Abstract
Traditional cross-coupling reactions, like Mizoroki-Heck Reaction and Suzuki Reaction, have revolutionized organic chemistry and are widely applied in modern organic synthesis. With the rapid development of C–H activation and asymmetric catalysis in recent years, enantioselective C–H activation/cross-coupling reactions have drawn much attention from researchers. This review summarizes recent advances in enantioselective C–H activation/Mizoroki-Heck Reaction and Suzuki Reaction, with emphasis on the structures and functions of chiral ligands utilized in different reactions.
1. Introduction
Over the past few decades, C–H bond activation was established as a credible and viable strategy in organic synthesis [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. On the basis of previous work, C–H activation/functionalization is employed as a dynamic strategy for the synthesis of a great deal of highly valuable natural products and other classes of compounds for pharmaceutics or research interests [19,20,21,22]. Chemists have long been captivated by such synthetic techniques, owing to the obvious merits. From a philosophical point of view, chemists regard C–H bonds as dormant equivalents of various pre-functionalized groups. Moreover, the direct modification of ubiquitously-existing C–H bonds successfully live up to the criteria of one perfect catalytic reaction that ought to be atom-economic and environmental-friendly [23]. Thus, it provides new synthetic disconnections in retrosynthetic analysis [24,25,26,27,28].
It is widely acknowledged that carbon-carbon (C–C) bond formation is fundamental and essential in organic chemistry. Numerous methods have been well developed to enable such C–C bond formation to proceed smoothly. Among these reactions, transition-metals (Pd, Rh, Ru, Cu, Zn, Sn, Mg, etc.) catalyzed cross-coupling reactions are efficient techniques to realize C–C bond formation, which can be well exemplified by Suzuki [29], Mizoroki-Heck [30], Sonogashira [31], Negishi [32], Stille [33], Kumada [34] coupling reaction, etc. These coupling reactions are renowned for their extraordinary utility, practicality, and reliability, and have been broadly utilized in many syntheses, involving pharmaceuticals, fine chemicals, agrochemicals, etc. Consequently, Richard Mizoroki-Heck, Ei-ichi Negishi, and Akira Suzuki jointly won the Nobel Prize in Chemistry 2010 for their excellent work of “palladium-catalyzed cross-coupling reactions in organic synthesis”, which furnished a novel way to achieve C–C bond formation that substantially accelerated the development of pharmaceutics and electronics industries [35]. Although lots of outstanding achievements in this field have been made, from an atom-economic and environmental-harmoniously perspective, there are still shortcomings in these known and powerful traditional cross-coupling reactions. For instance, the substrates must be pre-functionalized, such as using organic (pseudo) halides or organometallic reagents, to gain reactivity during cross-coupling processes. Subsequently, it would result in metal (pseudo) halide wastes, which are supposed to be evaded for atom-economic purpose. Therefore, it is a brilliant notion to develop new methods replacing the pre-functionalized substrates with raw arenes or hydrocarbons, which can achieve the C–H activation and C–C cross-coupling simultaneously. When compared with traditional cross-coupling reactions, the C–H activation/C–C cross coupling reactions have obvious superiority in the aspect of atom-economy and environmental benignity.
With the rapid development of C–H activation/C–C cross-coupling reactions and the emerging of asymmetric catalysis, the enantioselective C–H activation/C–C cross-coupling reactions drew much more attention from researchers. It is a dynamic research frontier to achieve C–H activation/C–C cross-coupling reactions in a stereoselective manner. However, the direct asymmetric functionalizations of inert C–H bonds still remain challenging in current organic synthesis because of the poor reaction selectivity (regioselectivity and enantioselectivity). This is not odd due to the properties of C–H bond: (1) Substrates commonly contain diverse C–H bonds, which usually have high but comparable bond dissociation energy (BDE of C–H bonds are typically 90–110 kcal∙mol−1) within one molecular, rather than bearing a single targeted C–H bond [36,37,38,39,40]; (2) Higher reaction temperature is required for most of the reactions that are related to C–H activation in order to meet the high energy for the cleavage of C–H bonds, which would undoubtedly impose a detrimental effect on the asymmetric induction or coordination between the chiral ligands and the transition metals. Despite the difficulty in controlling stereoselectivity of C–H cleavage, tremendous progresses have been made in transition-metal catalyzed enantioselective C–H activation in the past few decades [41,42,43,44,45,46]. Among these highly efficient synthetic methodologies, complex chiral ligands or neighboring directing groups are usually needed. For instance, the Pd-catalyzed desymmetrization of prochiral C–H bonds has emerged as a promising strategy that can lead to a wide range of corresponding coupling products.
To our knowledge, numerous articles and reviews have been reported previously [30,36,39,40,41,46,47,48,49,50,51,52], particularly in the field of enantioselective coupling via asymmetric cross-dehydrogenative-coupling (CDC) [40,51] and enantioselective metal carbenoid/nitrenoid insertion into unactivated C–H bonds [40,46,53,54]. As such, this review is meant to briefly highlight, discuss, and illustrate the latest progresses and encountered challenges on enantioselective C–H activation/Mizoroki-Heck reaction and Suzuki reaction with a focus on the origin of chirality. The corresponding mechanism and characteristic features of each part will be discussed in detail, meanwhile, emphasizing the structures and functions of chiral ligands utilized in different reactions.
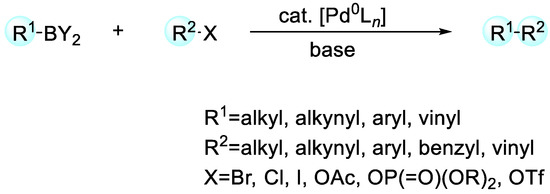
2. Enantioselective C–H Activation/Suzuki Reaction
Among different cross-coupling strategies, the Suzuki reaction between a C(sp2/sp3)-halide or C(sp2/sp3)-triflate with a C(sp2/sp3)-boronic acid or ester manifests remarkable performance in realizing C-C bond formation (Scheme 1). It has fascinated enormous attention since its first disclosure in 1979 and become a dynamic and powerful tool [29].
Scheme 1.
Traditional Suzuki coupling reaction.
The great influences of Suzuki reaction are attributed to its high tolerance of different functional groups and extraordinary efficiency under facile reaction conditions in various media. In addition, its prominent compatibility to diverse processes, including microwave [55] and continuous flow conditions [56,57], and the easy accessibility of organic boronic acid or ester coupling partners, which are usually stable to oxygen, water, as well as harsh reaction conditions, are also attractive to scientists.
In recent years, the direct asymmetric C–H activation/Suzuki cross-coupling reactions have substantially attracted the attention of synthetic chemists. Herein, we review the recent progresses on enantioselective C–H activation/Suzuki cross-coupling reactions of prochiral substrates.
In 2008, Li et al., developed the asymmetric C(sp3)–H arylation reactions of tetrahydroisoquinolines with aryl boronic acids [58]. The reaction proceeded smoothly with CuBr as the catalyst and T-HYDRO as the oxidant in DME. Interestingly, when chiral PhPyBox was added as the ligand, the desired product was obtained with 30% ee (Scheme 2). The addition of CuOTf instead of CuBr further increased the enantioselectivity to 44%.
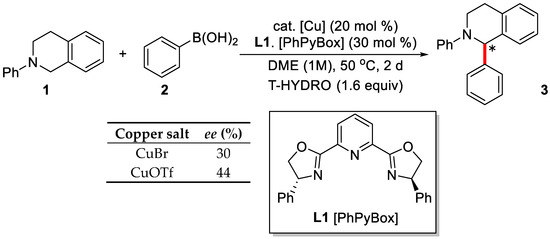
Scheme 2.
Copper-catalyzed oxidative C(sp3)–H bond arylation with aryl boronic acids. Reproduced from Reference [58].
There were two proposed mechanism pathways (Scheme 3) and path B is preferred. In pathway B, the copper-iminium ion intermediate is generated and the subsequent coupling with aryl boronic acids gives access to the desired product. Meanwhile, the introduction of chiral ligands could induce the enantioselective C–H activation process. However, the enantioselectivity still remains to be further improved for practical applications.
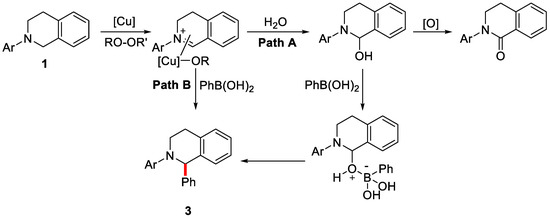
Scheme 3.
Proposed mechanism of the oxidative arylation reaction. Reproduced from Reference [58].
Since 2009, the direct arylation of inert C–H of heteroarenes catalyzed by Pd-catalysts has rapidly evolved as a reliable and practical method. In 2011, Itami et al., developed the Pd-catalyzed oxidative C4-selective C–H arylation of thiophenes and thiazoles enabled by boronic acids (Scheme 4) [59].
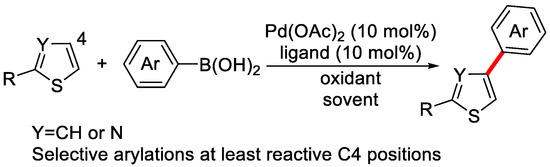
Scheme 4.
Selective arylation of heteroarenes via C–H activation [59].
This approach was further improved by Itami group in 2012 [60]. In order to access the hindered heterobiaryls, the screening of ligands was executed employing 4a and 5a (2,3-dimethylthiophene and 2-methylnaphthalenyl-1-boronic acid respectively) as substrates and PdII as the catalyst. Consequently, the desired coupling product was obtained with bisoxazolines ligands in the presence of Pd(OAc)2, and L3 was proved to be the most effective ligand (Scheme 5). Under the optimized reaction conditions, the scope of the substrates (thiophenes and arylboronic acids) was examined. A variety of different substrates were amenable for this system, delivering moderate yields of up to 84% and excellent C4 regioselectivities up to 99%. The successful synthesis of the tetra-ortho-substituted heterobiaryl 6aa manifested the high efficiency of this bisoxazoline-Pd catalytic system.
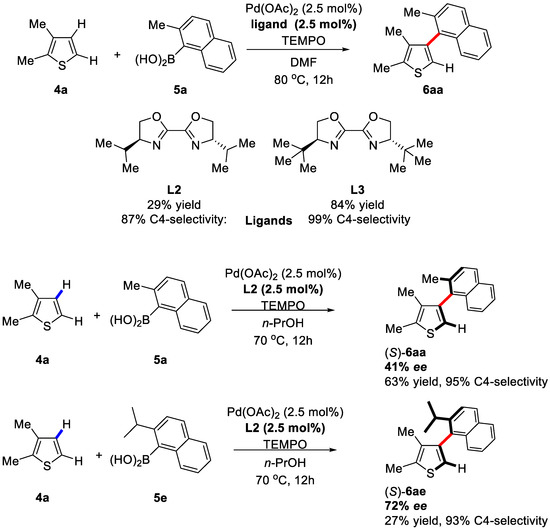
Scheme 5.
Generation of axially chiral biaryls via C–H activation/Suzuki reaction [60].
As the ligands delivering excellent performance (L2 and L3) are chiral, the enantioselective synthesis of axially chiral heterobiaryls was predicted to be possible. To have a better understanding of the axial chirality of the substituted heterobiaryls, the rotation energy of 3-methyl-4-(2-methylnaphthalen-1-yl)-thiophene (6ba) between two conformations was investigated by DFT calculations (Figure 1). It was found that the rotation energy was high enough for the two atropisomers to exist stably at room temperature.
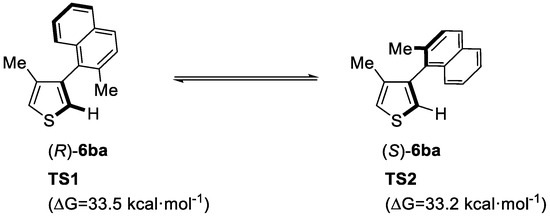
Figure 1.
Rotation energy of 6ba calculated at B3LYP/6-31G(d) level. Reproduced from Reference [60].
After investigating various conditions, impressively, the asymmetric induction truly occurred (Scheme 5). When the n-PrOH solution of 4a and 5a was exposed to Pd(OAc)2/L2 and TEMPO at 70 °C for 12 h under air, the product (S)-6aa was obtained with 41% ee and 63% yield. When a more sterically encumbered arylboronic acid 5e was used, the enantiometric excess of the corresponding product was improved to 72%, albeit with a lower yield. The absolute stereo-configuration of the asymmetric products was determined by X-ray crystallography.
In 2013, the same group revealed another version of this reaction [61]. In this paper, they utilized a PdII–sulfoxide–oxazoline/iron–phthalocyanine (FePc) dual catalyst system for the syntheses of sterically hindered heterobiaryls with air as the oxidant instead of using TEMPO as the stoichiometric co-oxidant (Scheme 6). It was proposed that the ligands (e.g., L4) took effect in the form of a Pd-sox complex, which exhibited higher reactivity in coupling hindered partners.
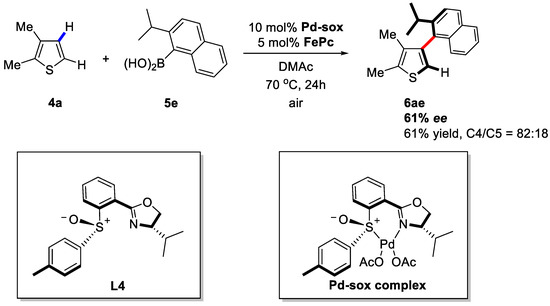
Scheme 6.
Enantioselective C–H activation/C–C coupling between heteroarenes and arylboronic acids [61].
In 2008, a huge advance was made by Yu and coworkers [62]. They developed the PdII/chiral mono-N-protected amino acid (MPAA) system and applied it in the desymmetrization reaction of prochiral substrates (Scheme 7).
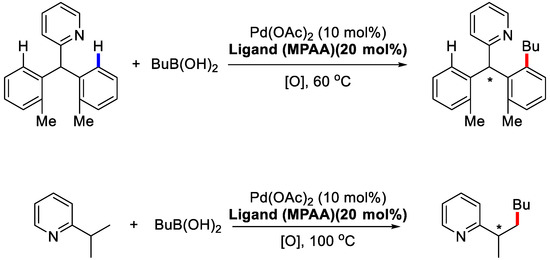
Scheme 7.
Desymmetrization of prochiral C(sp2)–H and C(sp3)–H bonds [62].
Detailed mechanism studies by Yu and coworkers suggested that using conformationally rigid chiral carboxylic acids as ligands as well as PdII catalyst might induce the enantioselective C–H activation process. Indeed, when Boc-protected chiral amino acids were used, the asymmetric induction took place. With compound 7 and butylboronic acid as model substrates, Boc-l-leucine afforded the corresponding product with 63% yield and 90% ee (Scheme 8). Intriguingly, when the Boc protecting group was removed or substituted by methyl group, the reactions failed to occur, which indicates that an electron-withdrawing group on the nitrogen atom is essential in maintaining the electrophilicity of PdII towards the C–H bond. Moreover, the esterification of the amino acid or the decrease of the nitrogen protecting group size resulted in significant declines of enantioselectivities. Thus, the bulkier menthoxycarbonyl protecting groups were introduced and ligand L13 was found to give the best results.
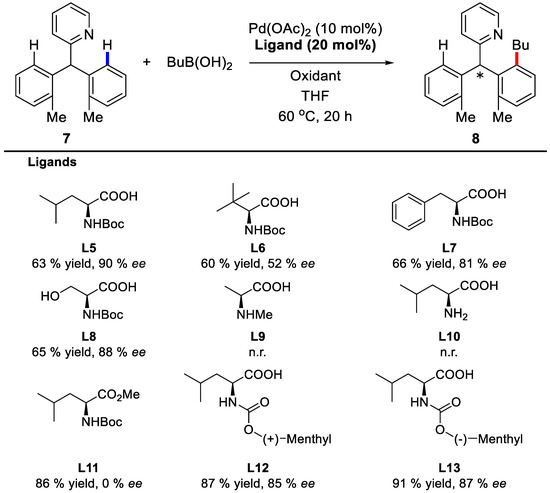
Scheme 8.
Influence of ligands on the enantioselectivities [62].
With the optimized condition in hand, different substrates and boronic acids were investigated and the products were obtained in good yields and moderate to excellent ee (Table 1). Furthermore, enantioselective alkylation of C(sp3)–H bonds, such as substrate 9, was also executed and the desired product was obtained with 38% yield and 37% ee (Scheme 9).

Table 1.
Investigation on substrate scope [62].
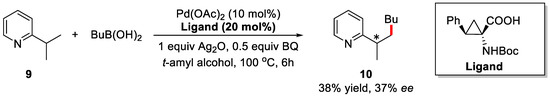
Scheme 9.
Enantioselective alkylation of C(sp3)–H bond [62].
In the proposed transition state, both the nitrogen atom and the carboxylate group of the amino acid ligand coordinate with the PdII center in a bidentate way, providing a chiral environment. Transition state 11a rather than 11b is preferred, in that the steric repulsion between the substituent on the newly generated chiral center (o-Tol) and the Boc group on the nitrogen center is minimized (Figure 2).
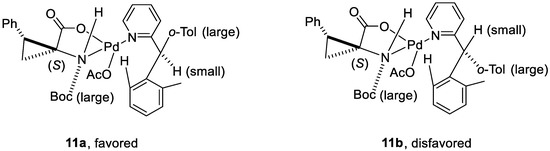
Figure 2.
Key intermediates in the proposed mechanistic model. Reproduced from Reference [62].
In 2011, Yu and coworkers reported the enantioselective C(sp3)–H activation of cyclopropanes catalyzed by PdII/MPAA (Scheme 10) [44].
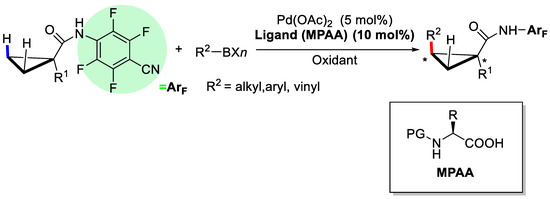
Scheme 10.
Asymmetric C–H activation/C–C coupling reaction of cyclopropane [44].
In this reaction, the amide derivative of 1-methylcyclopropanecarboxylic acid was utilized as the substrate and the electron-deficient arylamide group plays as a directing group. In the process of screening the chiral MPAA ligands, it was discovered that both the protecting group of the amine and the backbone of the amino acid were crucial for the enantioseectivities (Scheme 11). When the protecting group Boc was changed into TcBoc, the enantioselectivity increased dramatically to 78% from 31%, indicating that CCl3 might serve as a bulkier group and an electron-withdrawing group (EWG) simultaneously. Further screening revealed that phenylalanine derivative L23 was the best ligand, and up to 93% ee was achieved.
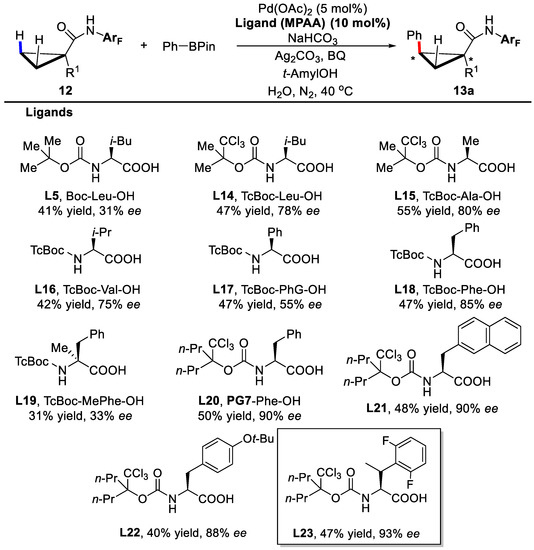
Scheme 11.
Screening of chiral mono-protected amino acid ligands [44].
With the optimized conditions, different cyclopropanes and organoboronic compounds were investigated and the products were obtained in good yields and good to excellent ee (Figure 3).
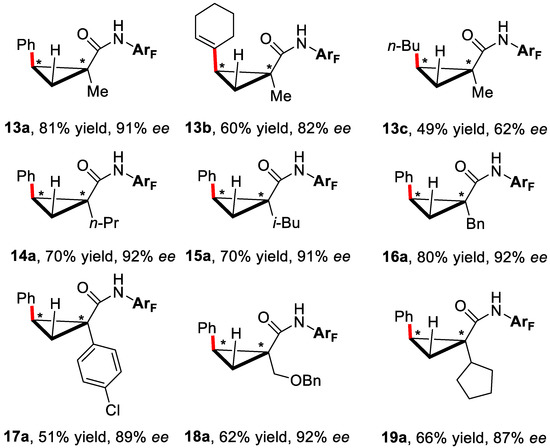
Figure 3.
The substrate scope of asymmetric cyclopropane C–H functionalization [44].
In 2014, the same group reported a further research for the arylation of methylene β-C(sp3)–H bonds of cyclobutanecarboxylic acid derivatives with arylboron reagents using palladium(II) catalyst with chiral mono-N-protected α-amino-O-methylhydroxamic acid (MPAHA) as the ligand (Scheme 12) [63]. This method provided a complementary protocol for the syntheses of enantioenriched cyclobutanes containing chiral quaternary stereocenters [64,65].
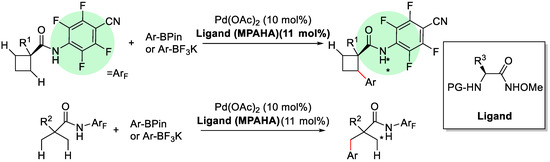
Scheme 12.
Enantioselective C(sp3)–H activation of cyclobutanes [63].
Similar to the precedent studies, MPAHA can generate a chiral complex with PdII catalyst and is the key to obtain appreciable yield and enantioselectivity. When O-methylhydroxamic acids were used as ligands instead of the previously used mono-protected amino acids, a significant boost of enantioselectivity was observed, which might derive from the stronger coordination between the ligand and the PdII center. Further evaluation revealed that the Boc protecting group and an aromatic side chain within the ligand were prone to elevate the enantioselectivities. Of the various ligands that were tested, L32 gave the best results (Scheme 13). Further optimization of the solvents, bases and catalysts eventually led to the desired product with 75% yield and 92% ee.
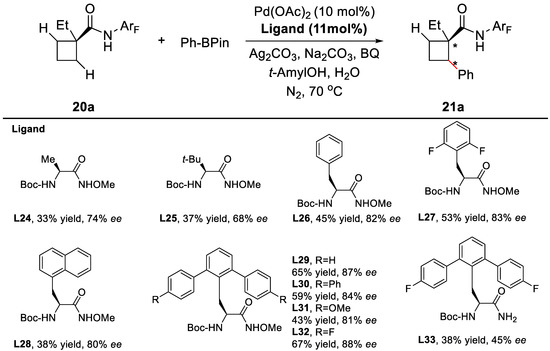
Scheme 13.
Designs of chiral ligands [63].
Under the optimized condition, the reaction was found to work well between a variety of arylboronic acid pinacol esters and various 1-substituted 1-cyclobutanecarboxylic acid derivatives (Scheme 14).
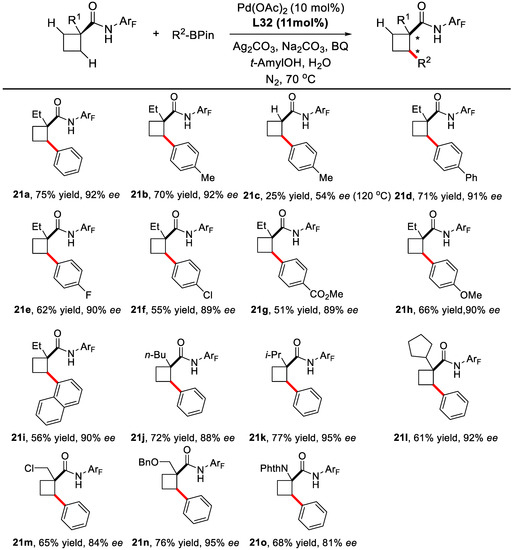
Scheme 14.
Substrates scope for C(sp3)–H activation of cyclobutanes [63].
It is well known that planar chiral ferrocenes are frequently applied as highly efficient catalysts or ligands in asymmetric synthesis [66,67,68,69,70,71]. Inspired by previous studies from Yu group [44,62], You et al., developed an enantioselective syntheses of planar chiral ferrocenes via palladium-catalyzed direct coupling with arylboronic acids in 2013 (Scheme 15) [72].
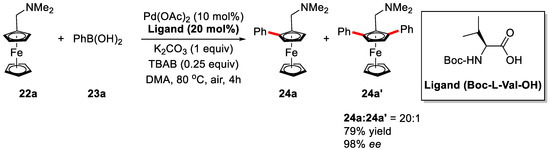
Scheme 15.
Synthesis of planar-Chiral Ferrocenes via enantioselective C–H activation [72].
In this work, dimethylaminomethylferrocene 22a and phenylboronic acid 23a were chosen as model substrates. The reaction proceeded smoothly in the presence of 10 mol % Pd(OAc)2, 20 mol % Boc-l-Val-OH, and 1 equiv of K2CO3 in DMA (Dimethylacetamide) at 80 °C under air, providing the desired product with 58% yield and 97% ee. The yield was further improved to 79% with 25 mol % TBAB as the additive when the reaction was performed at 60 °C.
With the optimized condition, various aminomethylferrocene derivatives and boronic acids were examined. Substituted arylboronic acids bearing either an electron-donating group or an electron-withdrawing group were well-tolerated and afforded the corresponding products in good yields and excellent enantioselectivities. Moreover, the reaction was also general for aminomethylferrocenes with different alkyl groups on the nitrogen atom (Scheme 16). In addition, a large scale reaction was executed smoothly, which further confirmed the practicality of this method.
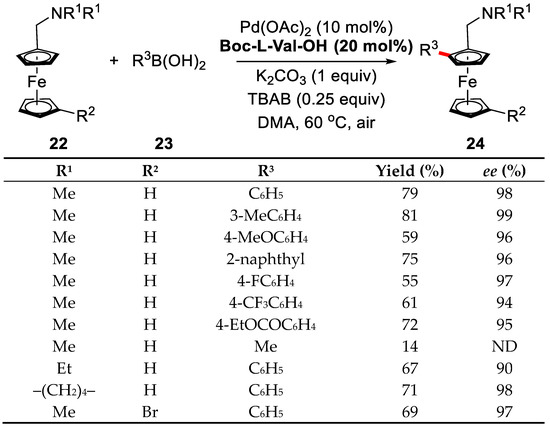
Scheme 16.
Investigation of substrate scope [72].
At last, the planar-chiral P,N-ligand L34 prepared from compound 24 was successfully utilized in the palladium-catalyzed allylic alkylation reaction (Scheme 17), which fully demonstrated the potential application of this novel protocol.
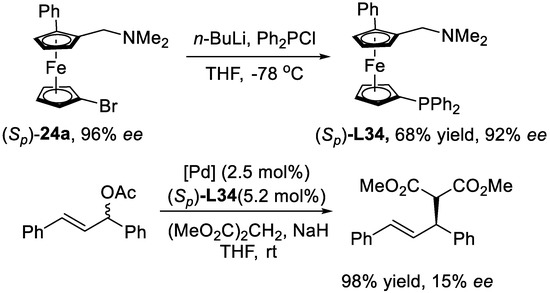
Scheme 17.
Application of the synthesized planar-chiral ferrocenes. Reproduced from Reference [72].
It is well known that chiral phosphorus compounds play important roles as ligands or organocatalysts in asymmetric synthesis [73,74,75,76]. In 2015, Han group reported the asymmetric syntheses of traditionally inaccessible P-stereogenic phosphinamides via Pd-catalyzed enantioselective C(sp2)–H functionalization (Scheme 18) [77].
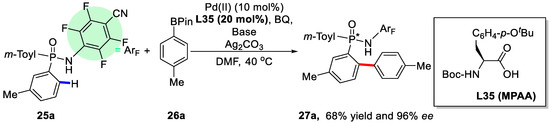
Scheme 18.
Enantioselective synthesis of P-stereogenic phosphinamides via asymmetric C–H arylation [77].
Similarly, chiral mono-N-protected amino acids (MPAA) were used as ligands in this reaction. The presence of the carbamate moiety employed as the N-protecting group and the carboxylic acid group within the ligand were essential to deliver the desired products with good enantioselectivities. Of the various chiral ligands tested, ligand L35 was found to be the optimal ligand, affording 68% yield and 96% ee (Scheme 18). The best reaction condition was found to be 10 mol % Pd(OAc)2, 20 mol % L35, 0.5 equiv of BQ, 1.5 equiv of Ag2CO3, 3.0 equiv of Li2CO3, and 40.0 equiv of H2O in anhydrous DMF at 40 °C under air. In addition, an array of substrates, including arylboronic esters decorated with different groups and different diarylphosphinamides, were subjected to this protocol and most of the reactions occurred efficiently (Scheme 19). Practically, this novel approach could be carried out in the gram scale with consistent efficiency.
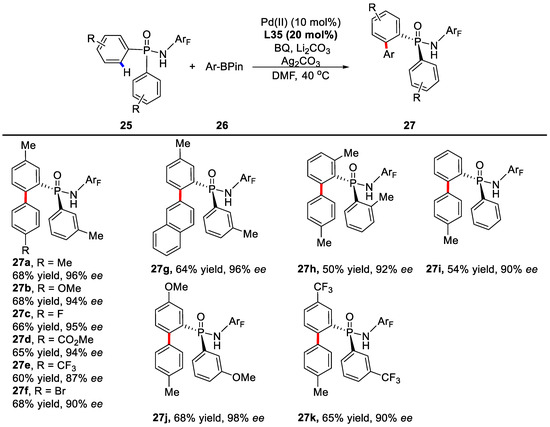
Scheme 19.
Evaluation of substrate scope [77].
Recently, the enantioselective ortho-C(sp2)–H coupling between para-nitrobenzenesulfonyl (nosyl) protected diarylmethylamines and arylboronic acid pinacol esters was established by Yu and coworkers (Scheme 20) [78].
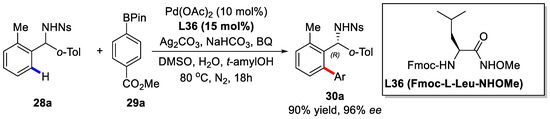
Scheme 20.
Enantioselective ortho-C–H activation of diarylmethylamines [78].
Herein, chiral mono-N-protected amino acids (MPAA) were adopted as chiral ligands at first. Further screening revealed that carbamate-protected, N-methoxyamide-substituted aliphatic amino acids were the best choice. When Fmoc-l-Leu-NHOMe was used, the desired product could be obtained in 90% yield and 96% ee. This method was further applied to a variety of different diarymethylamines with arylboronic acid pinacol esters as coupling partners. Under the optimized condition, most of the reactions proceeded smoothly with good yields and excellent enantioselectivities (Scheme 21 and Scheme 22).
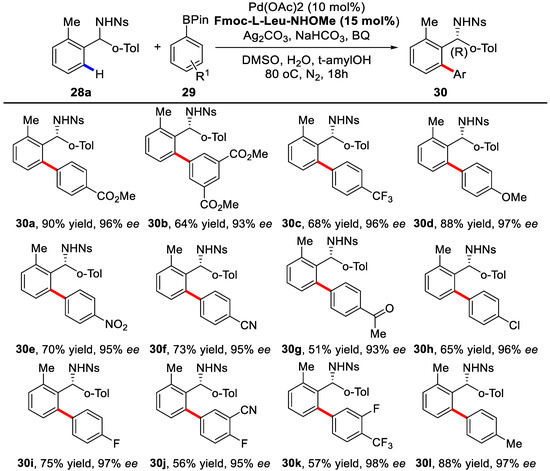
Scheme 21.
Reactions with different arylboronic acid pinacol esters [78].
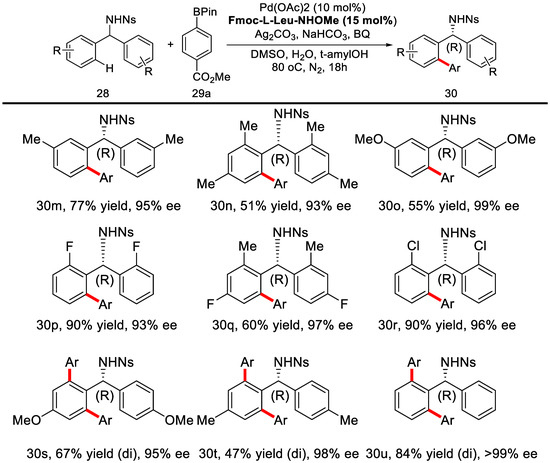
Scheme 22.
Reactions with different diarymethylamines [78].
A stereochemical model was proposed (Figure 4). It was assumed that the coordination between the imine moiety of the deprotonated anionic sulfonamide and PdII center promoted the stereoselective C–H activation followed by arylation.
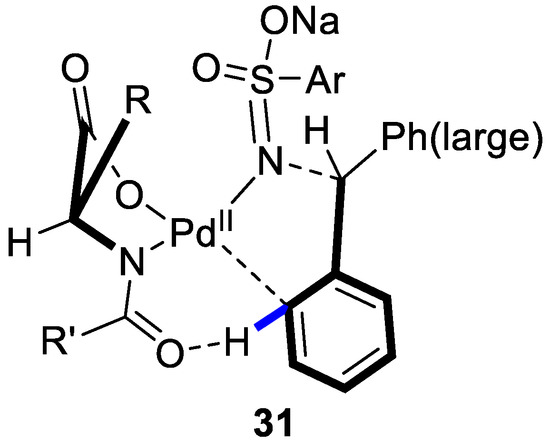
Figure 4.
Proposed transition state. Reproduced from Reference [78].
3. Enantioselective C–H Activation/Mizoroki-Heck Type Reaction
Another important type of cross coupling reaction is the Mizoroki-Heck reaction (Scheme 23), which exhibits extraordinary performance with high efficiency in assembling C–C bonds [79,80]. Herein, we will highlight the recent progresses in the field of enantioselective C–H activation concerning Mizoroki-Heck type reaction.

Scheme 23.
Mizoroki-Heck reaction.
Inspired by the excellent performances of monoprotected amino acids (MPAA) as ligands for enantioselective C–H activation [62], Yu and coworkers developed an enantioselective C–H olefination reaction of diphenylacetic acids using MPAA as chiral ligands [81].
Among the various chiral monoprotected α-amino acids examined, Boc-Ile-OH proved to be the best one. The yield could be improved to 73% (97% ee) with the preformed sodium salt of the starting material and KHCO3 as the base (Scheme 24).
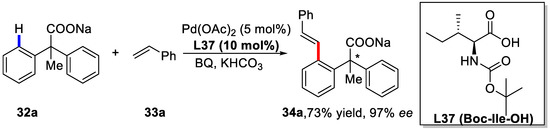
Scheme 24.
Enantioselective C–H olefination of diphenylacetic acids [81].
A broad range of styrenes with different substituents were inspected and it was found that styrenes with para and meta alkyl substituents gave higher enantioselectivities (92–97% ee). In addition, acrylate coupling partners were also tolerant to such condition, affording 99% ee. However, a mixture of the desired olefination product and the corresponding conjugated addition product was isolated (Scheme 25).
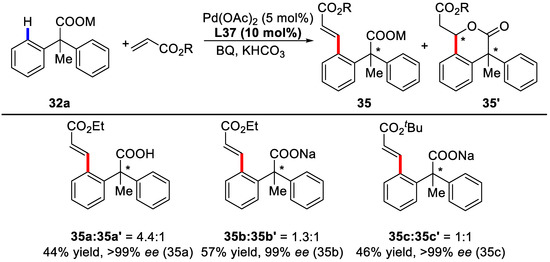
Scheme 25.
Acrylates employed as the coupling partners [81].
Different carboxylic acids were also treated with this strategy and most of them proceeded efficiently except for α-hydrogen containing substrate (58% ee). Besides, substrates containing electron-donating groups and moderately electron-withdrawing groups were well compatible to this procedure, although olefination of the latter gave lower yields (Scheme 26).
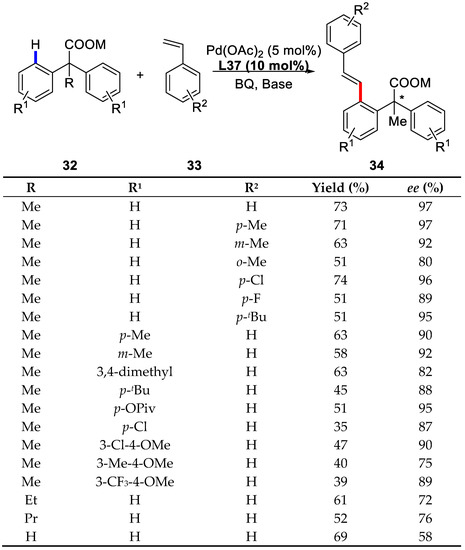
Scheme 26.
Enantioselective C–H olefination with substituted styrenes as coupling partners [81].
Yu also proposed a possible transition state for this enantioselective C–H olefination (Figure 5). A chiral carbon-Pd intermediate could be formed, followed by olefination to give the corresponding chiral product.
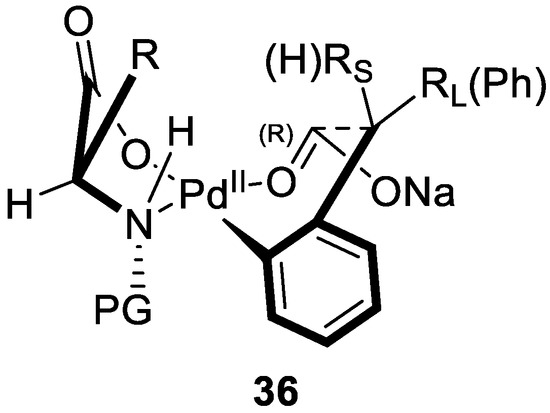
Figure 5.
The proposed transition state. Reproduced from Reference [81].
Based on the recent development of enantioselective C–H iodination using PdII/MPAA catalysts for kinetic resolution through C–H hydroxylation and iodination [82,83], Yu and coworkers developed a kinetic resolution method to achieve enantioselective C–H olefinations of α-hydroxy and α-amino phenylacetic acids utilizing PdII-catalyzed system in 2016 (Scheme 27) [84].
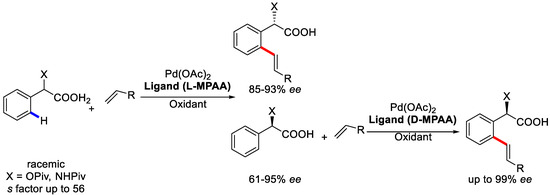
Scheme 27.
Enantioselective C–H olefination by kinetic resolution. Reproduced from Reference [84].
In this paper, Yu also employed mono-N-protected amino acid (MPAA) as ligands to enable enantioselective C–H bond olefinations. Of different MPAA ligands screened, Boc-l-Thr(Bz)-OH (L38) gave the best selectivity factor (s) [85] of 54 (90% ee and 45% yield). Notably, the loading of Pd(OAc)2 could be reduced to 5 mol % without a pronounced erosion of the selectivity, and the addition of 0.4 equivalent of olefin increased the enantioselectivity to 93%.
With the optimized reaction condition, a series of olefin coupling partners were subjected to this transformation and a broad range of electron-deficient olefins were well tolerated. Of note, acrylates were a good coupling partner affording s factors ranging from 46 to 54. Vinyl amides and vinyl phosphates also proceeded smoothly. A wide range of different substituted mandelic acid substrates were also successfully olefinated with reasonable s factors (Scheme 28).
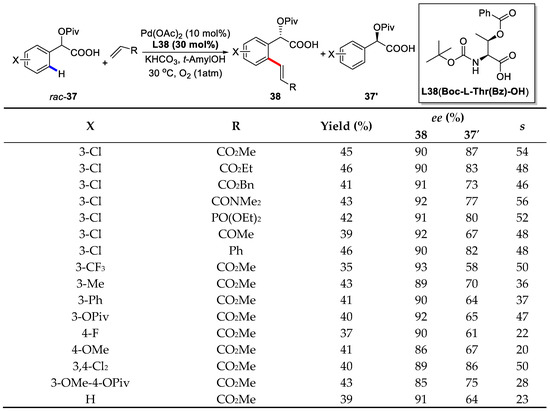
Scheme 28.
Enantioselective C–H olefination/kinetic resolution of mandelic acids [84].
Moreover, the scope of different substituted racemic phenylglycine substrates was investigated and most of them furnished the products with synthetically useful s factors (Scheme 29).
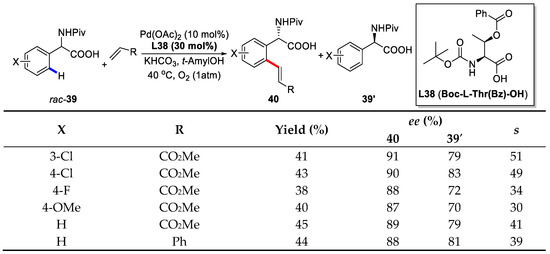
Scheme 29.
Enantioselective C–H olefination/kinetic resolution of phenylglycines [84].
Furthermore, for the remaining starting materials 37′ with 87% ee, a following olefination protocol using the opposite configuration ligand could afford the corresponding chiral product with 99% ee (Scheme 30).
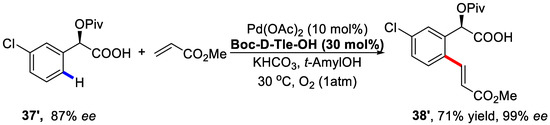
Scheme 30.
Enantioselective C–H olefination of the remaining starting material [84].
In analogy to previously mentioned mechanism, there are two proposed intermediates (Figure 6), TSS and TSR, respectively, and TSS is the favored configuration due to the less steric repulsion between the Boc group and larger OPiv moiety. This stereomodel can well rationalize the origin of the enantioselectivity.
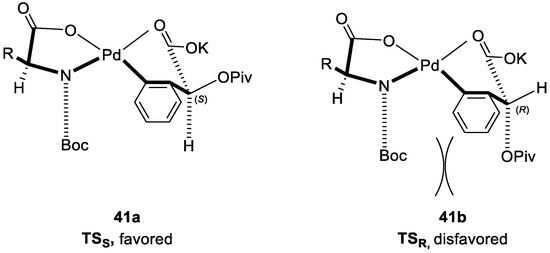
Figure 6.
The proposed intermediate state. Reproduced from Reference [84].
Another interesting finding on enantioselective C–H activation/Heck reaction was provided by Shi and coworkers in 2017 [86]. In this article, atroposelective synthesis of axially chiral biaryls by C–H olefination, utilizing the PdII/transient chiral auxiliary (TCA) as the catalytic system, was achieved. Of note, in this asymmetric C–H olefination reaction, the chiral free amino acid played as a transient chiral auxiliary (TCA), which promoted the C–H activation process instead of only serving as a simple chiral ligand. Of the various TCA examined, the L-tert-leucine (T1) was found to be the optimal. As expected, the other atropisomer was obtained, while the opposite TCA (D-tert-leucine) was employed (Scheme 31).
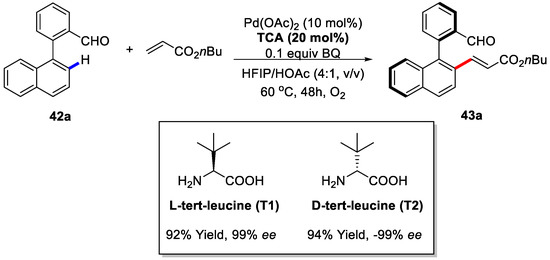
Scheme 31.
Optimization of the reaction [86].
With the optimized reaction condition, the substrate generality was inspected. A broad range of biaryls with different substituents were tolerated to this protocol. It ought to be noted that C–H olefination of biaryls with substituents at either 6- or 2′-position or less hindered substituents at both 6- and 2′-position proceeded successfully in a dynamic kinetic resolution (DKR) manner, giving enantioenriched products in good to excellent yields and excellent enantioselectivities (95 to >99% ee) (Scheme 32).
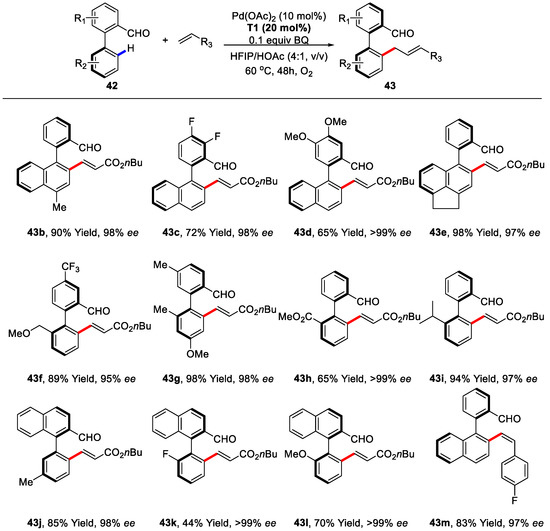
Scheme 32.
Substrate scope of biaryls concerning C–H olefination/DKR [86].
Nonetheless, for the biaryls bearing sterically more hindered substituents at both 6- and 2′-position, C–H olefination would occur through a kinetic resolution (KR) manner in excellent selectivities (Scheme 33). Moreover, various acrylates and styrenes were investigated and most of them were compatible to this method, except that electronrich styrenes were determined as inert coupling partners (Scheme 32 and Scheme 33).
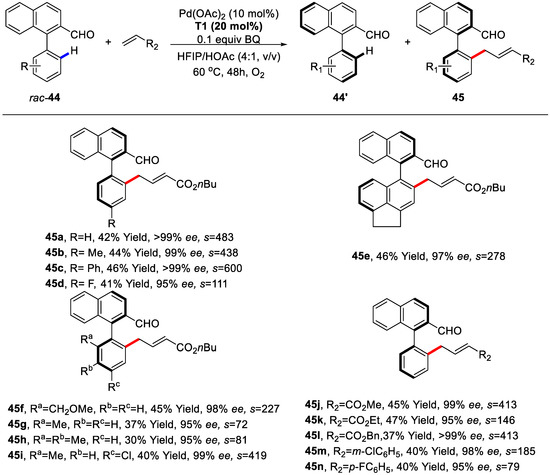
Scheme 33.
Substrate scope of biaryls concerning C–H olefination/KR [86].
As far as the mechanism was concerned, it was proposed that the chiral amino acid would react with the racemic substrate to give the imine intermediates A and B reversibly. Then, C–H cleavage of B took place selectively due to the minor steric repulsion, resulting in intermediate C with axially stereoenriched biaryl palladacycle. Then, intermediate C underwent a typical Heck reaction with olefin to afford intermediate D, which would be hydrolyzed to furnish the desired chiral biaryls (Ra)-E. Meanwhile, the Pd0 was reoxidised into PdII to close the catalytic cycle (Scheme 34).
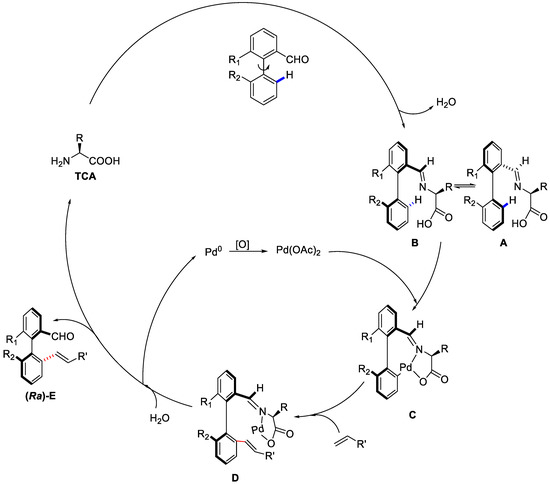
Scheme 34.
Hypothesis on the mechanism for the C–H olefination/DKR [86].
The PdII/MPAA catalysts were also applied in the enantioselective olefinations of N,N-dimethylaminomethylferrocene by Wu group (Scheme 35) [87].
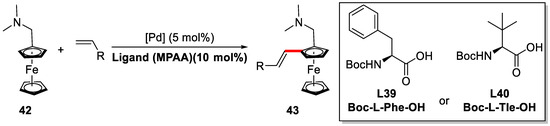
Scheme 35.
Pd-catalyzed direct C–H olefination of N,N-dimethylaminomethylferrocene [87].
In the proposed mechanism, the cyclopalladated complex A was first generated by palladium species through coordination with the substrate and the ligand. Then, intermediate A underwent a typical Heck reaction to furnish the desired product. It is worth noting that in this catalytic system, the N,N-dimethylaminomethylferrocenium C was generated in situ by air and served as a terminal oxidant to regenerate active PdII from the reduced Pd0 species, completing the catalytic cycle. Therefore, no external oxidant was needed (Scheme 36).
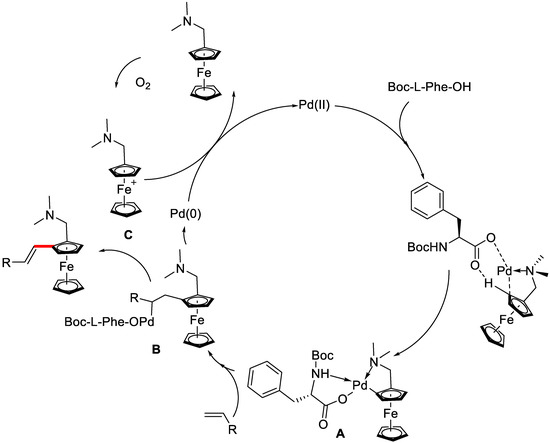
Scheme 36.
The proposed mechanism for the reaction. Reproduced from Reference [87].
N,N-dimethylaminomethylferrocene and butyl acrylate were chosen as the model substrates. It was found the introduction of mono-N-protected amino acids (MPAA) as ligands to PdII, not just induced the high enantioselectivities, but also dramatically increased the reaction yields. Both Boc-l-Phe-OH (L39) and Boc-l-Tle-OH (L40) gave excellent yields and enantioselectivities for the model reaction (Scheme 35). Under the optimized condition, various acrylates, styrenes, and even aliphatic olefins were tested (Scheme 37). All of them worked well with excellent enantioselectivities (up to 99%) and yields (up to 98%).
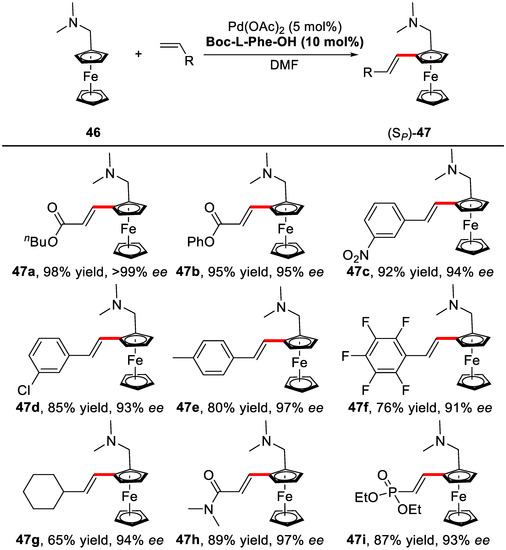
Scheme 37.
Substrate scope for the enantioselective C–H olefination reaction [87].
Another impressive development was revealed by You and coworkers [88]. They also employed the PdII/MPAA catalyst system in the enantioselective oxidative C–H/C–H cross-coupling of ferrocenes with heteroarenes to prepare planar chiral ferrocenes.
The Boc-l-Ile-OH proved to be the best ligand, affording the desired product in a C2 regioselective manner with 71% yield and nearly perfect 99% ee under the optimal reaction conditions with air as the oxidant. Subsequently, various ferrocene derivatives were evaluated and the reaction exhibited excellent tolerance towards different functional groups (Scheme 38).
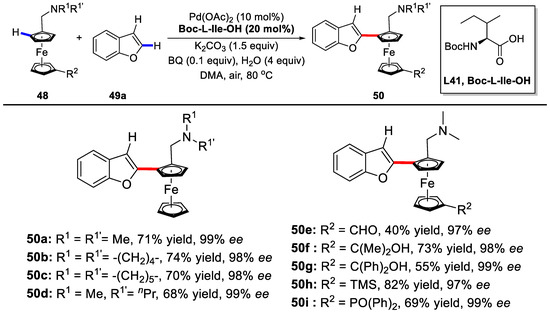
Scheme 38.
Enantioselective cross-coupling reactions of benzofurans [88].
Moreover, the scope of heteroarenes was explored as well. Various substituted benzofurans, furans, thiophenes, pyrroles, and indoles worked smoothly under this condition, providing impressive yields and enantioselectivities (up to 99%) (Scheme 39).
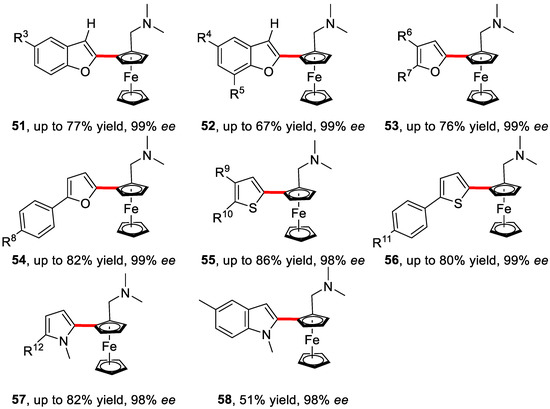
Scheme 39.
Enantioselective cross-coupling reactions with different heteroarenes [88].
Besides, the newly synthesized planar chiral ferrocenes could be further elaborated into useful ligands for asymmetric transformations (Scheme 40).
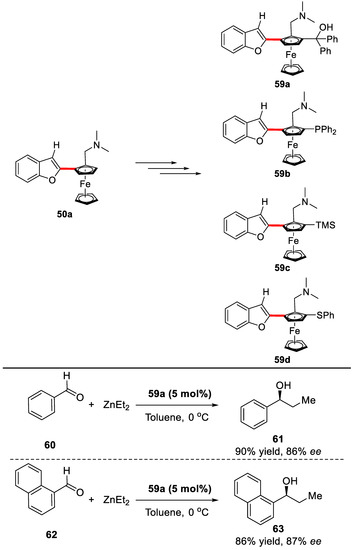
Scheme 40.
Applications of the protocol [88].
Another excellent work on synthesizing novel axially chiral biaryls by direct C–H bond olefination was fulfilled by You and coworkers [89]. This dehydrogenative coupling reaction was catalyzed by chiral Cp/rhodium complexes. Further screening revealed that catalysts with bulky substituents usually led to diminished yield and e.r. value (Scheme 41). Finally, the combination of the Cat.1, Ag2CO3 (1.0 equiv), and Cu(OAc)2 (20 mol %) in methanol was determined to be the optimal condition, affording the desired product with 94% yield and 90:10 e.r.
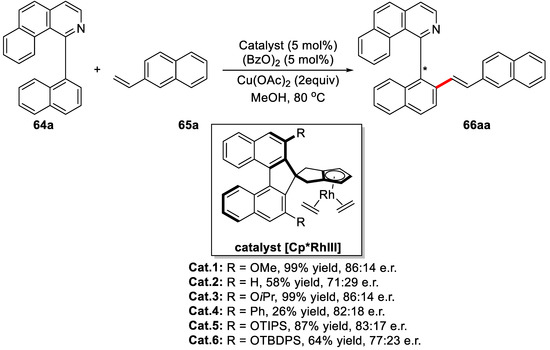
Scheme 41.
Catalyst screening for the enantioselective oxidative Heck coupling reaction [89].
Under the optimized condition, biaryl substrates bearing different EDG or EWG proceeded efficiently to generate the desired alkenylated products in moderate to excellent yields and enantioselectivities, with up to 97% conversion and 93:7 e.r. Additionally, different olefins were well tolerated to this reaction, including styrenes, acrylates, acrylamides, and vinyl phosphonate esters. Notably, ethylene was also introduced, which gave the desired product with 86:14 e.r. Moreover, the gram-scale reaction also worked well (Scheme 42).
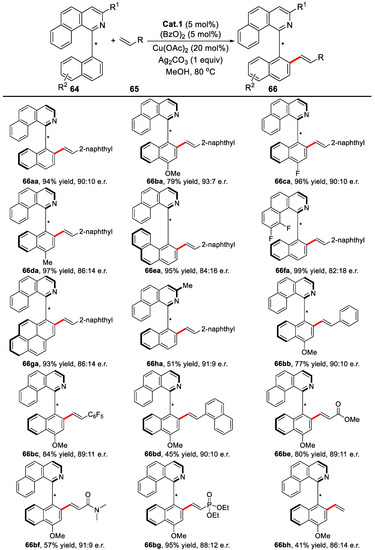
Scheme 42.
The investigation of substrate scope [89].
Finally, the product was successfully utilized as ligands in the rhodium-catalyzed conjugate addition of phenylboronic acid to cyclohexenone reaction (Scheme 43), which sufficiently demonstrated the potential of this method.
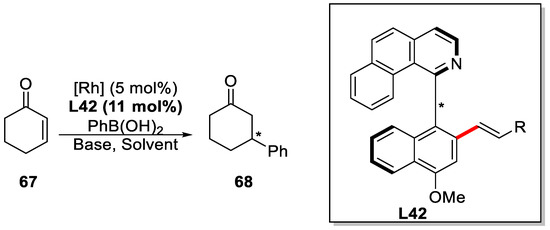
Scheme 43.
The application of enantioenriched biaryl products [89].
4. Summary
Although traditional cross-coupling reactions have revolutionized organic chemistry and are widely applied in modern organic synthesis, the need for prefunctionalized starting materials has prompted chemists to investigate more atom and step economic alternatives. Therefore, C–H activation has emerged as a powerful tool to achieve C–C bond formation, allowing for the transformation of otherwise unreactive C–H bonds, thus maximizing the overall operational efficiency and decreasing the amount of stoichiometric metallic waste. The combinations of C–H activation/C–C cross-coupling reactions provide unlimited possibilities for synthetic chemists to access complex molecules.
This review summarizes the recent development on eantioselective C–H activation/Mizoroki-Heck reaction and Suzuki reaction. Due to the low reactivity of the C–H bonds, and the selectivity problem rooted in the abundance of C–H bonds, these transformations are extremely difficult to achieve. However, thanks to the increased mechanistic studies, chemists continually develop a better understanding of the mechanical aspects ruling these transformations. The PdII/MPAA systems developed by Yu group have been utilized successfully and represents one of the most important progresses.
It should also be pointed out that the concept of toxic heavy metals and benign lighter metals should not be taken for granted. Recently, studies revealed that some palladium, rhodium compounds, which were often considered heavy and toxic, might be less toxic than lighter metals [90]. This may change our traditional views on the toxic effects of metal salts in favor of Pd-catalyzed C–H activation.
Even though apparent advancements in this area have been made, more general protocols are highly demanded. Much research efforts as far as to design new chiral catalysts and chiral ligands, expand the substrate scope, and improve the efficiency of these transformations are still needed before a more general, atom-economical, and more environmentally friendly process become the method of choice for chemists in industrial or academic settings.
Acknowledgments
We thank Shanghai Jiao Tong University, National “1000-Youth Talents Plan”, “1000 Talents Plan” of Zhejiang Province for financial support.
Author Contributions
Shuai Shi, Khan Shah Nawaz, Muhammad Kashif Zaman and Zhankui Sun analyzed the data and wrote the paper.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Mo, J.; Wang, L.; Liu, Y.; Cui, X. Transition-metal-catalyzed direct C–H functionalization under external-oxidant-free conditions. Synthesis 2015, 47, 439–459. [Google Scholar] [CrossRef]
- Li, S.-S.; Qin, L.; Dong, L. Rhodium-catalyzed C–C coupling reactions via double C–H activation. Org. Biomol. Chem. 2016, 14, 4554–4570. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J.-P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hopkinson, M.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Musaev, D.G.; Figg, T.M.; Kaledin, A.L. Versatile reactivity of Pd-catalysts: Mechanistic features of the mono-N-protected amino acid ligand and cesium-halide base in Pd-catalyzed C–H bond functionalization. Chem. Soc. Rev. 2014, 43, 5009–5031. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lan, J.; You, J. Oxidative C–H/C–H Coupling Reactions between Two (Hetero) arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A. Platinum-Catalyzed C–H Functionalization. Chem. Rev. 2017, 117, 8483–8496. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Glorius, F. CH bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wang, F.; Li, X. C–C, C–O and C–N bond formation via rhodium (iii)-catalyzed oxidative C–H activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Patureau, F.W.; Wencel-Delord, J.; Glorius, F. Cp* Rh-Catalyzed C—H Activations. Versatile Dehydrogenative Cross-Couplings of Csp2 C—H Positions with Olefins, Alkynes, and Arenes. ChemInform 2013, 44. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium (II)-catalyzed C–H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C–H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C–C and C–N bond formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O. Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds. Chem. Soc. Rev. 2011, 40, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Rech, J.C.; Yato, M.; Duckett, D.; Ember, B.; LoGrasso, P.V.; Bergman, R.G.; Ellman, J.A. Synthesis of Potent Bicyclic Bisarylimidazole c-Jun N-Terminal Kinase Inhibitors by Catalytic C−H Bond Activation. J. Am. Chem. Soc. 2007, 129, 490–491. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, S.J.; Tan, K.L.; Watzke, A.; Bergman, R.G.; Ellman, J.A. Total synthesis of (+)-Lithospermic acid by asymmetric intramolecular alkylation via catalytic C−H bond activation. J. Am. Chem. Soc. 2005, 127, 13496–13497. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Du Bois, J. A stereoselective synthesis of (−)-tetrodotoxin. J. Am. Chem. Soc. 2003, 125, 11510–11511. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wan, Z.; Kerr, R.G.; Davies, H.M. Synthetic and isolation studies related to the marine natural products (+)-elisabethadione and (+)-elisabethamine. J. Org. Chem. 2007, 72, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. Atom Economy-A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Wehn, P.M.; Du Bois, J. A Stereoselective Synthesis of the Bromopyrrole Natural Product (−)-Agelastatin A. Angew. Chem. 2009, 121, 3860–3863. [Google Scholar] [CrossRef]
- Chen, K.; Baran, P.S. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 2009, 459, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Asymmetric Synthesis of (−)-Incarvillateine Employing an Intramolecular Alkylation via Rh-Catalyzed Olefinic C−H Bond Activation. J. Am. Chem. Soc. 2008, 130, 6316–6317. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.L.; Hughes, C.C.; Trauner, D. Concise synthesis of (±)-rhazinilam through direct coupling. Org. Lett. 2005, 7, 5207–5209. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; Corey, E. A short synthetic route to (+)-austamide, (+)-deoxyisoaustamide, and (+)-hydratoaustamide from a common precursor by a novel palladium-mediated indole → dihydroindoloazocine cyclization. J. Am. Chem. Soc. 2002, 124, 7904–7905. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef]
- Jagtap, S. Heck Reaction—State of the Art. Catalysts 2017, 7, 267. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E.-I. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Heck, R.F.; Negishi, E.-I.; Suzuki, A. Nobel Prizes 2010. Angew. Chem. Int. Ed. 2010, 49, 8300. [Google Scholar]
- Wencel-Delord, J.; Colobert, F. Asymmetric C(sp2)–H Activation. Chem. A Eur. J. 2013, 19, 14010–14017. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.C. Organocatalytic C–H activation reactions. Beilstein J. Org. Chem. 2012, 8, 1374. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.-M.; Dai, L.-X.; You, S.-L. Enantioselective Palladium-Catalyzed Direct Alkylation and Olefination Reaction of Simple Arenes. Angew. Chem. Int. Ed. 2010, 49, 5826–5828. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Shi, B.-F.; Engle, K.M.; Maugel, N.; Yu, J.-Q. Transition metal-catalyzed C–H activation reactions: Diastereoselectivity and enantioselectivity. Chem. Soc. Rev. 2009, 38, 3242–3272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-L.; Wang, Y.; Luo, Y.-C.; Fu, X.-Z.; Xu, P.-F. Asymmetric C–H functionalization involving organocatalysis. Tetrahedron Lett. 2015, 56, 3703–3714. [Google Scholar] [CrossRef]
- Musaev, D.G.; Kaledin, A.; Shi, B.-F.; Yu, J.-Q. Key mechanistic features of enantioselective C–H bond activation reactions catalyzed by [(chiral mono-N-protected amino acid)–Pd (II)] complexes. J. Am. Chem. Soc. 2012, 134, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Wang, R. Catalytic Asymmetric Activation of a Csp3–H Bond Adjacent to a Nitrogen Atom: A Versatile Approach to Optically Active α-Alkyl α-Amino Acids and C1-Alkylated Tetrahydroisoquinoline Derivatives. Angew. Chem. Int. Ed. 2011, 50, 10429–10432. [Google Scholar] [CrossRef] [PubMed]
- Wasa, M.; Engle, K.M.; Lin, D.-W.; Yoo, E.J.; Yu, J.-Q. Pd (II)-catalyzed enantioselective C–H activation of cyclopropanes. J. Am. Chem. Soc. 2011, 133, 19598–19601. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, Z.-X. Enantioselective Rhodium-Catalyzed Allylic C–H Activation for the Addition to Conjugated Dienes. Angew. Chem. 2011, 123, 2192–2195. [Google Scholar] [CrossRef]
- Davies, H.M.; Manning, J.R. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.E.; Yu, J.-Q.; Musaev, D.G. Enantioselectivity Model for Pd-Catalyzed C–H Functionalization Mediated by the Mono-N-protected Amino Acid (MPAA) Family of Ligands. ACS Catal. 2017, 7, 4344–4354. [Google Scholar] [CrossRef]
- Motevalli, S.; Sokeirik, Y.; Ghanem, A. Rhodium-Catalysed Enantioselective C–H Functionalization in Asymmetric Synthesis. Eur. J. Org. Chem. 2016, 2016, 1459–1475. [Google Scholar] [CrossRef]
- Loxq, P.; Manoury, E.; Poli, R.; Deydier, E.; Labande, A. Synthesis of axially chiral biaryl compounds by asymmetric catalytic reactions with transition metals. Coord. Chem. Rev. 2016, 308, 131–190. [Google Scholar] [CrossRef]
- Qin, Y.; Lv, J.; Luo, S. Catalytic asymmetric α-C(sp3)–H functionalization of amines. Tetrahedron Lett. 2014, 55, 551–558. [Google Scholar] [CrossRef]
- Yang, L.; Huang, H. Asymmetric catalytic carbon–carbon coupling reactions via C–H bond activation. Catal. Sci. Technol. 2012, 2, 1099. [Google Scholar] [CrossRef]
- Wasa, M.; Engle, K.M.; Yu, J.-Q. Cross-Coupling of C(sp3)–H Bonds with Organometallic Reagents via Pd(II)/Pd(0) Catalysis. Isr. J. Chem. 2010, 50, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Davies, H.M. High symmetry dirhodium (II) paddlewheel complexes as chiral catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Balensiefer, T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Kaval, N.; Tomar, S.; Eycken, E.V.; Parmar, V.S. Transition metal-catalyzed carbon-carbon bond formation Suzuki, Heck, and Sonogashira reactions using microwave and microtechnology. Org. Process Res. Dev. 2008, 12, 468–474. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Immobilized Transition Metals as Catalysts for Cross-Couplings in Continuous Flow—A Critical Assessment of the Reaction Mechanism and Metal Leaching. ChemCatChem 2014, 6, 3286–3305. [Google Scholar] [CrossRef]
- Shu, W.; Pellegatti, L.; Oberli, M.A.; Buchwald, S.L. Continuous-Flow Synthesis of Biaryls Enabled by Multistep Solid-Handling in a Lithiation/Borylation/Suzuki-Miyaura Cross-Coupling Sequence. Angew. Chem. 2011, 123, 10853–10857. [Google Scholar] [CrossRef]
- Basle, O.; Li, C.-J. Copper-Catalyzed Oxidative sp3 C–H Bond Arylation with Aryl Boronic Acids. Org. Lett. 2008, 10, 3661–3663. [Google Scholar] [CrossRef] [PubMed]
- Kirchberg, S.; Tani, S.; Ueda, K.; Yamaguchi, J.; Studer, A.; Itami, K. Oxidative Biaryl Coupling of Thiophenes and Thiazoles with Arylboronic Acids through Palladium Catalysis: Otherwise Difficult C4-Selective C–H Arylation Enabled by Boronic Acids. Angew. Chem. Int. Ed. 2011, 50, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yamaguchi, J.; Studer, A.; Itami, K. Hindered biaryls by C–H coupling: Bisoxazoline-Pd catalysis leading to enantioselective C–H coupling. Chem. Sci. 2012, 3, 2165. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Kondo, H.; Yamaguchi, J.; Itami, K. Aromatic C–H coupling with hindered arylboronic acids by Pd/Fe dual catalysts. Chem. Sci. 2013, 4, 3753. [Google Scholar] [CrossRef]
- Shi, B.-F.; Maugel, N.; Zhang, Y.-H.; Yu, J.-Q. Pd(II)-catalyzed enantioselective activation of C(sp2)–H and C(sp3)–H bonds using monoprotected amino acids as chiral ligands. Angew. Chem. Int. Ed. Engl. 2008, 47, 4882–4886. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.-J.; Lin, D.-W.; Miura, M.; Zhu, R.-Y.; Gong, W.; Wasa, M.; Yu, J.-Q. Palladium(II)-catalyzed enantioselective C(sp(3))–H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc. 2014, 136, 8138–8142. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M. Bioactive cyclobutane-containing alkaloids. J. Nat. Med. 2008, 62, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, K.; Takahashi, K.; Tsuda, E. SNF4435C and D, Novel Immunosuppressants Produced by a Strain of Streptomyces spectabilis. J. Antibiot. 2001, 54, 541–547. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fu, G.C. Applications of planar-chiral heterocycles as ligands in asymmetric catalysis. Acc. Chem. Res. 2006, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Arae, S.; Ogasawara, M. Catalytic asymmetric synthesis of planar-chiral transition-metal complexes. Tetrahedron Lett. 2015, 56, 1751–1761. [Google Scholar] [CrossRef]
- Gómez Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.C. Asymmetric catalysis with “planar-chiral” derivatives of 4-(dimethylamino) pyridine. Acc. Chem. Res. 2004, 37, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Colacot, T.J. A concise update on the applications of chiral ferrocenyl phosphines in homogeneous catalysis leading to organic synthesis. Chem. Rev. 2003, 103, 3101–3118. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.-X.; Tu, T.; You, S.-L.; Deng, W.-P.; Hou, X.-L. Asymmetric catalysis with chiral ferrocene ligands. Acc. Chem. Res. 2003, 36, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-W.; Shi, Y.-C.; Gu, Q.; Zhao, Z.-L.; You, S.-L. Enantioselective synthesis of planar chiral ferrocenes via palladium-catalyzed direct coupling with arylboronic acids. J. Am. Chem. Soc. 2013, 135, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Mahlau, M.; List, B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem. Int. Ed. 2013, 52, 518–533. [Google Scholar] [CrossRef] [PubMed]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-J.; Guan, J.; Wu, G.-J.; Xu, P.; Gao, L.-X.; Han, F.-S. Pd(II)-catalyzed enantioselective synthesis of P-stereogenic phosphinamides via desymmetric C–H arylation. J. Am. Chem. Soc. 2015, 137, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Laforteza, B.N.; Chan, K.S.; Yu, J.-Q. Enantioselective ortho-C–H cross-coupling of diarylmethylamines with organoborons. Angew Chem Int Ed Engl 2015, 54, 11143–11146. [Google Scholar] [CrossRef] [PubMed]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J., Jr. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Shi, B.-F.; Zhang, Y.-H.; Lam, J.K.; Wang, D.-H.; Yu, J.-Q. Pd(II)-catalyzed enantioselective C–H olefination of diphenylacetic acids. J. Am. Chem. Soc. 2010, 132, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Xiao, K.-J.; Yu, J.-Q. Room-temperature enantioselective C–H iodination via kinetic resolution. Science 2014, 346, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-W.; Gu, Q.; You, S.-L. Pd (II)-catalyzed intermolecular direct C–H bond Iodination: An efficient approach toward the synthesis of axially chiral compounds via kinetic resolution. ACS Catal. 2014, 4, 2741–2745. [Google Scholar] [CrossRef]
- Xiao, K.-J.; Chu, L.; Yu, J.-Q. Enantioselective C–H Olefination of alpha-Hydroxy and alpha-Amino Phenylacetic Acids by Kinetic Resolution. Angew. Chem. Int. Ed. Engl. 2016, 55, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kagan, H.; Fiaud, J. Kinetic resolution. Top. Stereochem 1988, 18, 21. [Google Scholar]
- Yao, Q.-J.; Zhang, S.; Zhan, B.-B.; Shi, B.-F. Atroposelective Synthesis of Axially Chiral Biaryls by Palladium-Catalyzed Asymmetric C–H Olefination Enabled by a Transient Chiral Auxiliary. Angew. Chem. Int. Ed. Engl. 2017, 56, 6617–6621. [Google Scholar] [CrossRef] [PubMed]
- Pi, C.; Li, Y.; Cui, X.; Zhang, H.; Han, Y.; Wu, Y. Redox of ferrocene controlled asymmetric dehydrogenative Heck reaction via palladium-catalyzed dual C–H bond activation. Chem. Sci. 2013, 4, 2675. [Google Scholar] [CrossRef]
- Gao, D.-W.; Gu, Q.; You, S.-L. An Enantioselective Oxidative C–H/C–H Cross-Coupling Reaction: Highly Efficient Method to Prepare Planar Chiral Ferrocenes. J. Am. Chem. Soc. 2016, 138, 2544–2547. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; You, S.-L. Construction of axial chirality by rhodium-catalyzed asymmetric dehydrogenative Heck coupling of biaryl compounds with alkenes. Angew. Chem. Int. Ed. Engl. 2014, 53, 13244–13247. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts. Angew. Chem. Int. Ed. Engl. 2016, 55, 12150–12162. [Google Scholar] [CrossRef] [PubMed]


















































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).