Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane

 ,
,  ,
,  and
and 
Abstract
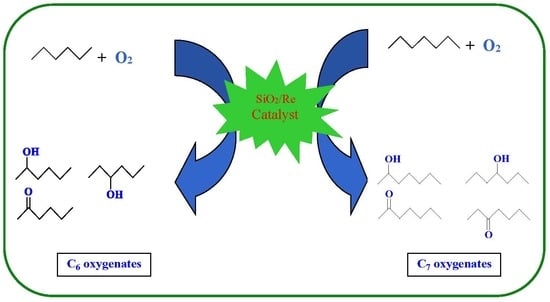
1. Introduction
2. Results
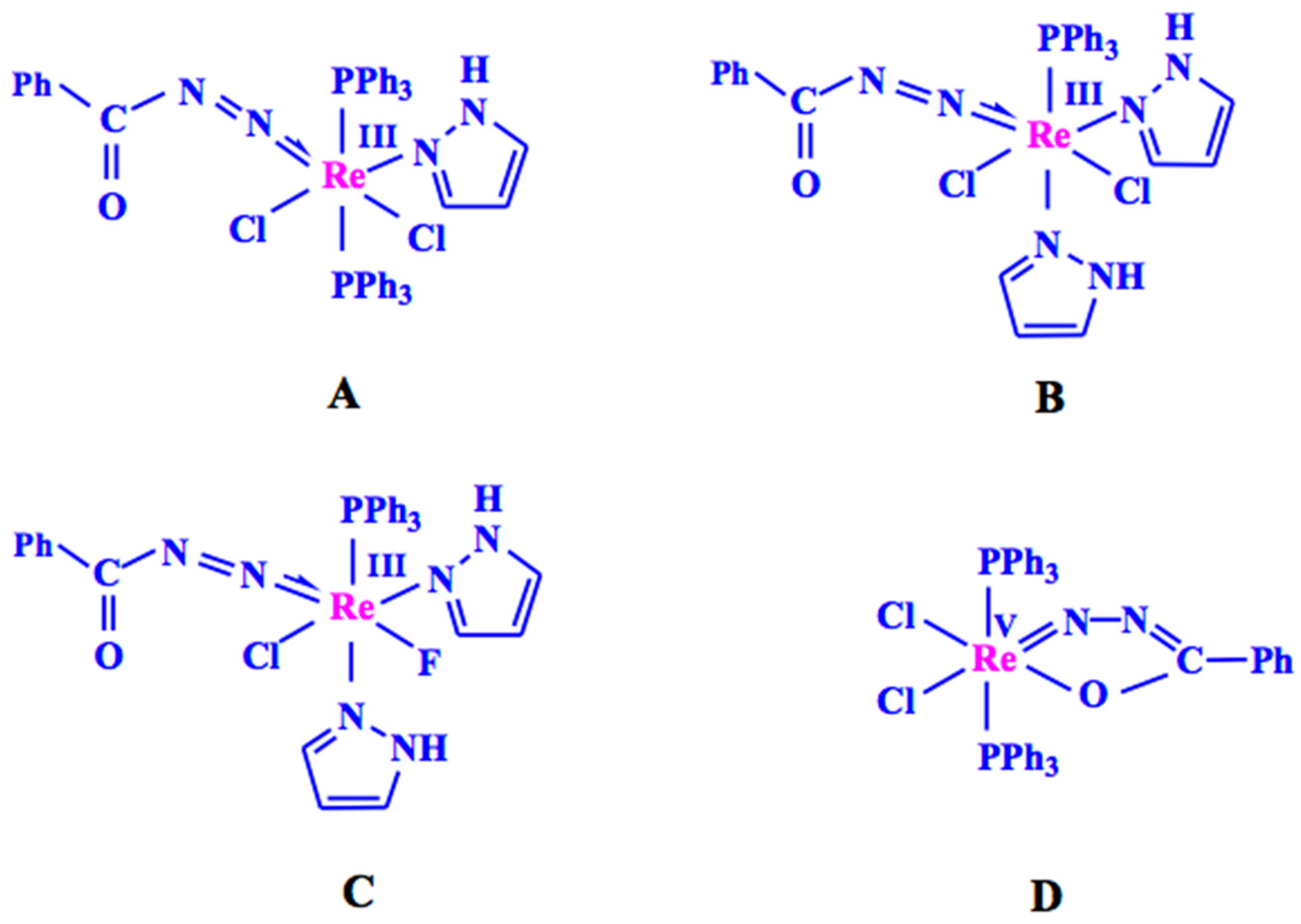
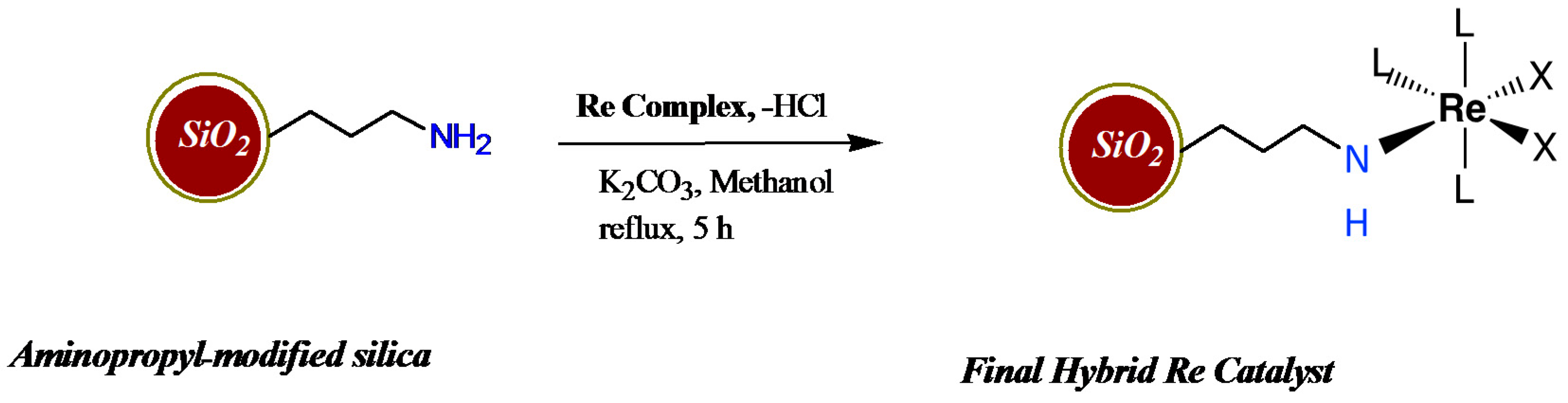
2.1. Catalyst Synthesis and Characterization
2.2. Catalytic Batch Oxidation of Alkanes
2.3. Optimization of Reaction Conditions for the Oxidations
2.3.1. Effect of the Temperature
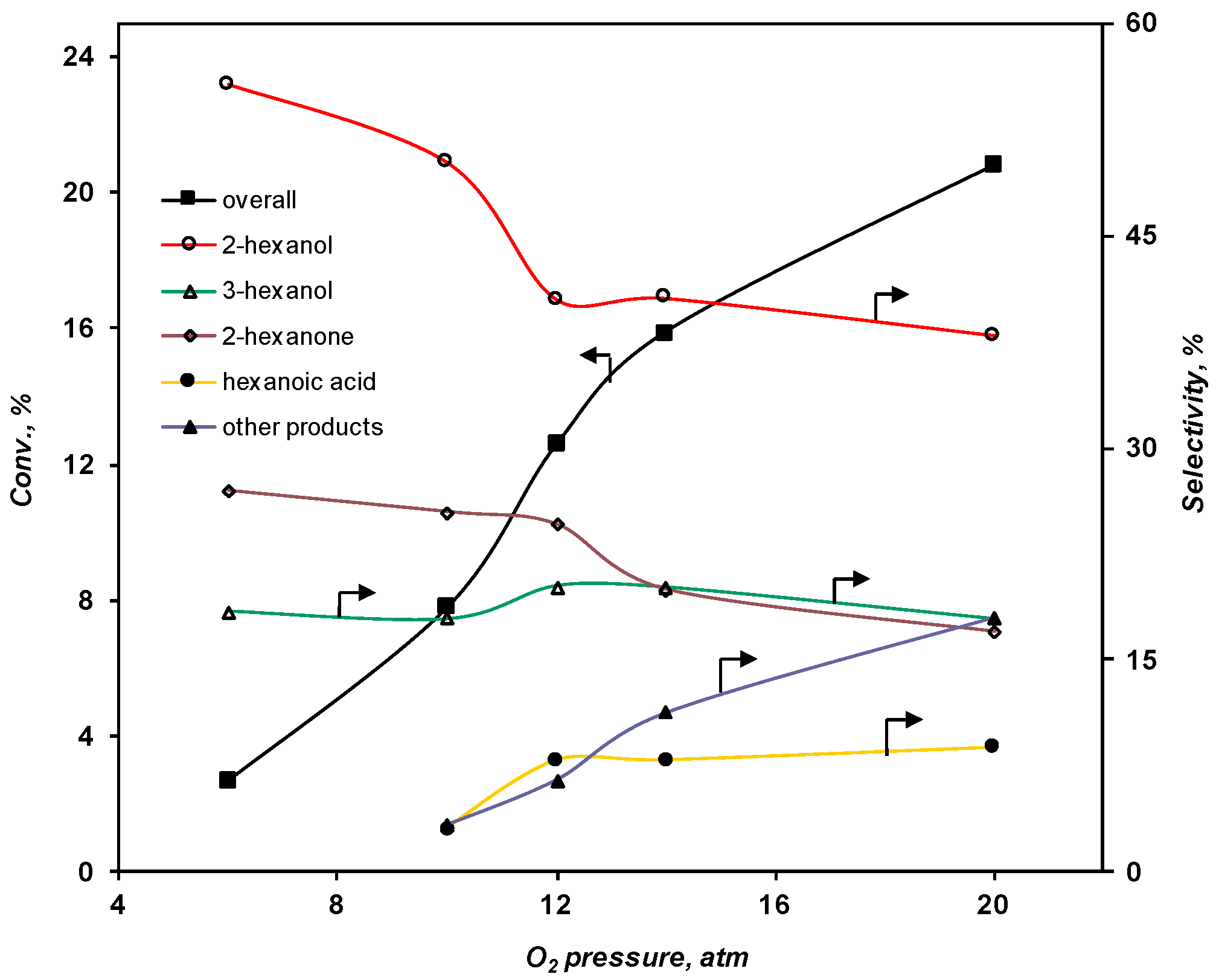
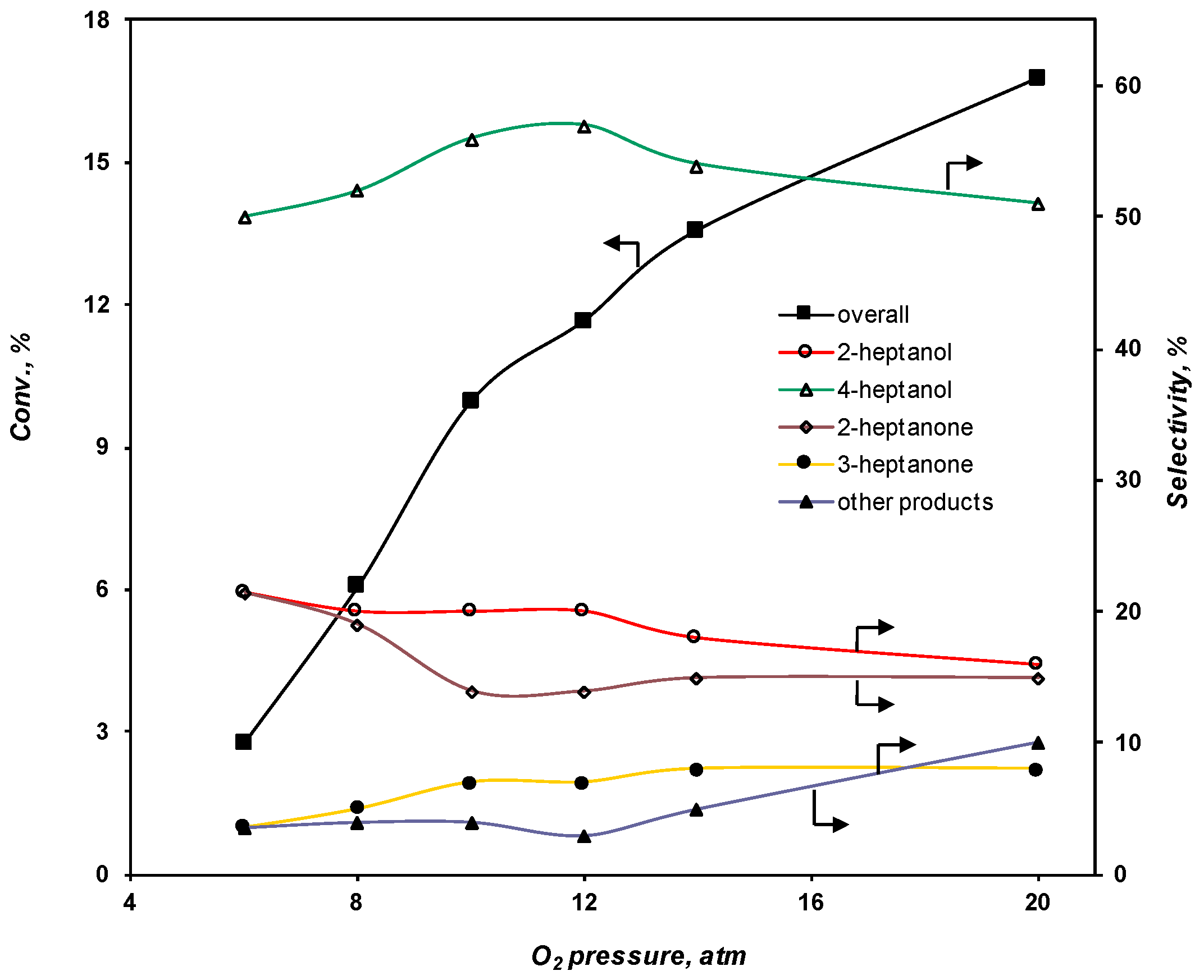
2.3.2. Effect of the O2 Pressure
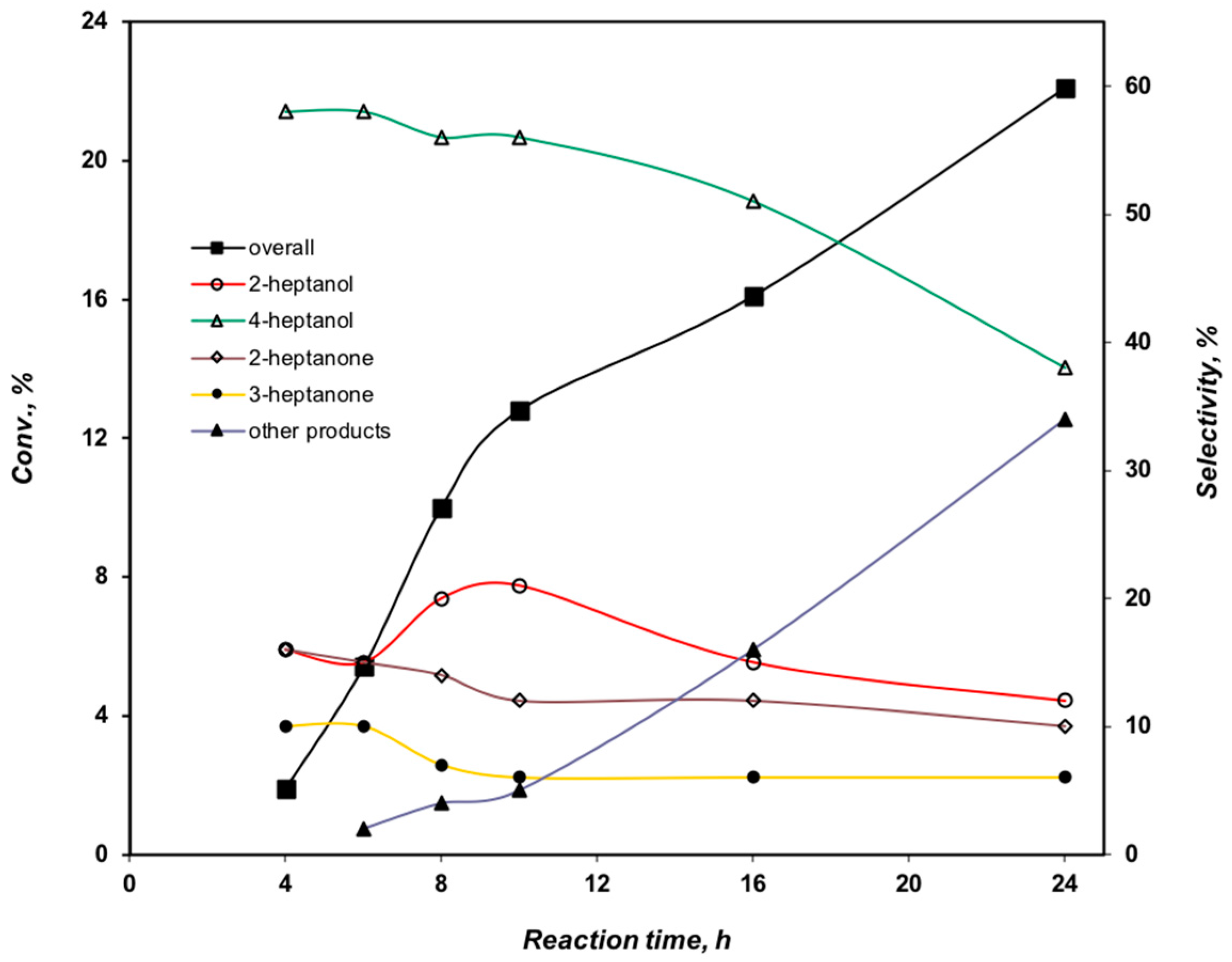
2.3.3. Effect of the Reaction Time
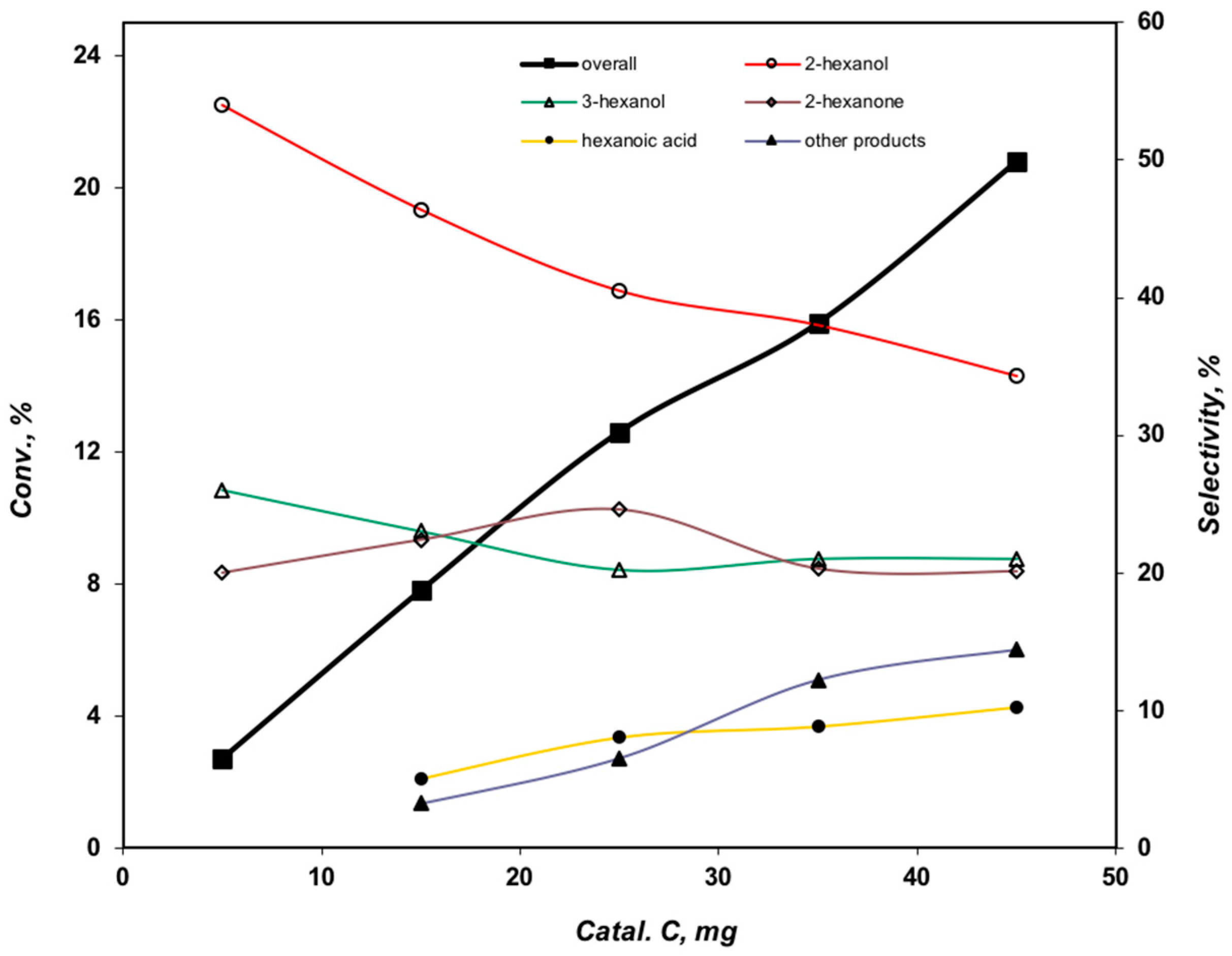
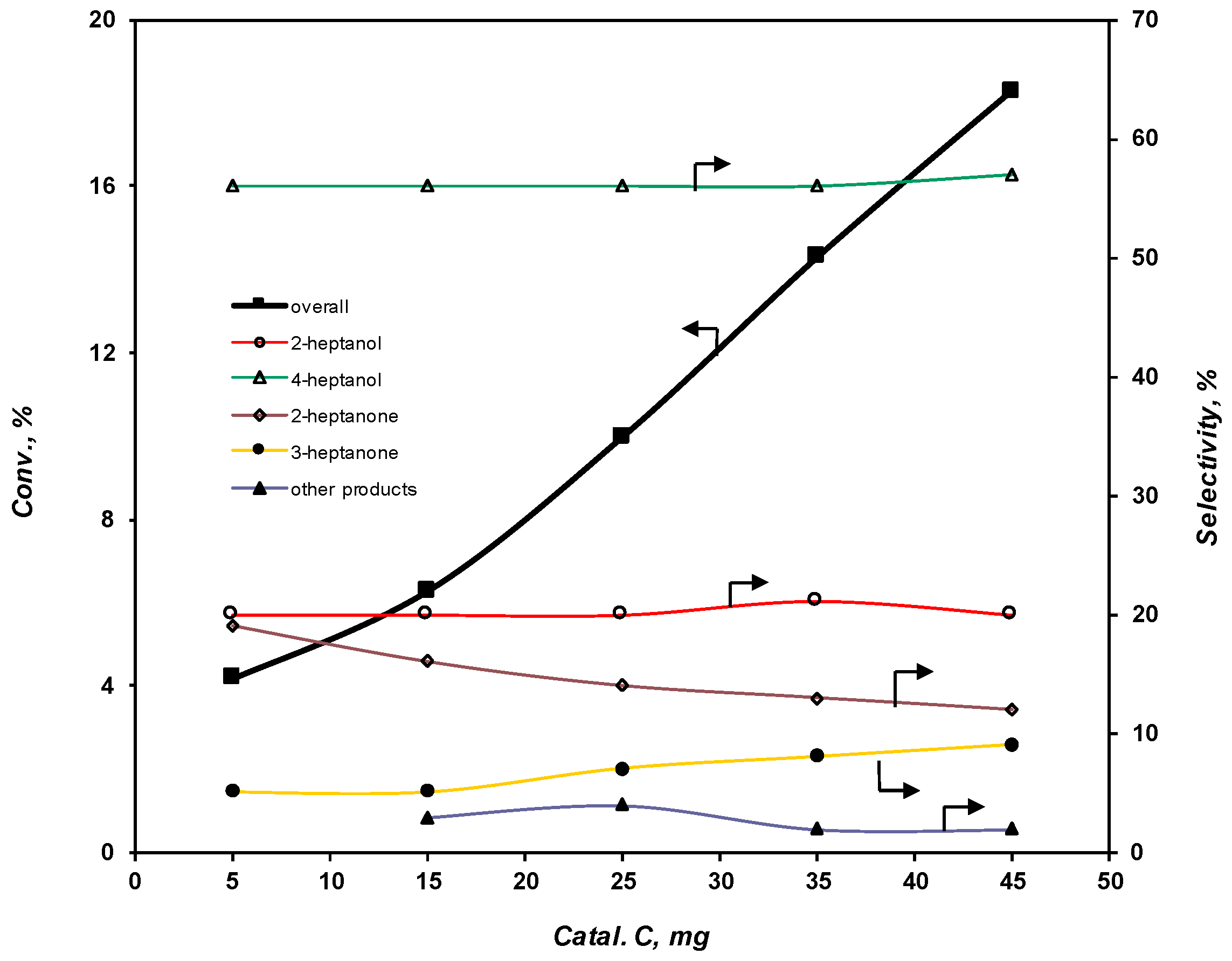
2.3.4. Effect of the Catalyst Amount
2.3.5. Effect of the Co-Catalyst
2.4. Catalyst Stability and Recyclability
2.5. Proposed Reaction Mechanism for n-Alkanes Oxidation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hodnett, B.K. Heterogeneous Catalytic Oxidation: Fundamental and Technological Aspects of the Selective and Total Oxidation of Organic Compounds; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Centi, G.; Cavani, F.; Trifiro, F. Selective Oxidation by Heterogeneous Catalysis; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001. [Google Scholar]
- Kroschwitz, J.I. Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: New York, NY, USA, 2014. [Google Scholar]
- Ullmann’s Encyclopedia of Industrial Chemistry, 6th ed.; Wiley-VCH: Weinheim, Germany, 1999–2016; Volume 11, pp. 41–49.
- Clerici, M.G.; Ricci, M.; Strukul, G. Metal-Catalysis in Industrial Organic Processes; Chiusoli, G.P., Maitlis, P.M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2006. [Google Scholar]
- Martins, L.M.D.R.S. C-homoscorpionate oxidation catalysts—Electrochemical and catalytic activity. Catalysts 2017, 7, 12. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-soluble C-scorpionate complexes: Catalytic and biological applications. Eur. J. Inorg. Chem. 2016, 15–16, 2236–2252. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Mishra, G.S.; da Silva, M.F.G.; Wanke, R.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. CuII complexes bearing the 2,2,2-tris(1-pyrazolyl)ethanol or 2,2,2-tris(1-pyrazolyl)ethylmethanesulfonate scorpionates. X-Ray structural characterization and application in the mild catalytic peroxidative oxidation of cyclohexane. Dalton Trans. 2009, 42, 9207–9215. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, M.L.; Kozlov, Y.N.; Mandelli, D.; Pombeiro, A.J.L.; Shul’pin, G.B. Mechanism of Al3+-Catalyzed Oxidations of Hydrocarbons: Dramatic Activation of H2O2 toward O−O Homolysis in Complex [Al(H2O)4(OOH)(H2O2)]2+ Explains the Formation of HO• Radicals. Inorg. Chem. 2011, 50, 3996–4005. [Google Scholar] [CrossRef] [PubMed]
- Bäckwall, J.-E. Modern Oxidation Methods; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Olah, G.A.; Molnar, A. Hydrocarbon Chemistry, 3rd ed.; Wiley: New York, NY, USA, 1995; p. 10. [Google Scholar]
- Yasutaka, I.; Sakaguchi, S.; Iwahama, T. Innovation of Hydrocarbon oxidation with molecular oxygen and related reactions. Adv. Synth. Catal. 2001, 243, 393–427. [Google Scholar] [CrossRef]
- Mishra, G.S.; Kumar, A. Silica gel supported [1,4-bis(salicylidene amino)-phenylene] vanadium oxo complex catalyst for the oxidation of n-heptane using molecular oxygen. J. Mol. Catal. A Chem. 2003, 192, 275–280. [Google Scholar] [CrossRef]
- Pérez, P.J. (Ed.) Alkane C-H Activation by Single Site Metal Catalysts; Springer: Berlin/Heidelberg, Germany, 2012; p. 143. [Google Scholar]
- Machado, K.; Mishra, J.; Suzuki, S.; Mishra, G.S. Synthesis of superparamagnetic carbon nanotubes immobilized Pt and Pd pincer complexes: Highly active and selective catalysts towards cyclohexane oxidation with dioxygen. Dalton Trans. 2014, 43, 17475–17482. [Google Scholar] [CrossRef] [PubMed]
- Machado, K.; Tavares, P.B.; Mishra, G.S. Synthesis and application of FeIII, NiII and MnII complexes anchored to HMS as efficient catalysts for cycloalkane oxyfunctionalization. J. Mol. Catal. A Chem. 2014, 383–384, 159–166. [Google Scholar] [CrossRef]
- Mishra, G.S.; Alegria, E.C.B.; Martins, L.M.D.R.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Cyclohexane oxidation with dioxygen catalyzed by supported pyrazole rhenium complexes. J. Mol. Catal. A Chem. 2008, 285, 92–100. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Tris(pyrazol-1yl)methane metal complexes for catalytic mild oxidative functionalizations of alkanes, alkenes and ketones. Coord. Chem. Rev. 2014, 265, 74–88. [Google Scholar] [CrossRef]
- Anisia, K.S.; Kumar, A. Oxidation of n-heptane with molecular oxygen using heterogeneous catalyst formed by covalently binding [1,2-bis(salicylideneamino)-phenylene] zirconium complex to modified silica gel. J. Mol. Catal. A Chem. 2004, 219, 319–326. [Google Scholar] [CrossRef]
- Machado, K.; Freire, C.; Kumar, A.; Tavares, P.B.; Mishra, G.S. Single site anchored novel pentacoordinate Schiff-base CoII complexes over SBA-15 for selective oxidation (O2) of n-alkanes and kinetic study. Polyhedron 2014, 69, 119–126. [Google Scholar] [CrossRef]
- Stekrova, M.; Zdenkova, R.; Vesely, M.; Vyskocilova, E.; Cerveny, L. Immobilization of methyltrioxorhenium on mesoporous aluminosilicate materials. Materials 2014, 7, 2650–2668. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.P.; Schmidt, L.D. C6 oxygenates from n-hexane in a single-gauze reactor. Chem. Eng. Sci. 2000, 55, 5693–5703. [Google Scholar] [CrossRef]
- Zhan, B.-Z.; Modén, B.; Dakka, J.; Santiesteban, J.G.; Iglesia, E. Catalytic oxidation of n-hexane on Mn-exchanged zeolites: Turnover rates, regioselectivity, and spatial constraints. J. Catal. 2007, 245, 316–325. [Google Scholar] [CrossRef]
- Stoylkova, T.Y.; Chanev, C.D.; Lechert, H.T.; Bezouhanova, C.P. Oxidative conversion of n-heptane over molecular sieves. Appl. Catal. A Gen. 2000, 203, 121–126. [Google Scholar] [CrossRef]
- Mishra, G.S.; Machado, K.; Kumar, A. Highly selective n-alkanes oxidation to ketones with molecular oxygen catalyzed by SBA-15 supported rhenium catalysts. J. Ind. Eng. Chem. 2014, 20, 2228–2235. [Google Scholar] [CrossRef]
- Mishra, G.S.; Pombeiro, A.J.L. Oxyfunctionalization of n-pentane and n-hexane by oxovanadium complexes supported on carbamated modified silica gel. Appl. Catal. A Gen. 2006, 304, 185–194. [Google Scholar] [CrossRef]
- Mishra, G.S.; Kumar, A. Selective oxidation of linear alkanes by Schiff base ligand 1,4-bis(salicylidene amino)-phenylene vanadium complex bonded on modified silica gel. Kinet. Catal. 2004, 45, 394–399. [Google Scholar] [CrossRef]
- Alegria, E.C.B.; Martins, L.M.D.R.S.; Haukka, M.; Pombeiro, A.J.L. Rhenium Complexes of Tris(pyrazolyl)methanes and Sulfonate Derivative. Dalton Trans. 2006, 4954–4961. [Google Scholar] [CrossRef] [PubMed]
- Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Syntheses and Properties of Re(III) Complexes Derived from Hydrotris(1-pyrazolyl)methanes. Molecular Structure of [ReCl2(HCpz3)(PPh3)][BF4]. J. Organomet. Chem. 2005, 690, 1947–1958. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Smoleński, P.; Kuznetsov, M.L.; Pombeiro, A.J.L. Oxorhenium complexes bearing the water-soluble tris(pyrazol-1-yl)methanesulfonate, 1,3,5-triaza-7-phosphaadamantane or related ligands, as catalysts for the Baeyer-Villiger oxidation of ketones. Inorg. Chem. 2013, 52, 4534–4546. [Google Scholar] [CrossRef] [PubMed]
- Parshall, G.W.; Shive, L.W.; Cotton, F.A. Inorganic Syntheses; John Wiley & Sons: New York, NY, USA, 1977; Volume 17, pp. 110–112. [Google Scholar] [CrossRef]
- Alegria, E.C.B.; Kirillova, M.V.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Pyrazole and trispyrazolylmethane rhenium complexes as catalysts for ethane and cyclohexane oxidations. Appl. Catal. A Gen. 2007, 317, 43–52. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L. Characterization of Coordination Compounds by Electrochemical Parameters. Eur. J. Inorg. Chem. 2007, 1473–1482. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Martins, A.; Alegria, E.C.B.A.; Carvalho, A.P.; Pombeiro, A.J.L. Efficient cyclohexane oxidation with hydrogen peroxide catalysed by a C-scorpionate iron(II) complex immobilized on desilicated MOR zeolite. Appl. Catal. A 2013, 464–465, 43–50. [Google Scholar] [CrossRef]
- Milunovic, M.N.M.; Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Pombeiro, A.J.L.; Krachler, R.; Trettenhahn, G.; Turta, C.; Shova, S.; Arion, V.B. Hexanuclear and undecanuclear iron(III) carboxylates as catalyst precursors for cyclohexane oxidation. Dalton Trans. 2013, 42, 14388–14401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martins, L.M.D.R.S.; de Peixoto Almeida, M.; Carabineiro, S.A.C.; Figueiredo, J.L.; Pombeiro, A.J.L. Heterogenisation of a C-scorpionate Fe(II) complex in carbon materials for cyclohexane oxidation with hydrogen peroxide. ChemCatChem 2013, 5, 3847–3856. [Google Scholar] [CrossRef]
- Sutradhar, M.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Vanadium complexes: Recent progress in oxidation catalysis. Coord. Chem. Rev. 2015, 301–302, 200–239. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Pombeiro, A.J.L. Radical formation in the [MeReO3]-catalyzed aqueous peroxidative oxidation of alkanes: A theoretical mechanistic study. Inorg. Chem. 2009, 48, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. C–H functionalization: Thoroughly tuning ligands at a metal ion, a chemist can greatly enhance catalyst’s activity and selectivity. Dalton Trans. 2013, 42, 12794–12818. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.F.S.; Luzyanin, K.V.; Kirilova, M.V.; Silva, M.F.C.G.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Novel scorpionate and pyrazole dioxovanadium complexes, catalysts for carboxylation and peroxidative oxidation of alkanes. Adv. Synth. Catal. 2010, 352, 171–187. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Kozlov, Y.N.; Nizova, G.V.; Süss-Fink, G.; Stanislas, S.; Kitaygorodskiy, A.; Kulikova, V.S. Oxidations by the reagent “O2–H2O2–vanadium derivative–pyrazine-2-carboxylic acid”. Part 12.1 Main features, kinetics and mechanism of alkane hydroperoxidation. J. Chem. Soc. Perkin Trans. 2001, 2, 1351–1371. [Google Scholar]
- Shul’pin, G.B.; Kozlov, Y.N.; Shul’pina, L.S.; Kudinov, A.R.; Mandelli, D. Extremely Efficient Alkane Oxidation by a New Catalytic Reagent H2O2/Os3(CO)12/Pyridine. Inorg. Chem. 2009, 48, 10480–10482. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Kozlov, Y.N.; Shul’pina, L.S.; Petrovskiy, P.V. Oxidation of alkanes and alcohols with hydrogen peroxide catalyzed by complex Os3(CO)10(µ-H)2. Appl. Organomet. Chem. 2010, 24, 464–472. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkane | Run | Catalyst | Conversion (%) | ||||
| 2-Hexanol | 3-Hexanol | 2-Hexanone | Hexanoic Acid | Total | |||
| n-hexane | 1 | A | 3.0 | 1.5 | 1.7 | 0.6 | 7.4 |
| 2 | B | 3.9 | 2.0 | 2.2 | 0.8 | 9.3 | |
| 3 | C | 5.1 | 2.5 | 3.1 | 1.0 | 12.6 | |
| 4 | D | 2.1 | 1.1 | 1.3 | 0.4 | 5.2 | |
| n-heptane | 2-Heptanol | 4-Heptanol | 2-Heptanone | 3-Heptanone | |||
| 5 | A | 1.1 | 3.0 | 1.0 | 0.4 | 5.9 | |
| 6 | B | 1.6 | 4.0 | 1.3 | 0.6 | 8.2 | |
| 7 | C | 2.0 | 5.5 | 1.4 | 0.7 | 10.0 | |
| 8 | D | 0.8 | 2.2 | 0.6 | 0.3 | 4.0 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, G.S.; Alegria, E.C.B.A.; Pombeiro, A.J.L.; Martins, L.M.D.R.S. Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane. Catalysts 2018, 8, 114. https://doi.org/10.3390/catal8030114
Mishra GS, Alegria ECBA, Pombeiro AJL, Martins LMDRS. Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane. Catalysts. 2018; 8(3):114. https://doi.org/10.3390/catal8030114
Chicago/Turabian StyleMishra, Gopal S., Elisabete C. B. A. Alegria, Armando J. L. Pombeiro, and Luísa M. D. R. S. Martins. 2018. "Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane" Catalysts 8, no. 3: 114. https://doi.org/10.3390/catal8030114
APA StyleMishra, G. S., Alegria, E. C. B. A., Pombeiro, A. J. L., & Martins, L. M. D. R. S. (2018). Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane. Catalysts, 8(3), 114. https://doi.org/10.3390/catal8030114