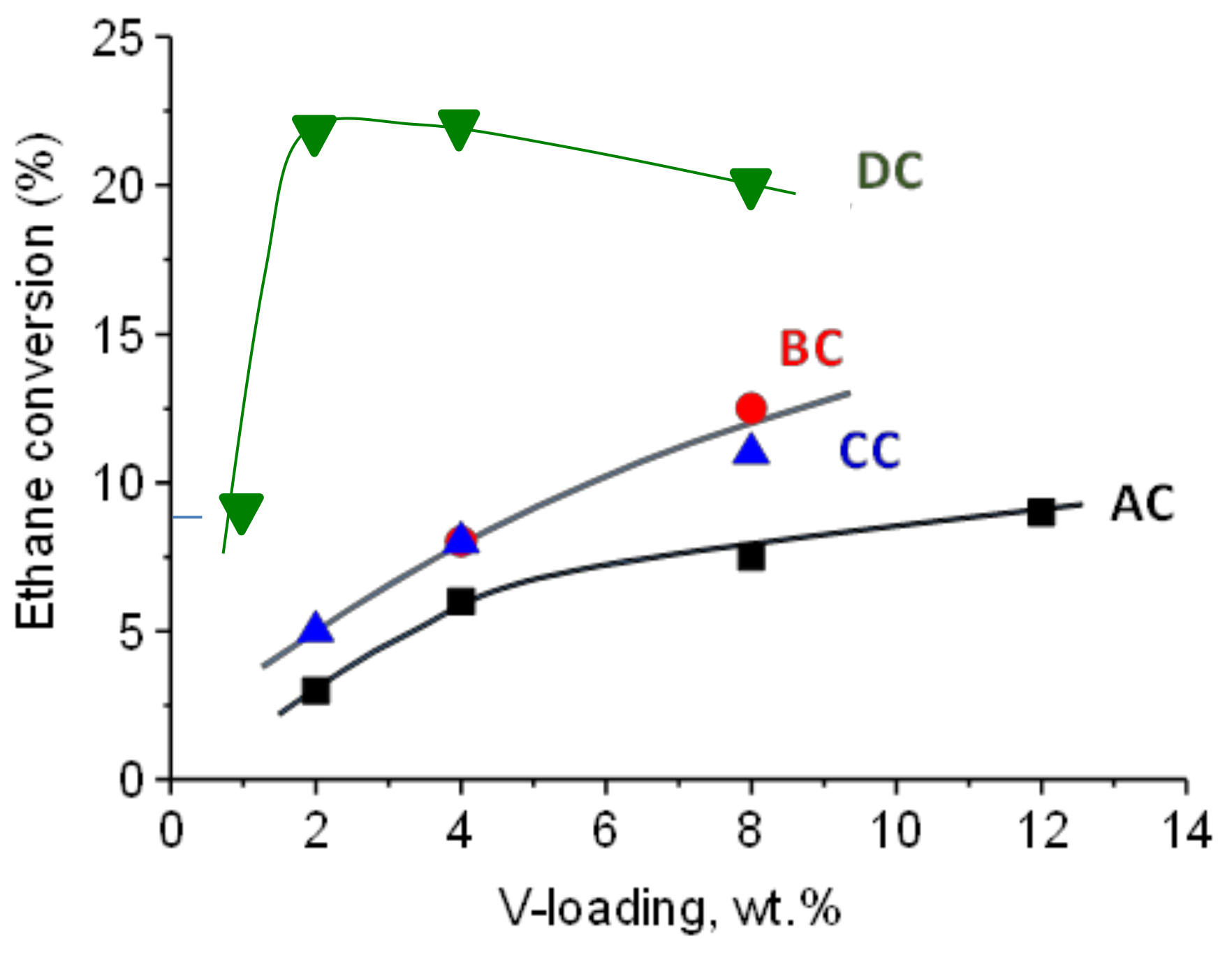
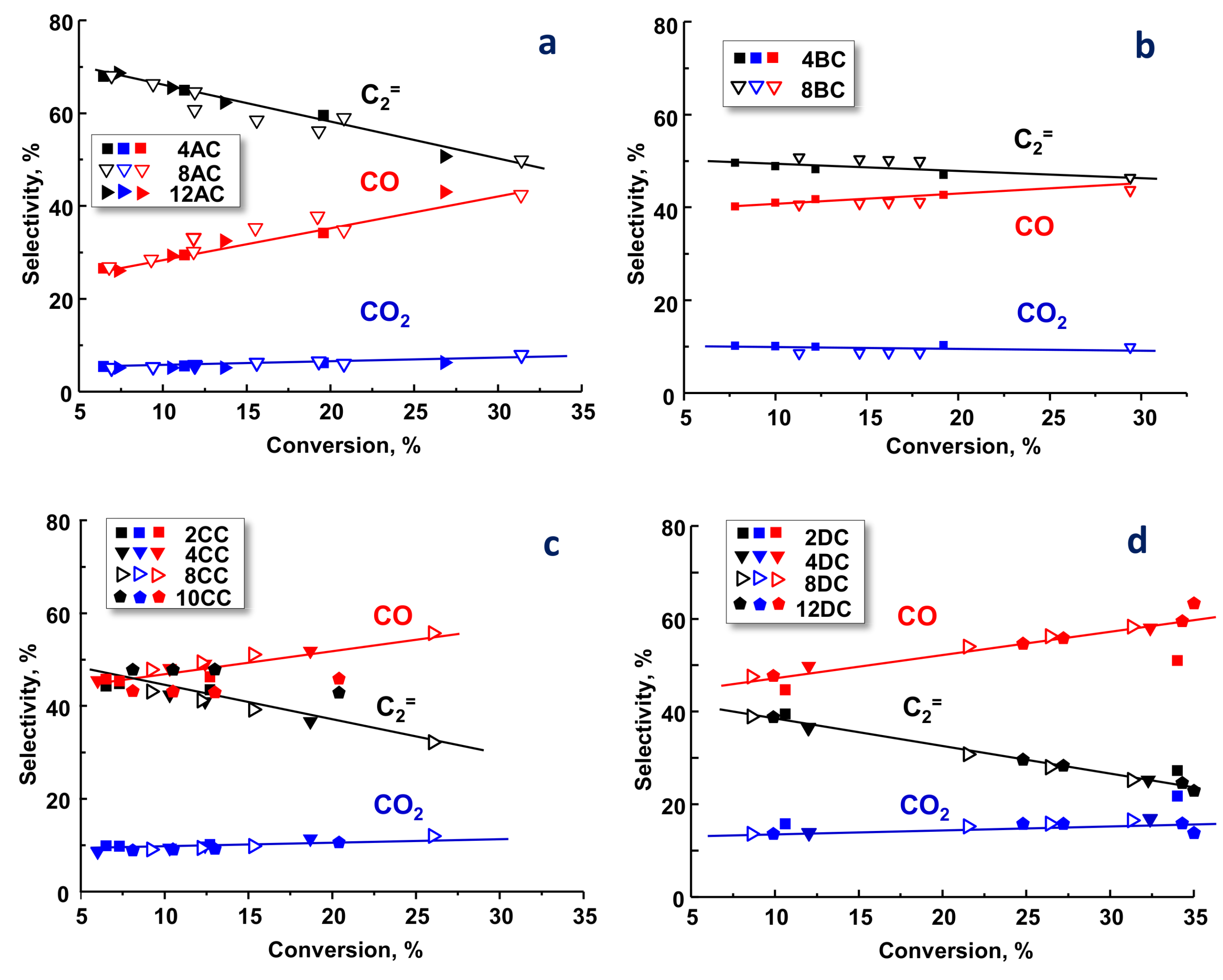
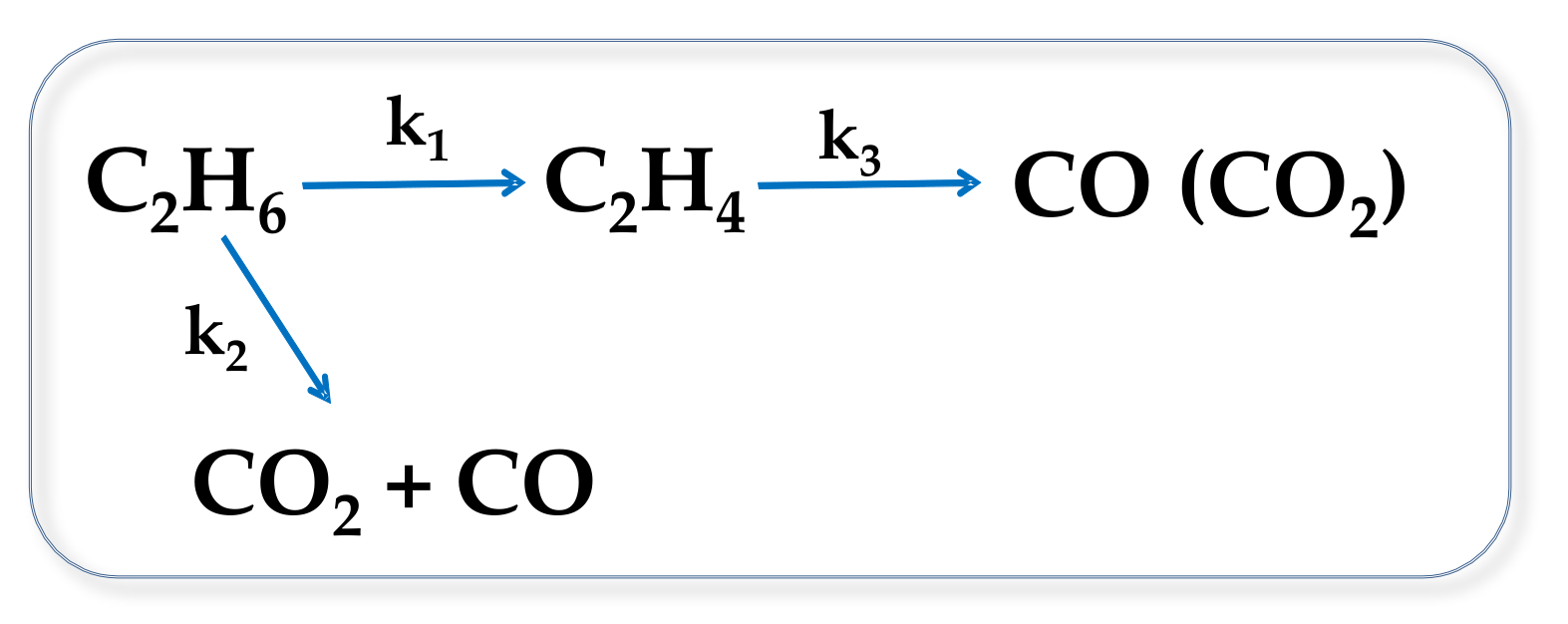
2.2. Catalytic Results on ODH of Ethane
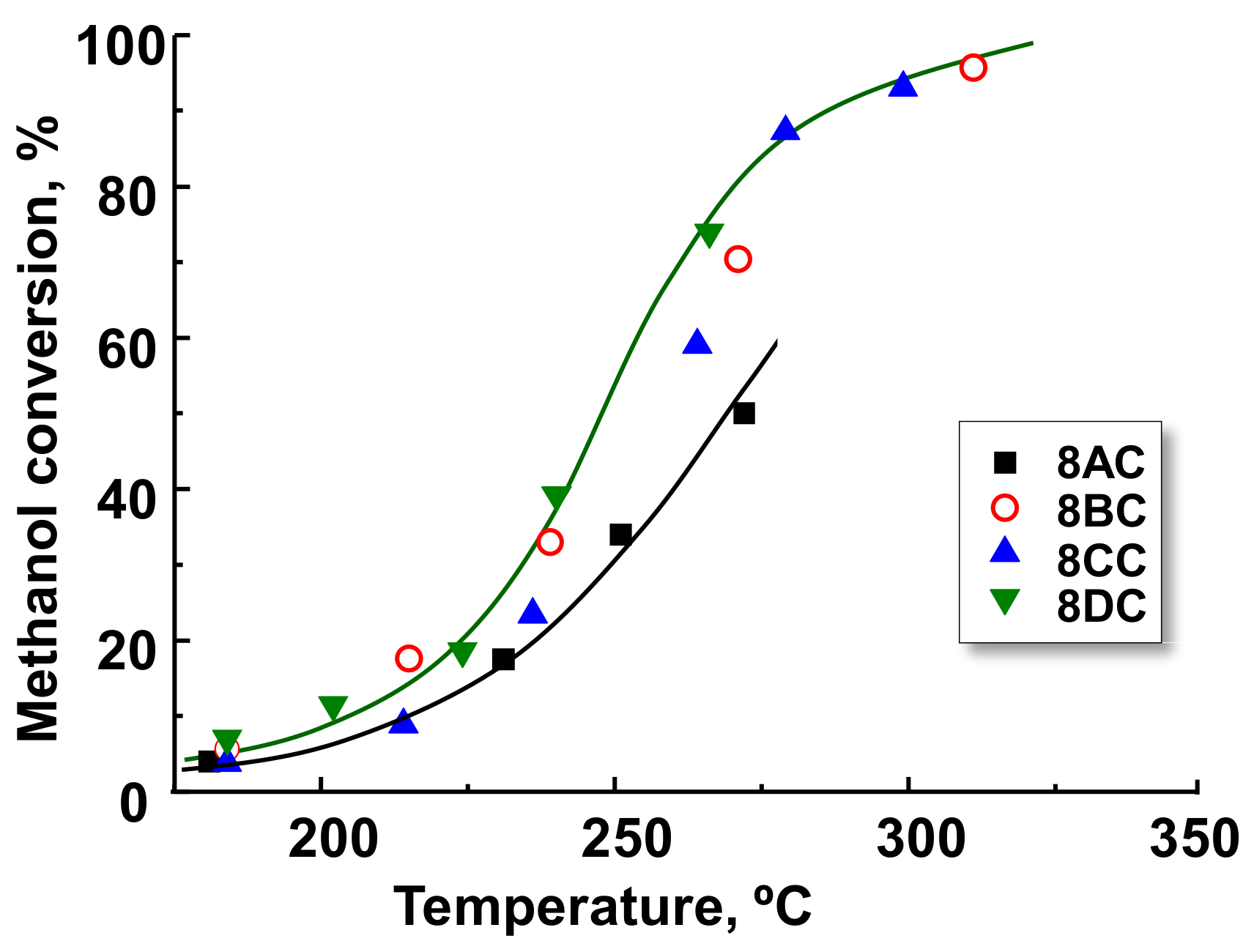
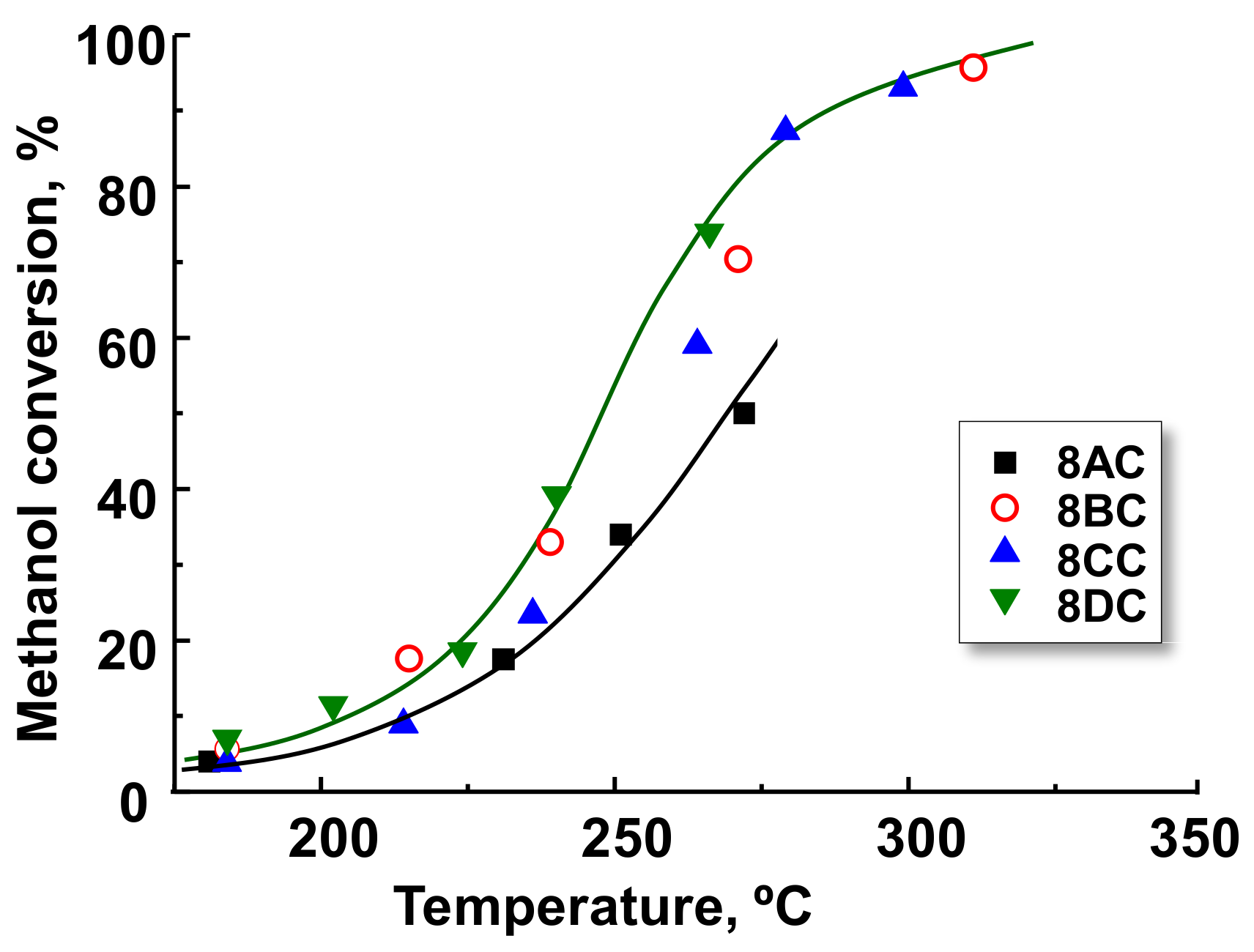
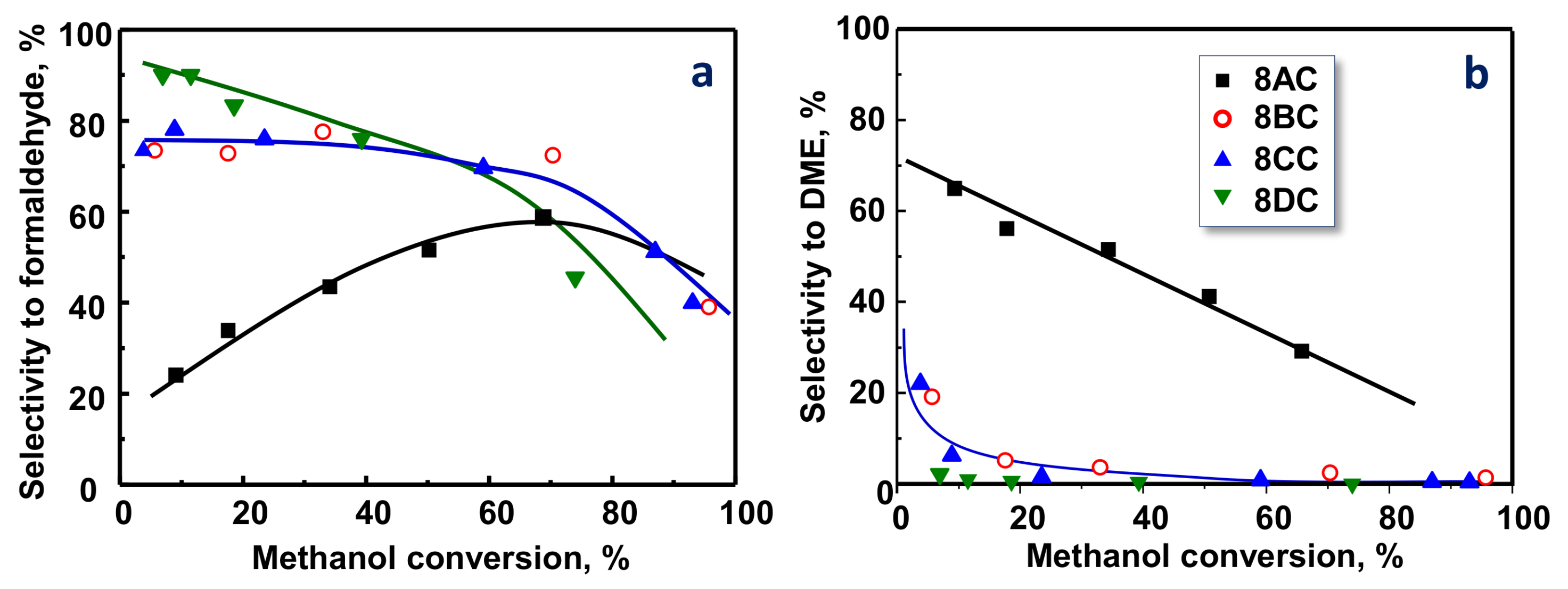
2.3. Selective Aerobic Transformation of Methanol
2.4. General Remarks
3. Materials and Methods
3.1. Preparation of Metal Oxide Support
3.2. Preparation of Catalyst
3.3. Characterization Techniques
3.4. Catalytic Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chieregato, A.; López Nieto, J.M.; Cavani, F. Mixed oxides catalysts with vanadium as the key element for gas phase reactions. Coord. Chem. Rev. 2015, 301–302, 3–23. [Google Scholar] [CrossRef]
- López Nieto, J.M. Selective oxidative activation of light alkanes. From supported vanadia to multicomponent bulk V-containing catalysts. Top. Catal. 2006, 41, 3–15. [Google Scholar] [CrossRef]
- Wachs, I.E. Catalysis science of supported vanadium oxide catalysts. Dalton Trans. 2013, 42, 11762–11769. [Google Scholar] [CrossRef] [PubMed]
- James, O.O.; Mandal, S.; Alele, N.; Chowdhury, B.; Maity, S. Lower alkanes dehydrogenation: Strategies and reaction routes to corresponding alkenes. Fuel Process. Technol. 2016, 149, 239–255. [Google Scholar] [CrossRef]
- Blasco, T.; López Nieto, J.M. Oxidative dehydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A Gen. 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Kung, H.H.; Kung, M.C. Oxidative dehydrogenation of alkanes over vanadium-magnesium-oxides. Appl. Catal. A Gen. 1997, 157, 105–116. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiró, F. Some aspects that affect the selective oxidation of paraffins. Catal. Today 1997, 36, 431–439. [Google Scholar] [CrossRef]
- Banares, M.A. Supported metal oxide and other catalysts for ethane conversion: A review. Catal. Today 1999, 51, 319–348. [Google Scholar] [CrossRef]
- Bhasin, M.M.; McCain, J.H.; Vora, B.V.; Imai, T.; Pujado, P.R. Dehydrogenation and oxydehydrogenation of paraffins to olefins. Appl. Catal. A Gen. 2001, 221, 397–419. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane. How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Gartner, C.A.; Van Veen, A.C.; Lercher, J.A. Oxidative dehydrogenation of ethane: Common principles and mechanistic aspects. ChemCatChem 2013, 5, 3196–3217. [Google Scholar] [CrossRef]
- Galli, A.; Lopez Nieto, J.M.; Dejoz, A.; Vazquez, M.I. The effect of potassium on the selective oxidation of n-butane and ethane over Al2O3-supported vanadia catalysts. Catal. Lett. 1995, 34, 51–58. [Google Scholar] [CrossRef]
- Argyle, M.D.; Chen, K.; Bell, A.T.; Iglesia, E. Ethane oxidative dehydrogenation pathways on vanadium oxide catalysts. J. Phys. Chem. B 2002, 106, 5421–5427. [Google Scholar] [CrossRef]
- Dinse, A.; Ozarowski, A.; Hess, Ch.; Schomacker, R.; Dinse, K.P. Potential of high frequency EPR for investigation of supported vanadium oxide. J. Phys. Chem. C 2008, 112, 17664–17671. [Google Scholar] [CrossRef]
- Chen, K.D.; Bell, A.T.; Iglesia, E. The relationship between the electronic and redox properties of dispersed metal oxides and their turnover rates in oxidative dehydrogenation reactions. J. Catal. 2002, 209, 35–42. [Google Scholar] [CrossRef]
- Lopez Nieto, J.M.; Soler, J.; Concepcion, P.; Herguido, J.; Menendez, M.; Santamaria, J. Oxidative dehydrogenation of alkanes over V-based catalysts: Influence of redox properties on catalytic performance. J. Catal. 1999, 185, 324–332. [Google Scholar] [CrossRef]
- Argyle, M.D.; Chen, K.; Iglesia, E.; Bell, A.T. In situ UV-visible spectroscopic measurement of kinetic parameters and active sites for catalytic oxidation of alkanes on vanadium oxides. J. Phys. Chem. B 2005, 109, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Khodakov, A.; Yang, J.; Bell, A.T.; Iglesia, E. Isotopic tracer kinetic studies of oxidative dehydrogenation pathways on vanadium oxide catalysts. J. Catal. 1999, 186, 325–333. [Google Scholar] [CrossRef]
- Mul, G.; Banares, M.A.; Garcia Cortez, G.; Linden, B.; Khatib, S.J.; Moulijn, J.A. Multitrack and operando Raman G-C study of oxidative dehydrogenation of propane over alumina-supported vanadium oxide catalysts. Phys. Chem. Chem. Phys. 2003, 5, 4378–4383. [Google Scholar] [CrossRef]
- Olthof, B.; Khodakov, A.; Bell, A.T.; Iglesia, E. Effects of support composition and pretreatment conditions on the structure of vanadia dispersed on SiO2, Al2O3, TiO2, ZrO2 and HfO2. J. Phys. Chem. B 2000, 104, 1516–1528. [Google Scholar] [CrossRef]
- Blasco, T.; Galli, A.; Lopez Nieto, J.M.; Trifiro, F. Oxidative dehydrogenation of ethane and n-butane on VOx/Al2O3 catalysts. J. Catal. 1997, 169, 203–211. [Google Scholar] [CrossRef]
- Lin, X.; Hoel, C.A.; Sachtler, W.M.H.; Poeppelmeier, K.R.; Weitz, E. Oxidative dehydrogenation (ODH) of ethane with O2 as oxidant on selected transition metal-loaded zeolites. J. Catal. 2009, 265, 54–62. [Google Scholar] [CrossRef]
- Al-Ghamdi, S.A.; De Lasa, H. Propylene production via propane oxidative dehydrogenation over VOx/γ-Al2O3 catalyst. Fuel 2014, 128, 120–140. [Google Scholar] [CrossRef]
- Solsona, B.; Dejoz, A.; Garcia, T.; Concepción, P.; Lopez Nieto, J.M.; Vázquez, M.I.; Navarro, M.T. Molybdenum-vanadium supported on mesoporous alumina catalysts for the oxidative dehydrogenation of ethane. Catal. Today 2006, 117, 228–233. [Google Scholar] [CrossRef]
- Chen, S.; Ma, F.; Xu, A.; Wang, L.; Chen, F.; Lu, W. Study on the structure, acidic properties of V-Zr nanocrystal catalysts in the oxidative dehydrogenation of propane. Appl. Surf. Sci. 2014, 289, 316–325. [Google Scholar] [CrossRef]
- Elbadawi, A.H.; Ba-Shammakh, M.S.; Al-Ghamdi, S.; Razzak, S.A.; Hossain, M.M. Reduction kinetics and catalytic activity of VOx/γ-Al2O3-ZrO2 for gas phase oxygen free ODH of ethane. Chem. Eng. J. 2016, 284, 448–457. [Google Scholar] [CrossRef]
- Rostom, S.; de Lasa, H. Propane Oxidative Dehydrogenation Using Consecutive Feed Injections and Fluidizable VOx/γAl2O3 and VOx/ZrO2−γAl2O3 Catalysts. Ind. Eng. Chem. Res. 2017, 56, 13109–13124. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A.A. Ni−Nb−O Mixed Oxides as Highly Active and Selective Catalysts for Ethene Production via Ethane Oxidative Dehydrogenation. Part I: Characterization and Catalytic Performance. J. Catal. 2006, 237, 162–174. [Google Scholar] [CrossRef]
- Skoufa, Z.; Heracleous, E.; Lemonidou, A.A. Mechanism and Nature of Active Sites over NiO-Based Catalysts via Isotopic Labeling and Methanol Sorption Studies. J. Catal. 2015, 322, 118–129. [Google Scholar] [CrossRef]
- Ipsakis, D.; Heracleous, E.; Silvester, L.; Bukur, D.B.; Lemonidou, A.A. Reduction and Oxidation Kinetic Modeling of NiO-based Oxygen Transfer Materials. Chem. Eng. J. 2017, 308, 840–852. [Google Scholar] [CrossRef]
- Delgado, D.; Solsona, B.; Ykrelef, A.; Rodriguez-Gomez, A.; Caballero, A.; Rodriguez-Aguado, E.; Rodriguez-Castellon, E.; Lopez Nieto, J.M. Redox and Catalytic Properties of Promoted NiO Catalysts for the Oxidative Dehydrogenation of Ethane. J. Phys. Chem. C 2017, 121, 25132–25142. [Google Scholar] [CrossRef]
- Solsona, B.; Concepcion, P.; Lopez Nieto, J.M.; Dejoz, A.; Cecilia, J.A.; Agouram, S.; Soriano, M.D.; Torres, V.; Jimenez-Jimenez, J.; Rodriguez Castellon, E. Nickel Oxide Supported on Porous Clay Heterostructures as Selective Catalysts for the Oxidative Dehydrogenation of Ethane. Catal. Sci. Technol. 2016, 6, 3419–3429. [Google Scholar] [CrossRef]
- Lopez Nieto, J.M.; Botella, P.; Vazquez, M.I.; Dejoz, A. The selective Oxidative Dehydrogenation of Ethane over Hydrothermally Synthesized MoVTeNb Catalysts. Chem. Commun. 2002, 1906–1907. [Google Scholar] [CrossRef]
- Lopez Nieto, J.M.; Botella, P.; Vazquez, M.I.; Dejoz, A. Method for the Oxidative Dehydrogenation of Ethane. US Patent 7,319,179 B2, 15 January 2008. [Google Scholar]
- Gartnet, C.; Van Veen, A.C.; Lercher, J.A. Oxidative Dehydrogenation of Ethane on Dynamically Rearranging Supported Chloride Catalysts. J. Am. Chem. Soc. 2014, 136, 12691–12701. [Google Scholar] [CrossRef] [PubMed]
- Tatibouet, J.M. Methanol oxidation as a catalytic surface probe. Appl. Catal. A Gen. 1997, 148, 213–252. [Google Scholar] [CrossRef]
- Forzatti, P.; Tronconi, E.; Elmi, A.S.; Busca, G. Methanol oxidation over vanadia-based catalysts. Appl. Catal. A Gen. 1997, 157, 387–408. [Google Scholar] [CrossRef]
- Wachs, I.E.; Chen, Y.; Jehng, J.M.; Briand, L.E.; Tanaka, T. Molecular structure and reactivity of the group V metal oxides. Catal. Today 2003, 78, 13–24. [Google Scholar] [CrossRef]
- Shah, P.R.; Baldychev, I.; Vohs, J.M.; Gorte, R.J. Comparison of redox isotherms for vanadia supported on zirconia and titania. Appl. Catal. A Gen. 2009, 361, 13–17. [Google Scholar] [CrossRef]
- Baldychev, I.; Gorte, R.J.; Vohs, J.M. The impact of redox properties on the reactivity of V2O5/Al2O3 catalysts. J. Catal. 2010, 269, 397–403. [Google Scholar] [CrossRef]
- Hess, C. Nanostructured vanadium oxide model catalysts for selective oxidation reactions. Chem. Phys. Chem. 2009, 10, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Zoelle, A.; Yang, Y.; Rioux, R.M.; Hamilton, N.G.; Amakawa, K.; Nielsen, P.K.; Trunschke, A. Surface roughness effects in the catalytic behavior of vanadia supported on SBA-15. J. Catal. 2014, 312, 170–178. [Google Scholar] [CrossRef]
- Wang, N.L.; Qiu, J.E.; Wu, J.; You, K.Y.; Luo, H.A. A comparison of the redox properties of bulk vanadium mixed oxide catalysts. Catal. Lett. 2015, 145, 1792–1797. [Google Scholar] [CrossRef]
- Beck, B.; Harth, M.; Hamilton, N.G.; Carrero, C.; Uhlrich, J.J.; Trunschke, A.; Shaikhutdinov, S.; Schubert, H.; Freund, H.J.; Schlögl, R.; et al. Partial oxidation of ethanol on vanadia catalysts on supporting oxides with different redox properties compare to propane. J. Catal. 2012, 296, 120–131. [Google Scholar] [CrossRef]
- Kim, T.; Wachs, I.E. CH3OH oxidation over well-defined supported V2O5/Al2O3 catalysts. Influence of vanadium oxide loading and surface vanadium-oxygen functionalities. J. Catal. 2008, 255, 197–205. [Google Scholar] [CrossRef]
- Baldychev, I.; Vohs, J.M.; Gorte, R.J. The effect of support on redox properties and methanol-oxidation activity of vanadia catalysts. Appl. Catal. A Gen. 2011, 391, 86–91. [Google Scholar] [CrossRef]
- Soriano, M.D.; Chieregato, A.; Zamora, S.; Basile, F.; Cavani, F.; López Nieto, J.M. Promoted hexagonal tungsten bronzes as selective catalysts in the aerobic transformation of alcohols: Glycerol and methanol. Top. Catal. 2016, 59, 178–185. [Google Scholar] [CrossRef]
- Hu, F.; Wu, X.; Wanga, Y.; Lai, X. Ultrathin γ-Al2O3 nanofibers with large specific surface area and their enhanced thermal stability by Si-doping. RSC Adv. 2015, 5, 54053–54058. [Google Scholar] [CrossRef]
- Zang, F.; Chupas, P.J.; Lui, S.L.A.; Hanson, J.C.; Caliebe, W.A.; Lee, P.L.; Chan, S.W. In situ study in the crystallization from amorphous to cubic zirconium oxide: Rietveld and reverse Monte Carlo Analyses. Chem. Mater. 2007, 19, 3118–3126. [Google Scholar] [CrossRef]
- Pieck, C.L.; Del Val, S.; Lopez Granados, M.; Banares, M.A.; Fierro, J.L.G. Bulk and Surface structures of V2O5/ZrO2 systems and their relevance for o-xylene oxidation. Langmuir 2002, 18, 2642–2648. [Google Scholar] [CrossRef]
- Soriano, M.D.; Rodríguez-Castellón, E.; García-González, E.; López Nieto, J.M. Catalytic behavior of NaV6O15 bronze for partial oxidation of hydrogen sulfide. Catal. Today 2014, 238, 62–68. [Google Scholar] [CrossRef]
- Zhao, C.; Wachs, I.E. Selective oxidation of propylene over model supported V2O5 catalysts. Influence of surface coverage and oxide support. J. Catal. 2008, 257, 181–189. [Google Scholar] [CrossRef]
- Martinez-Huerta, M.V.; Gao, X.; Tian, H.; Wachs, I.E.; Fierro, J.L.G.; Banares, M.A. Oxidative dehydrogenation of ethane to ethylene over alumina-supported vanadium oxide catalysts. Relationship between molecular structure and chemical reactivity. Catal. Today 2006, 118, 279–287. [Google Scholar] [CrossRef]
- Khodakov, A.; Yang, J.; Su, S.; Iglesia, E.; Bell, A.T. Structure and properties of vanadium oxide-zirconia catalysts for propane oxidative dehydrogenation. J. Catal. 1998, 177, 343–351. [Google Scholar] [CrossRef]
- Kanervo, J.M.; Harlin, M.E.; Krause, A.O.I.; Bañares, M.A. Characterisation of alumina-supported vanadium oxide catalysts by kinetic analysis of H2-TPR data. Catal. Today 2003, 78, 171–180. [Google Scholar] [CrossRef]
- Deo, G.; Wachs, I.E. Reactivity of supported vanadium oxide Catalysts: The partial oxidation of methanol. J. Catal. 1994, 146, 232–334. [Google Scholar] [CrossRef]
- Gao, X.; Banares, M.A.; Wachs, I.E. Ethane and n-butane oxidation over supported vanadium oxide catalysts: An in situ UV-visible diffuse reflectance spectroscopic investigation. J. Catal. 1999, 188, 325–331. [Google Scholar] [CrossRef]
- Silversmit, G.; Depla, D.; Poelman, H.; Marin, G.B.; De Gryse, R. Determination of the V2p XPS binding energies for different vanadium oxidation states (V5+ to V0+). J. Electron. Spectrosc. 2004, 135, 167–175. [Google Scholar] [CrossRef]
- Hess, C.; Tzolova-Müller, G.; Herbert, R. The influence of water on the dispersion of vanadia supported on silica SBA-15, a combined XPS and Raman study. J. Phys. Chem. C 2007, 111, 9471–9479. [Google Scholar] [CrossRef]
- Chen, K.; Bell, A.T.; Iglesia, E. Kinetics and mechanism of oxidative dehydrogenation of propane on vanadium, molybdenum and tungsten oxides. J. Phys. Chem. B 2000, 104, 1292–1299. [Google Scholar] [CrossRef]
- Elbadawi, A.H.; Ba-Shammakh, M.S.; Al-Ghamdi, S.; Razzak, S.A.; Hossain, M.M.; de Lasa, H.I. A fluidizable VOx/γ-Al2O3-ZrO2 catalyst for the ODH of ethane to ethylene operating in a gas phase oxygen free environment. Chem. Eng. Sci. 2016, 145, 59–70. [Google Scholar] [CrossRef]
- Solsona, B.; Blasco, T.; Lopez Nieto, J.M.; Peña, M.L.; Rey, F.; Vidal Moya, A. Vanadium oxide supported on mesoporous MCM-41 as selective catalysts in the oxidative dehydrogenation of alkanes. J. Catal. 2001, 203, 443–452. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET | Support | V-Loading | TPR-Experiments | |
|---|---|---|---|---|---|
| (m2/g) | (wt% V-Atoms) b | TMC (°C) c | H2-Uptake (mmolH2/gcat) d | ||
| 4AC | 136 | Al2O3 | 4 (0.53) | 478 | 0.5 |
| 8AC | 159 | Al2O3 | 8 (0.83) | 470 | 0.6 |
| 12AC | 115 | Al2O3 | 12 (1.61) | 597 | 2.0 |
| 4BC | 177 | Al-Zr-O (Al/Zr = 75/25) | 4 (0.41) | 454 | 0.6 |
| 8BC | 143 | Al-Zr-O (Al/Zr = 75/25) | 8 (0.93) | 477 | 3.7 |
| 2CC | 88 | Al-Zr-O (Al/Zr = 25/75) | 2 (0.43) | 461 | 0.5 |
| 4CC | 46 | Al-Zr-O (Al/Zr = 25/75) | 4 (1.57) | 420 | 0.5 |
| 8CC | 45 | Al-Zr-O (Al/Zr = 25/75) | 8 (1.95) | 409/602 | 0.5 |
| 12CC | 70 | Al-Zr-O (Al/Zr = 25/75) | 12 (2.66) | 426/605 | 1.5 |
| 1DC | 34 | ZrO2 | 1 (0.57) | 392 | 0.2 |
| 2DC | 33 | ZrO2 | 2 (1.13) | 391 | 0.3 |
| 4DC | 24 | ZrO2 | 4 (2.85) | 379 | 0.8 |
| 8DC | 28 | ZrO2 | 8 (4.73) | 396/583 | 2.2 |
| 12DC | 23 | ZrO2 | 12 (8.11) | 573 | 1.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benomar, S.; Massó, A.; Solsona, B.; Issaadi, R.; López Nieto, J.M. Vanadium Supported on Alumina and/or Zirconia Catalysts for the Selective Transformation of Ethane and Methanol. Catalysts 2018, 8, 126. https://doi.org/10.3390/catal8040126
Benomar S, Massó A, Solsona B, Issaadi R, López Nieto JM. Vanadium Supported on Alumina and/or Zirconia Catalysts for the Selective Transformation of Ethane and Methanol. Catalysts. 2018; 8(4):126. https://doi.org/10.3390/catal8040126
Chicago/Turabian StyleBenomar, Souhila, Amada Massó, Benjamín Solsona, Rachid Issaadi, and Jose M. López Nieto. 2018. "Vanadium Supported on Alumina and/or Zirconia Catalysts for the Selective Transformation of Ethane and Methanol" Catalysts 8, no. 4: 126. https://doi.org/10.3390/catal8040126
APA StyleBenomar, S., Massó, A., Solsona, B., Issaadi, R., & López Nieto, J. M. (2018). Vanadium Supported on Alumina and/or Zirconia Catalysts for the Selective Transformation of Ethane and Methanol. Catalysts, 8(4), 126. https://doi.org/10.3390/catal8040126