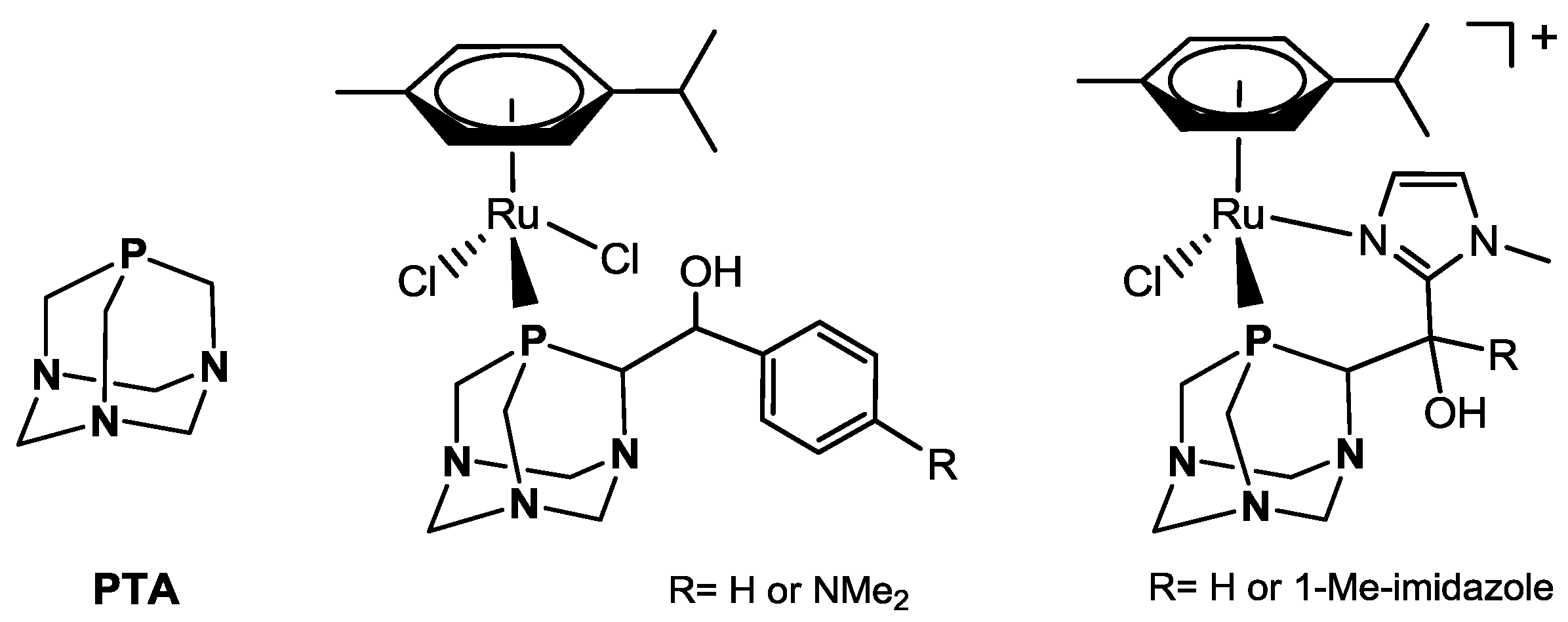
Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase †




Abstract
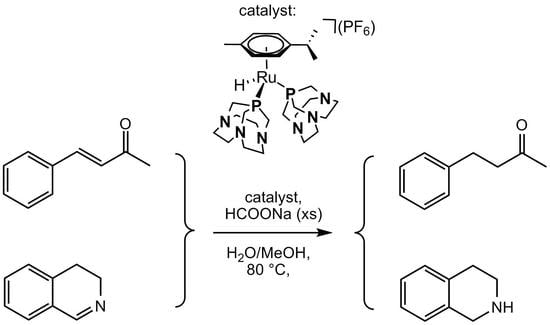
:
1. Introduction
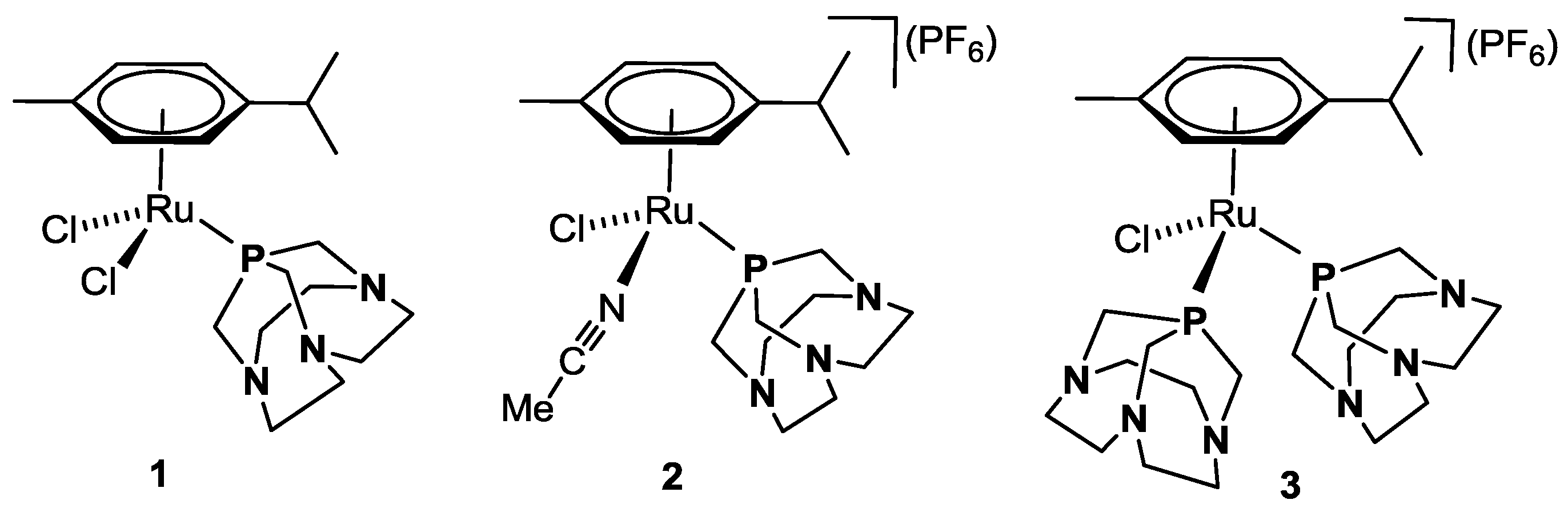
2. Results
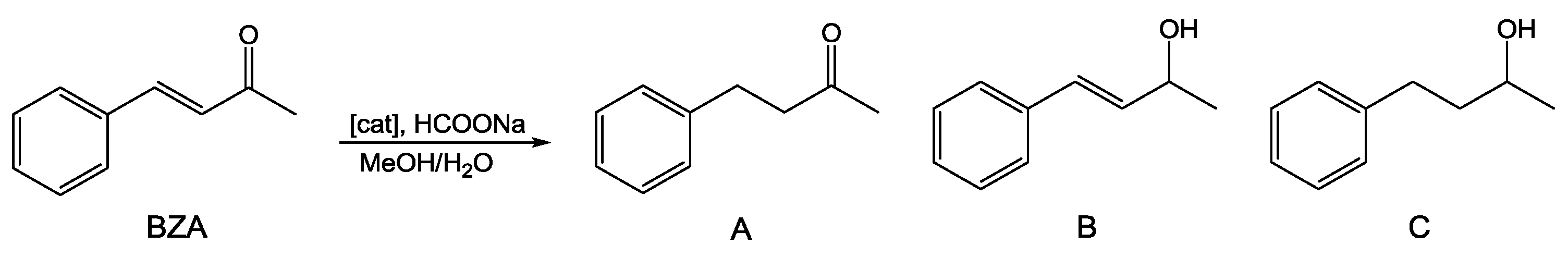
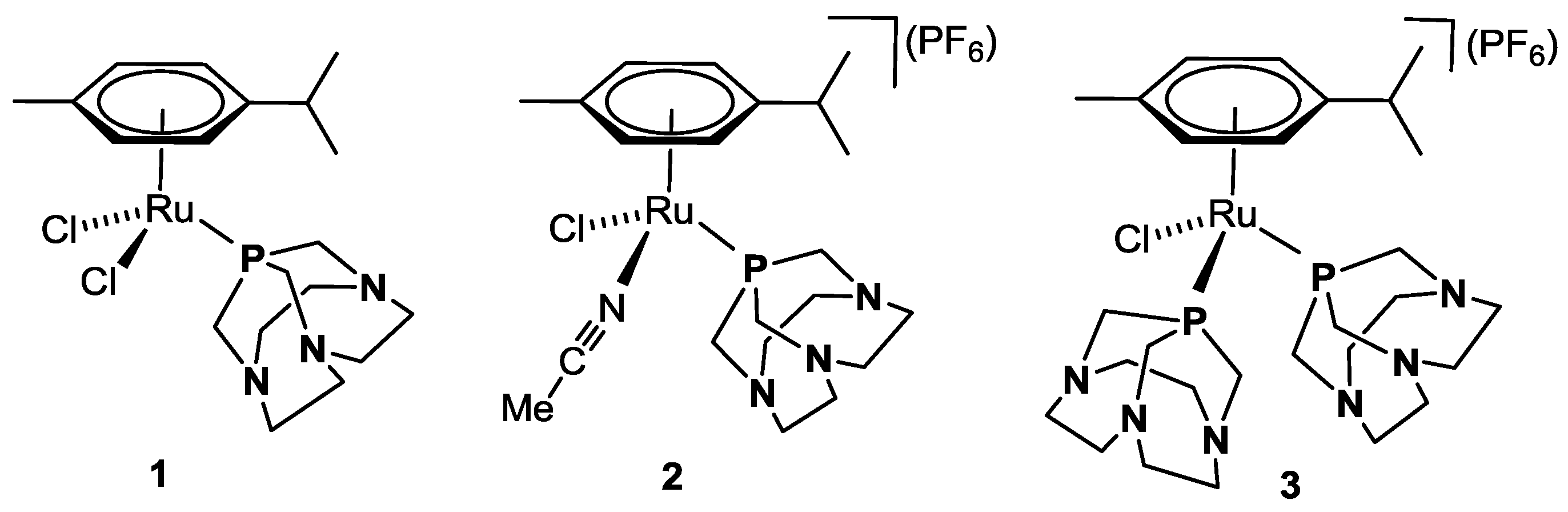
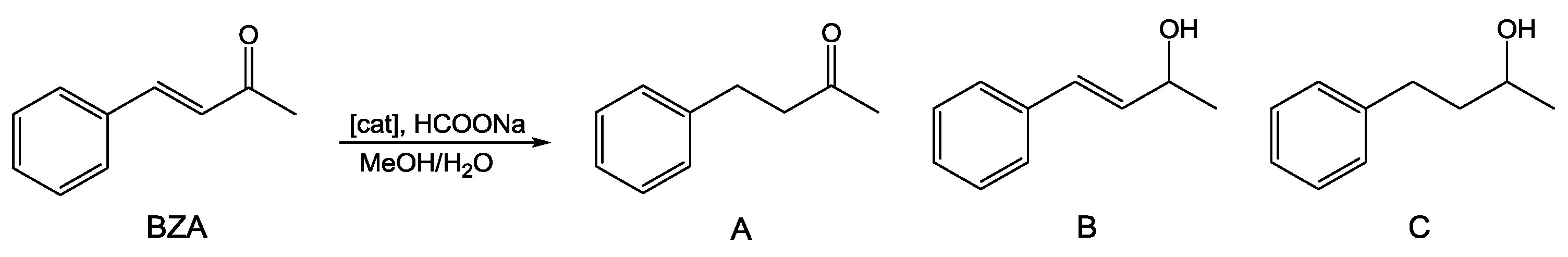
2.1. Catalytic Transfer Hydrogenation of BZA
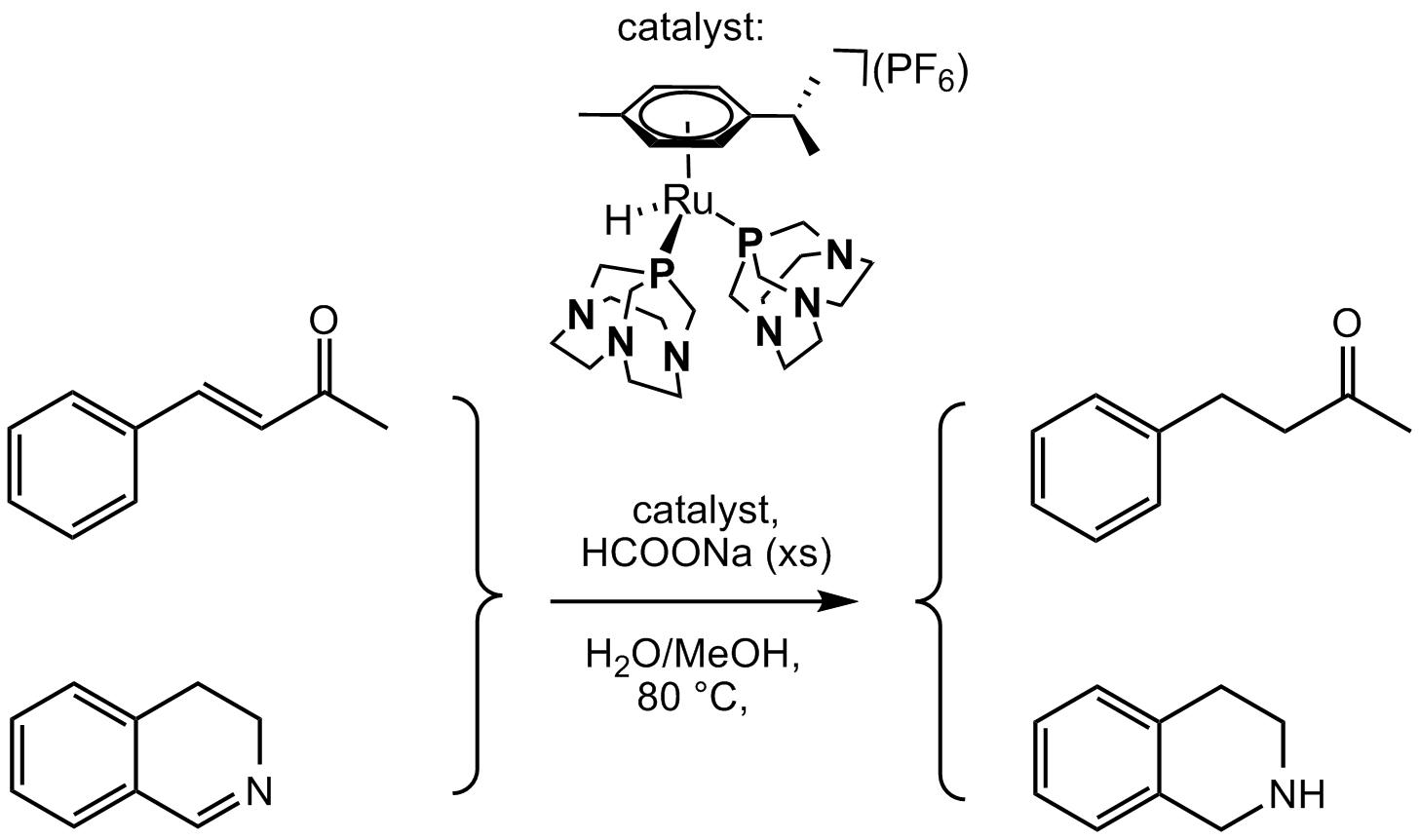
2.2. Transfer Hydrogenation of Cyclic Imines
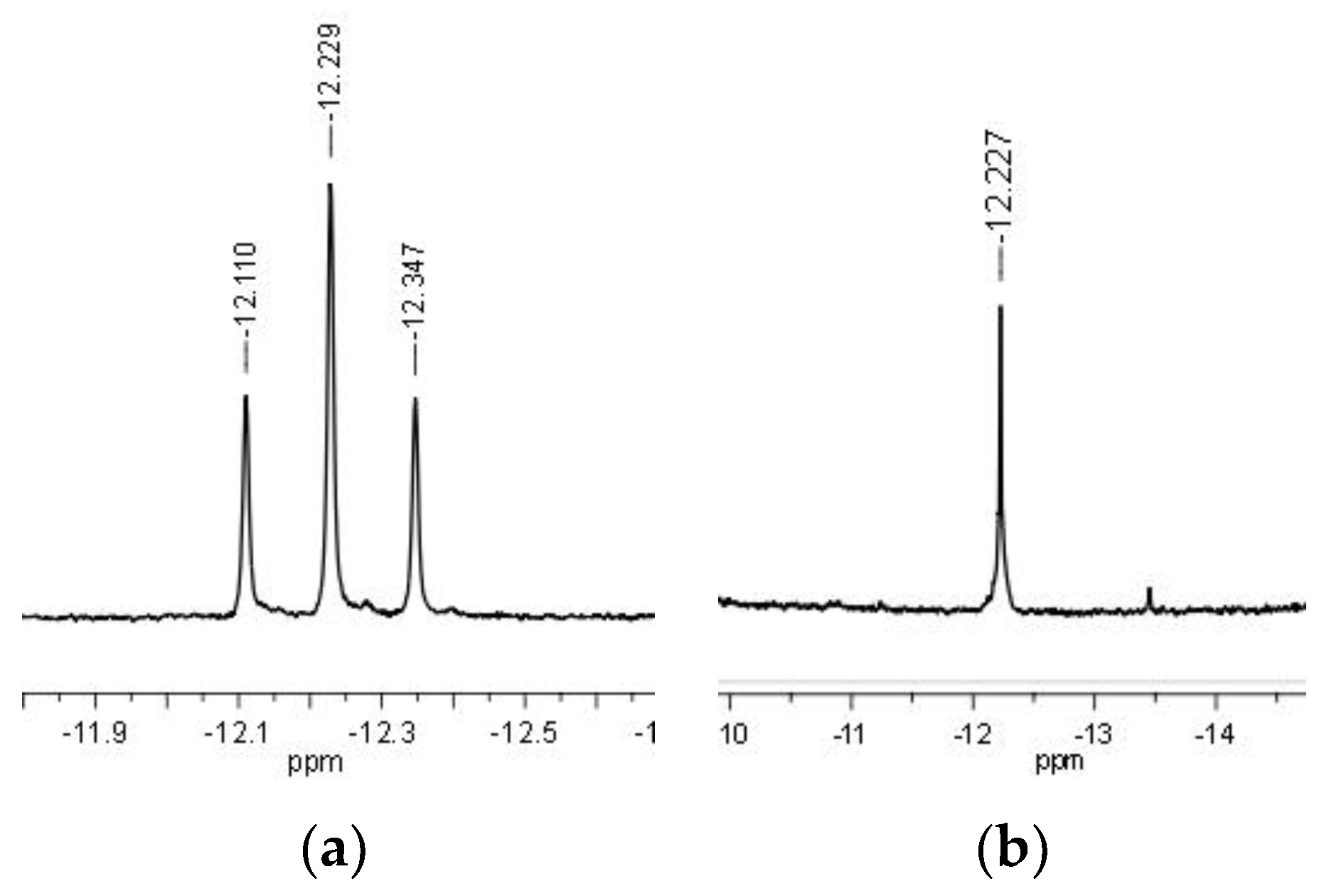
2.3. NMR Mechanistic Studies under Pseudo-Catalytic Conditions
3. Experimental Section
3.1. Materials and Methods
3.2. Transfer Hydrogenation Tests
3.3. NMR Scale Experiments
3.4. Synthesis of κP-[RuH(η6-p-cymene)(CAP)2](PF6) 4
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morris, R.H. Ruthenium and Osmium. In The Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 3; pp. 45–70. ISBN 978-3-527-31161-3. [Google Scholar]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Evans, D.; Osborn, J.A.; Jardine, F.H.; Wilkinson, G. Homogeneous Hydrogenation and Hydroformylation using Ruthenium Complexes. Nature 1965, 208, 1203–1204. [Google Scholar] [CrossRef]
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Zanotti-Gerosa, A.; Hems, W.; Groarke, M.; Hancock, F. Ruthenium-Catalysed Asymmetric Reduction of Ketones. Platin. Met. Rev. 2005, 49, 158–165. [Google Scholar] [CrossRef]
- Clarke, M.L.; Dìaz-Valenzuela, M.B.; Slawin, A.M.Z. Hydrogenation of Aldehydes, Esters, Imines, and Ketones Catalyzed by a Ruthenium Complex of a Chiral Tridentate Ligand. Organometallics 2007, 26, 16–19. [Google Scholar] [CrossRef]
- James, B.R. Synthesis of chiral amines catalyzed homogeneously by metal complexes. Catal. Today 1997, 37, 209–221. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.E. Ruthenium-Catalyzed Transfer Hydrogenation of Imines by Propan-2-ol in Benzene. Chem. Eur. J. 2002, 8, 2955–2961. [Google Scholar] [CrossRef]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef]
- Ito, J.-I.; Nishiyama, H. Recent topics of transfer hydrogenation. Tetrahedron Lett. 2014, 55, 3133–3146. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, K.H. Hydrophilic ligands and their application in aqueous-phase metal-catalyzed reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Lorenzini, F. Developments in the chemistry of tris(hydroxymethyl)phosphine. Coord. Chem. Rev. 2010, 254, 420–430. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA). Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA) and derivatives. Part II. The quest for tailored ligands, complexes and related applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA) and derivatives. Part III. Variations on a theme: Novel architectures, materials and applications. Coord. Chem. Rev. 2018, 355, 328–361. [Google Scholar] [CrossRef]
- Akbayeva, D.N.; Gonsalvi, L.; Oberhauser, W.; Peruzzini, M.; Vizza, F.; Brüggeller, P.; Romerosa, A.; Sava, G.; Bergamo, A. Synthesis, catalytic properties and biological activity of new water soluble ruthenium cyclopentadienyl PTA complexes [(C5R5)RuCl(PTA)2] (R= H, Me; PTA= 1,3,5-triaza-7-phosphaadamantane). Chem. Commun. 2003, 264–265. [Google Scholar] [CrossRef]
- Bolaño, S.; Gonsalvi, L.; Zanobini, F.; Vizza, F.; Bertolasi, V.; Romerosa, A.; Peruzzini, M. Water soluble ruthenium cyclopentadienyl and aminocyclopentadienyl PTA complexes as catalysts for selective hydrogenation of α,β-unsaturated substrates (PTA = 1,3,5-triaza-7-phosphaadamantane). J. Mol. Catal. A Chem. 2004, 224, 61–70. [Google Scholar] [CrossRef]
- Dyson, P.J.; Ellis, D.J.; Laurenczy, G. Minor Modifications to the Ligands Surrounding a Ruthenium Complex Lead to Major Differences in the Way in which they Catalyse the Hydrogenation of Arenes. Adv. Synth. Catal. 2003, 345, 211–215. [Google Scholar] [CrossRef]
- Krogstad, D.A.; Guerriero, A.; Ienco, A.; Manca, G.; Peruzzini, M.; Reginato, G.; Gonsalvi, L. Imidazolyl-PTA Derivatives as Water-Soluble (P,N) Ligands for Ruthenium-Catalyzed Hydrogenations. Organometallics 2011, 30, 6292–6302. [Google Scholar] [CrossRef]
- Guerriero, A.; Oberhauser, W.; Riedel, T.; Peruzzini, M.; Dyson, P.J.; Gonsalvi, L. New Class of Half-Sandwich Ruthenium(II) Arene Complexes Bearing the Water-Soluble CAP Ligand as an in Vitro Anticancer Agent. Inorg. Chem. 2017, 56, 5514–5518. [Google Scholar] [CrossRef] [PubMed]
- Britvin, S.N.; Lotnyk, A. Water-Soluble Phosphine Capable of Dissolving Elemental Gold: The Missing Link between 1,3,5-triaza-7-phosphaadamantane (PTA) and Verkade’s Ephemeral Ligand. J. Am. Chem. Soc. 2015, 137, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Britvin, S.N.; Rumyantsev, A.M.; Zobnina, A.E.; Padkina, M.V. Between Adamantane and Atrane: Intrabridgehead Interactions in the Cage-Like Phosphane Related to a Novel Tris(homoadamantane) Ring System. Chem. Eur. J. 2016, 22, 14227–14235. [Google Scholar] [CrossRef] [PubMed]
- Fleury-Brégeot, N.; de la Fuente, V.; Castillón, S.; Claver, C. Highlights of Transition Metal-Catalyzed Asymmetric Hydrogenation of Imines. ChemCatChem 2010, 2, 1346–1371. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Canivet, J.; Süss-Fink, G. Water-soluble arene ruthenium catalysts containing sulfonated diamine ligands for asymmetric transfer hydrogenation of α-aryl ketones and imines in aqueous solution. Green Chem. 2007, 9, 391–397. [Google Scholar] [CrossRef]
- Barbaro, P.; Gonsalvi, L.; Guerriero, A.; Liguori, F. Facile heterogeneous catalytic hydrogenation of C=N and C=O bonds in neat water: Anchoring of water-soluble metal complexes onto ion-exchange resins. Green Chem. 2012, 14, 3211–3219. [Google Scholar] [CrossRef]
- Antkiewicz-Michaluk, L.; Wąsik, A.; Michaluk, J. 1-Methyl-1,2,3,4-Tetrahydroisoquinoline, an Endogenous Amine with Unexpected Mechanism of Action: New Vistas of Therapeutic Application. Neurotox. Res. 2014, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.J.; Mebi, C.A. Aqueous organometallic chemistry: Synthesis, structure and reactivity of the water-soluble metal hydride CpRu(PTA)2H. Organometallics 2004, 23, 5317–5323. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]-decane): A Water Soluble Compound That Exhibits pH Dependent DNA Binding Providing Selectivity for Diseased Cells. Chem. Commun. 2001, 2, 1396–1397. [Google Scholar] [CrossRef]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, A.K. (η6-Hexamethylbenzene)Ruthenium Complexes. Inorg. Synth. 1982, 21, 74–78. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, Y.; Wang, R.; Wu, G.; Chen, P. Lithium amidoborane, a highly chemoselective reagent for the reduction of α,β-unsaturated ketones to allylic alcohols. Org. Biomol. Chem. 2012, 10, 367–371. [Google Scholar] [CrossRef] [PubMed]
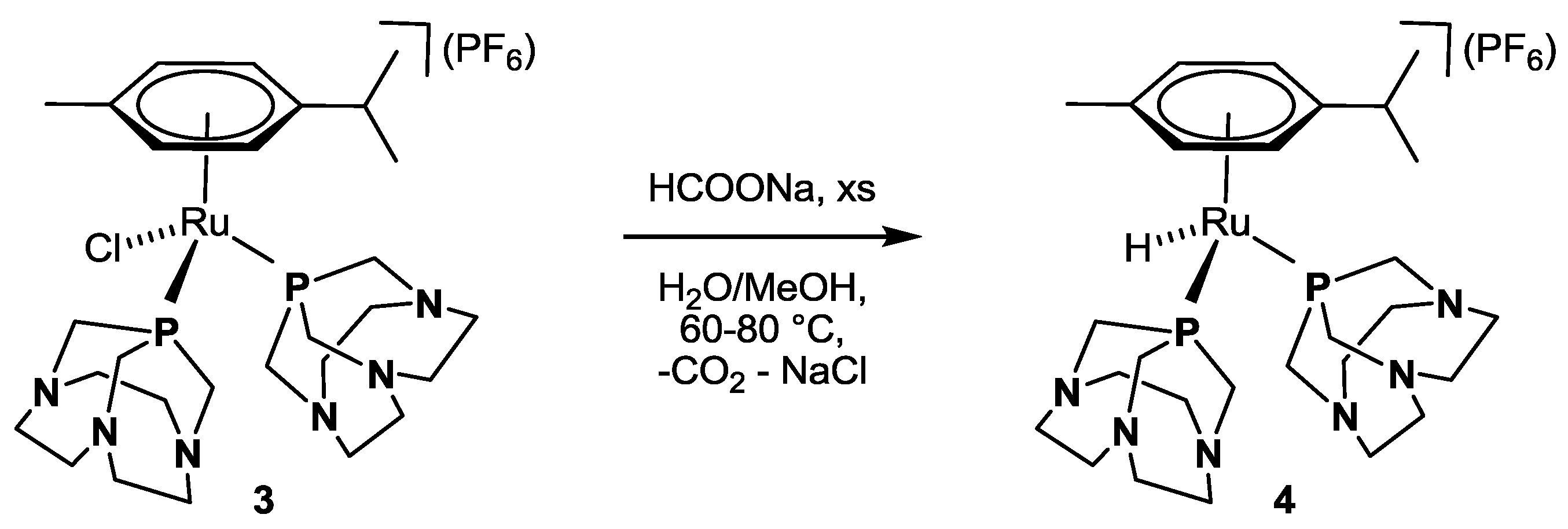


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
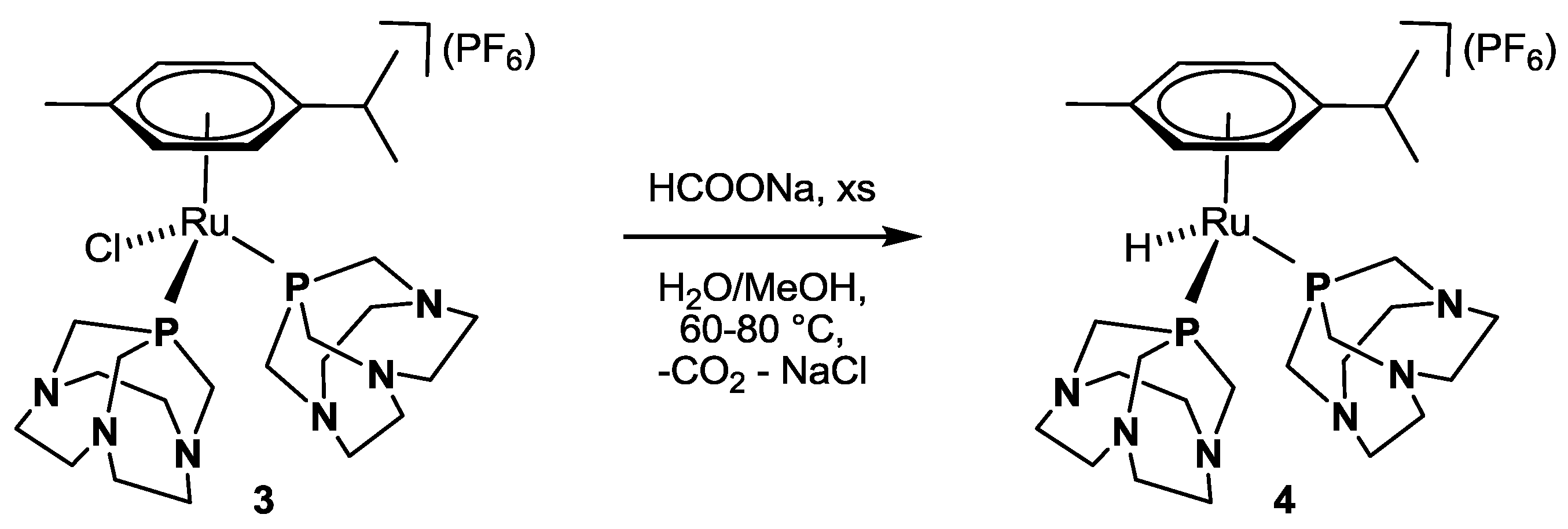{kind=link}
| Entry | Catalyst | T (°C) | % Conv. | Time (h) | Yield A (%) b | Yield B (%) b | Yield C (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 60 | 60.0 | 24 | 45.3 | 13.0 | 1.7 |
| 2 c | 1 | 60 | 18.7 | 24 | 14.2 | 4.4 | 0.1 |
| 3 | 1 | 80 | 99.4 | 4 | 82.0 | 5.2 | 12.2 |
| 4 c | 1 | 80 | 76.3 | 4 | 61.7 | 12.8 | 1.8 |
| 5 | 2 | 80 | 82.6 | 4 | 68.9 | 11.5 | 2.2 |
| 6 c | 2 | 80 | 51.9 | 4 | 40.4 | 10.8 | 0.7 |
| 7 | 3 | 80 | 71.8 | 24 | 52.3 | 15.8 | 3.7 |
| 96.0 | 48 | 68.7 | 13.5 | 13.8 | |||
| 8 c | 3 | 80 | 66.5 | 24 | 46.6 | 17.8 | 2.1 |
| 92.5 | 48 | 73.5 | 13.0 | 6.0 | |||
| 9 | RAPTA-C | 80 | 99.5 | 24 | 93.2 | 3.2 | 3.1 |
| 10 c | RAPTA-C | 80 | 30.0 | 24 | 23.8 | 6.0 | 0.2 |
| Entry | T (°C) | Solvent | Substrate | Product b | Yield c | Time (h) |
|---|---|---|---|---|---|---|
| 1 | 80 | H2O | 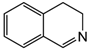 | 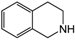 | 39.9 | 24 |
| 3,4-dihydroisoquinoline | 1,2,3,4-tetrahydroisoquinoline | |||||
| 2 | 60 | MeOH/H2O 1:1 |  | 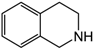 | 10.9 | 24 |
| 3 | 80 | MeOH/H2O 1:1 | 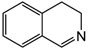 | 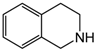 | 74.1 92.5 | 24 48 |
| 4 | 80 | MeOH/H2O 1:1 | 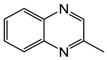 | 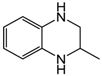 | 0.4 | 24 |
| 2-methylquinoxaline | 2-methyl-1,2,3,4-tetrahydroquinoxaline | |||||
| 5 | 80 | MeOH/H2O 1:1 | 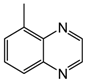 |  | 0.0 | 48 |
| 5-methylquinoxaline | 5-methyl-1,2,3,4-tetrahydroquinoxaline | |||||
| 6 | 80 | MeOH/H2O 1:1 | 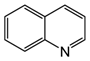 | 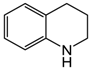 | 1.5 3.0 | 24 48 |
| quinoline | 1,2,3,4-tetrahydroquinoline |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase. Catalysts 2018, 8, 88. https://doi.org/10.3390/catal8020088
Guerriero A, Peruzzini M, Gonsalvi L. Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase. Catalysts. 2018; 8(2):88. https://doi.org/10.3390/catal8020088
Chicago/Turabian StyleGuerriero, Antonella, Maurizio Peruzzini, and Luca Gonsalvi. 2018. "Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase" Catalysts 8, no. 2: 88. https://doi.org/10.3390/catal8020088
APA StyleGuerriero, A., Peruzzini, M., & Gonsalvi, L. (2018). Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase. Catalysts, 8(2), 88. https://doi.org/10.3390/catal8020088