AuPd/3DOM TiO2 Catalysts: Good Activity and Stability for the Oxidation of Trichloroethylene



Abstract
:1. Introduction
2. Results and Discussion
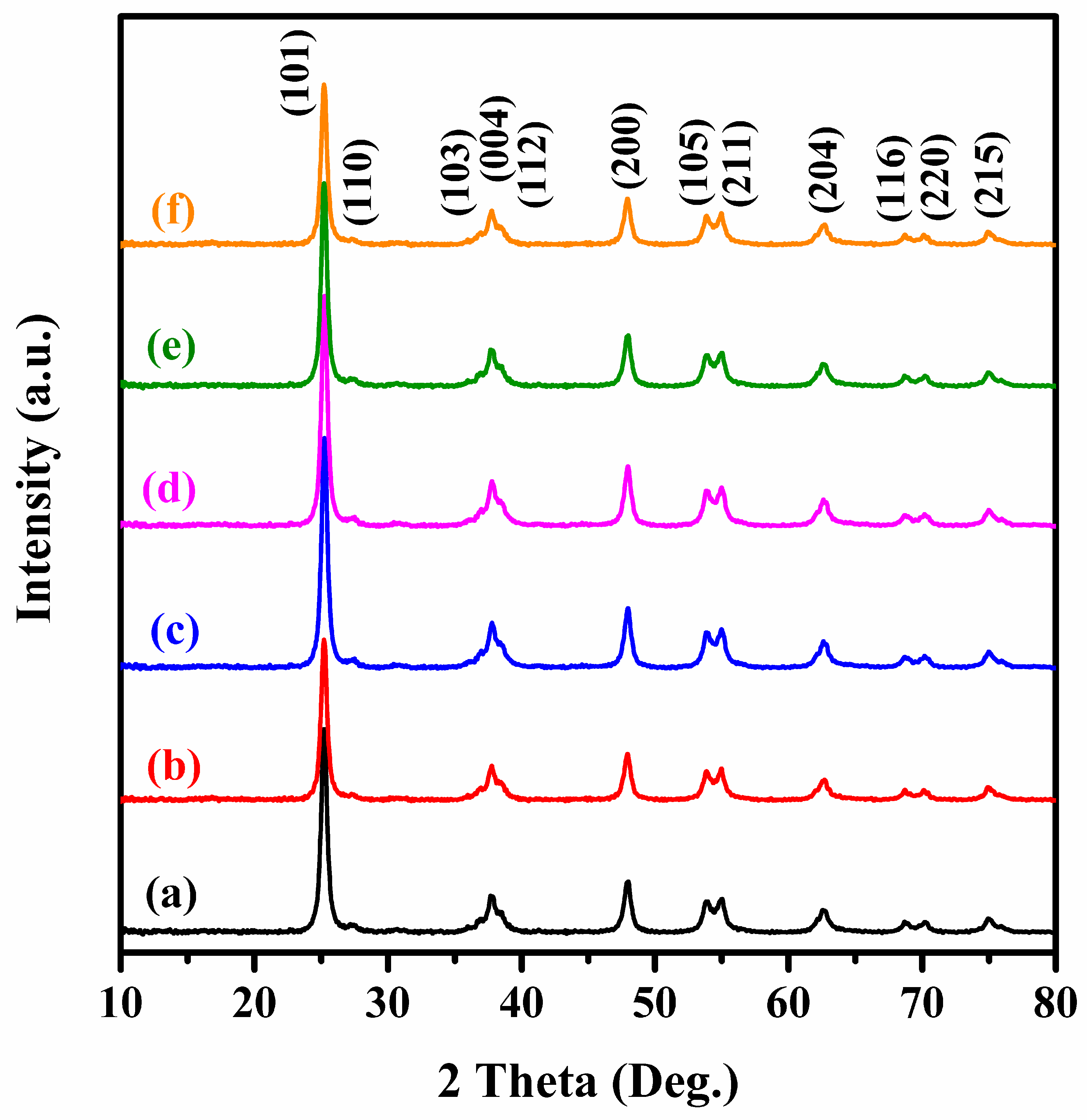
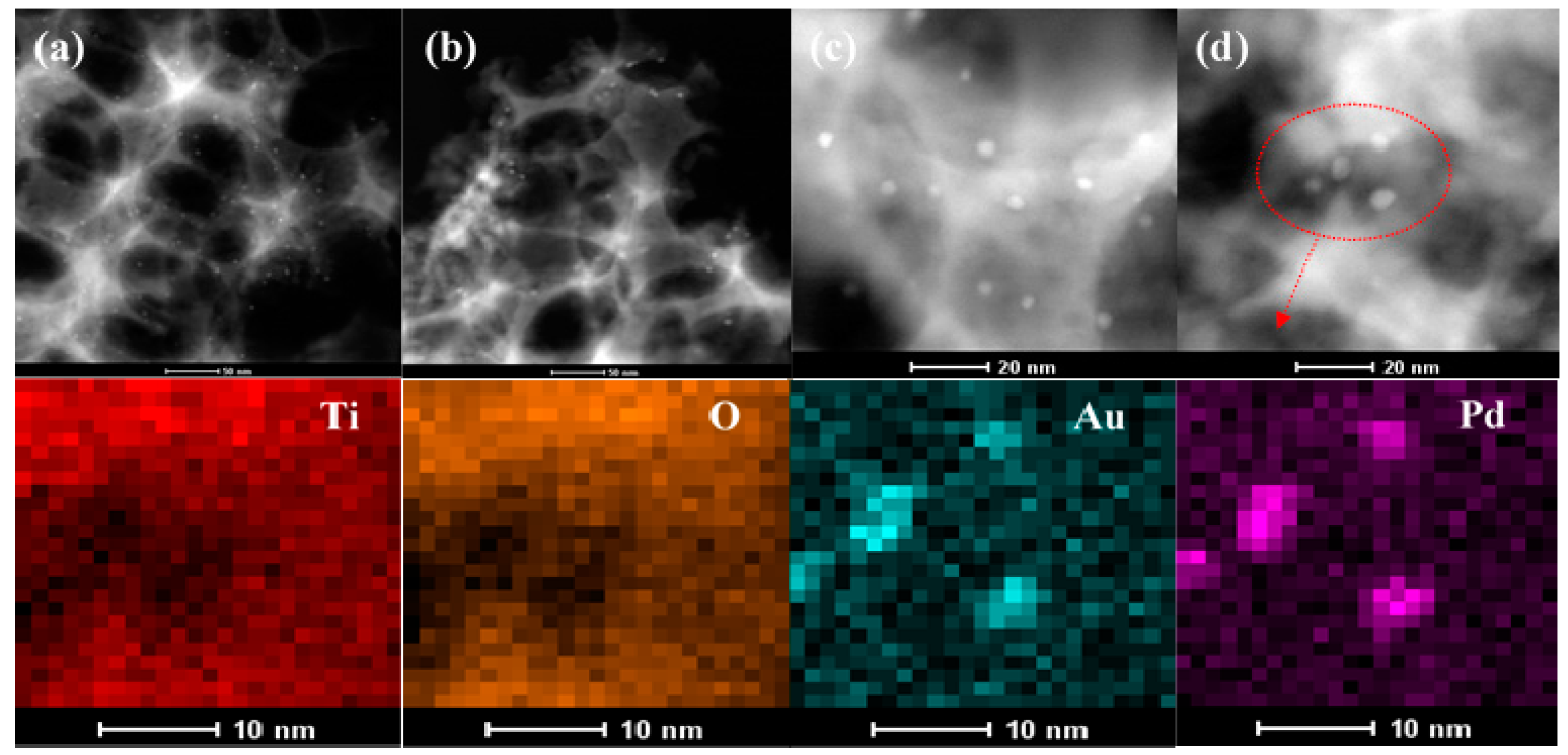
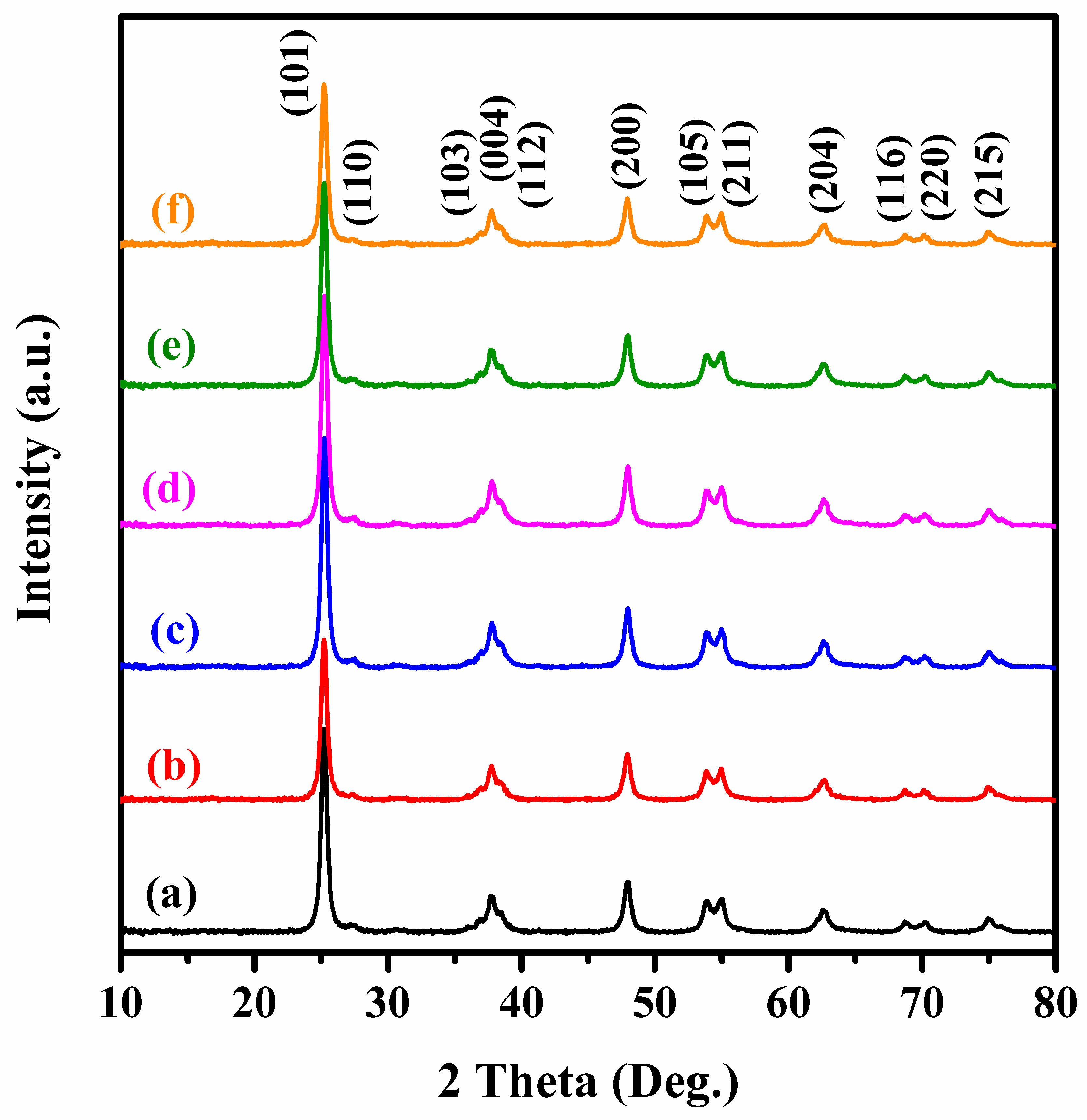
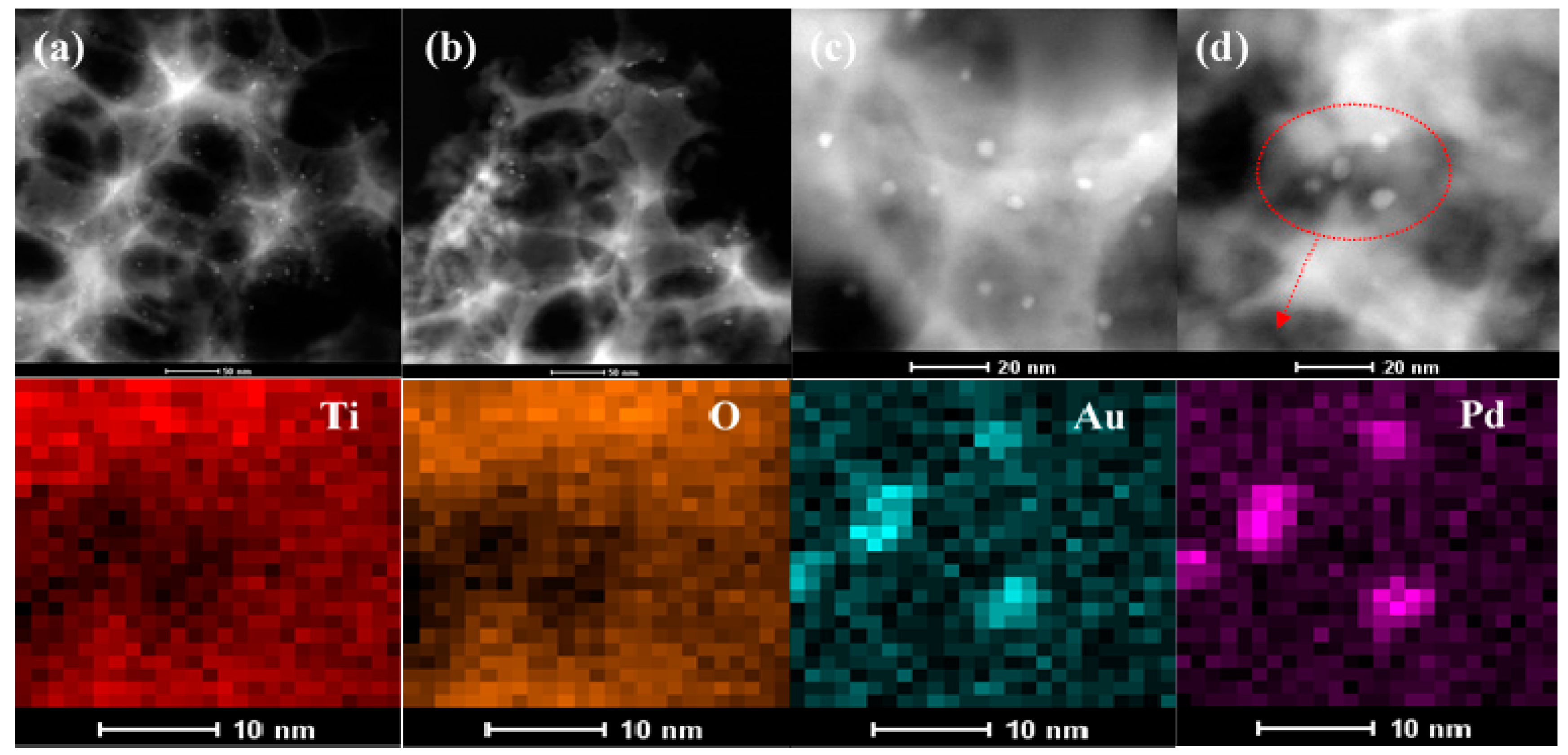
2.1. Crystal Phase, Morphology, Pore Structure, and Surface Area
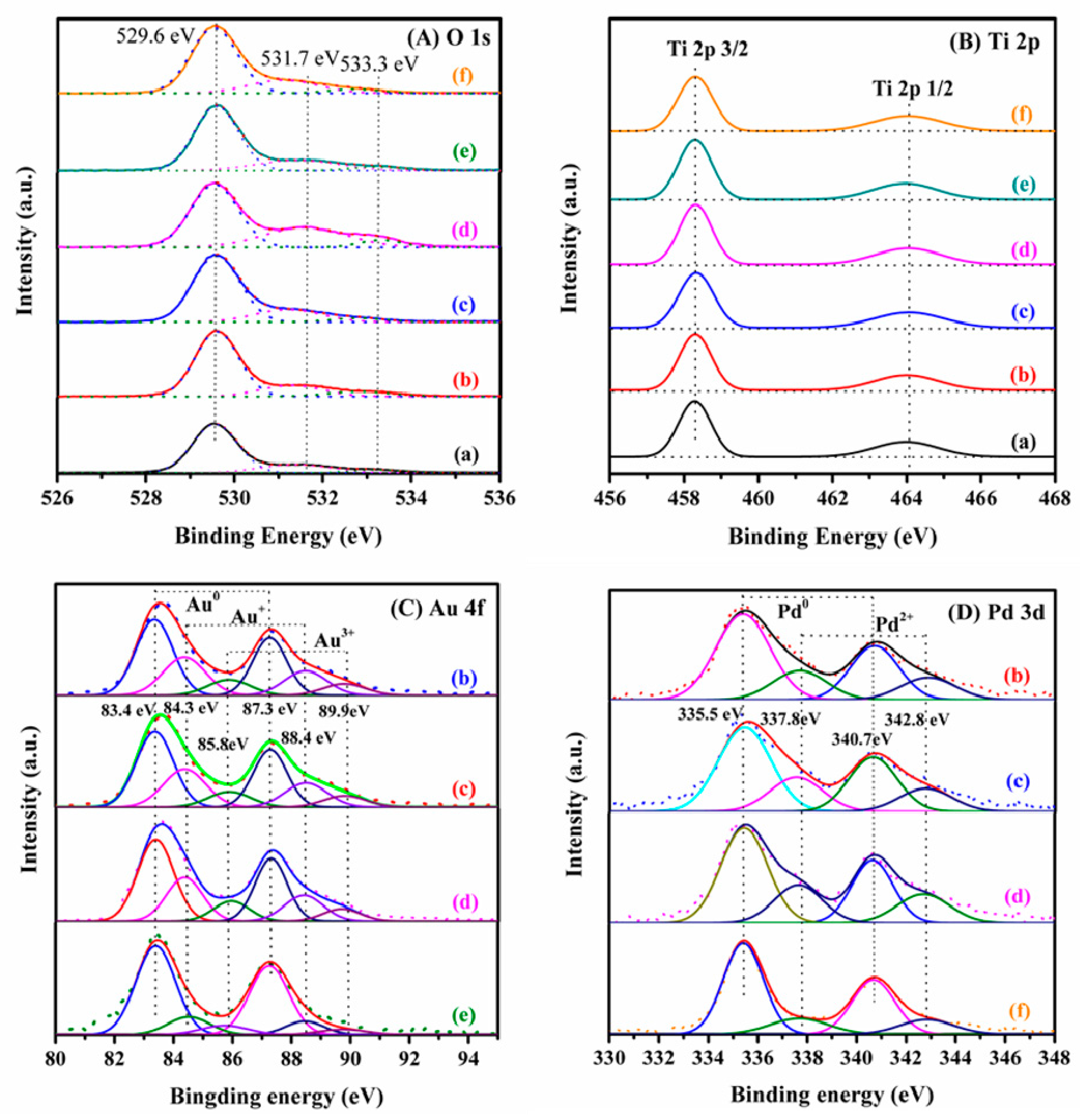
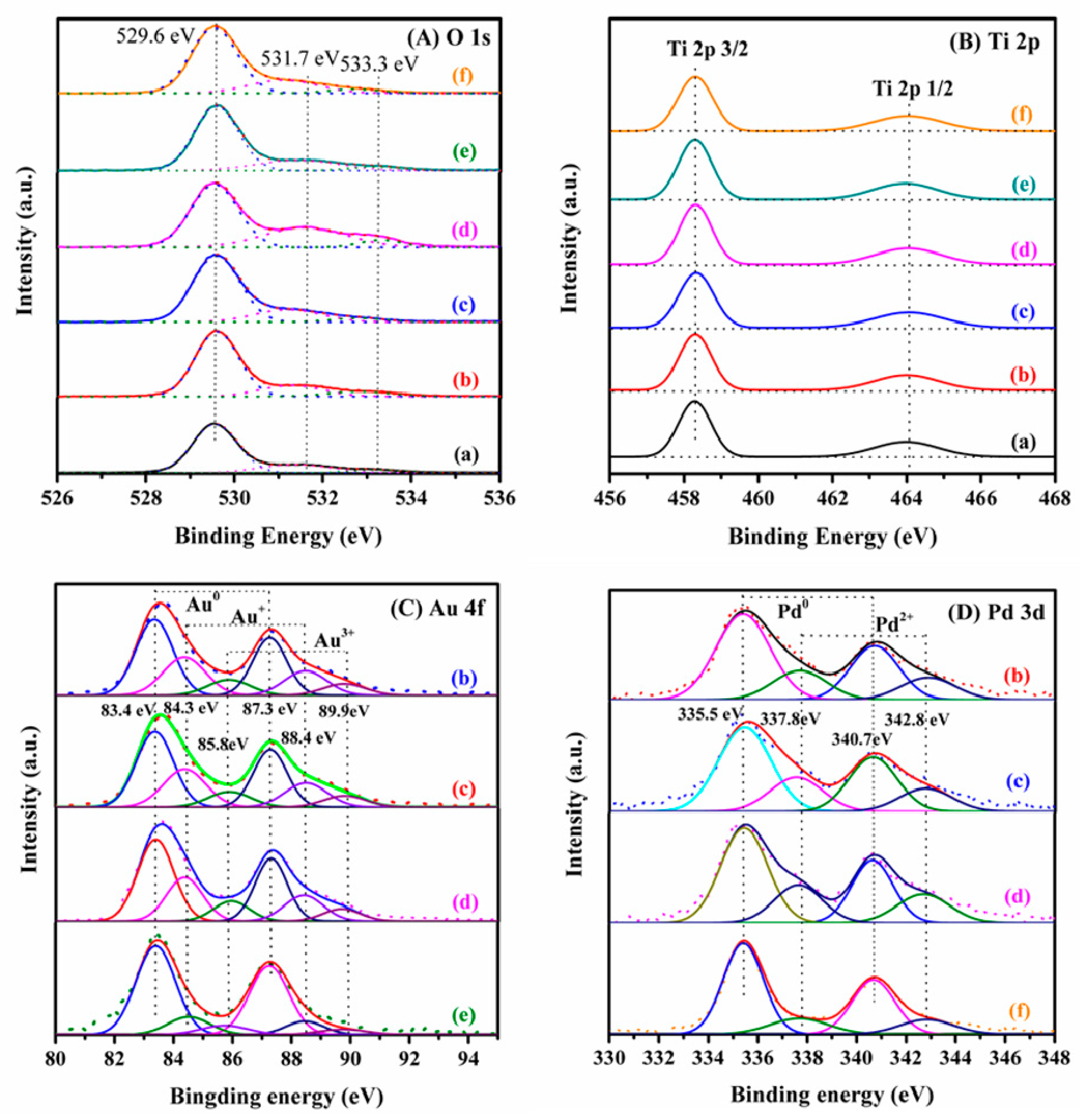
2.2. Metal Oxidation State and Surface Element Composition
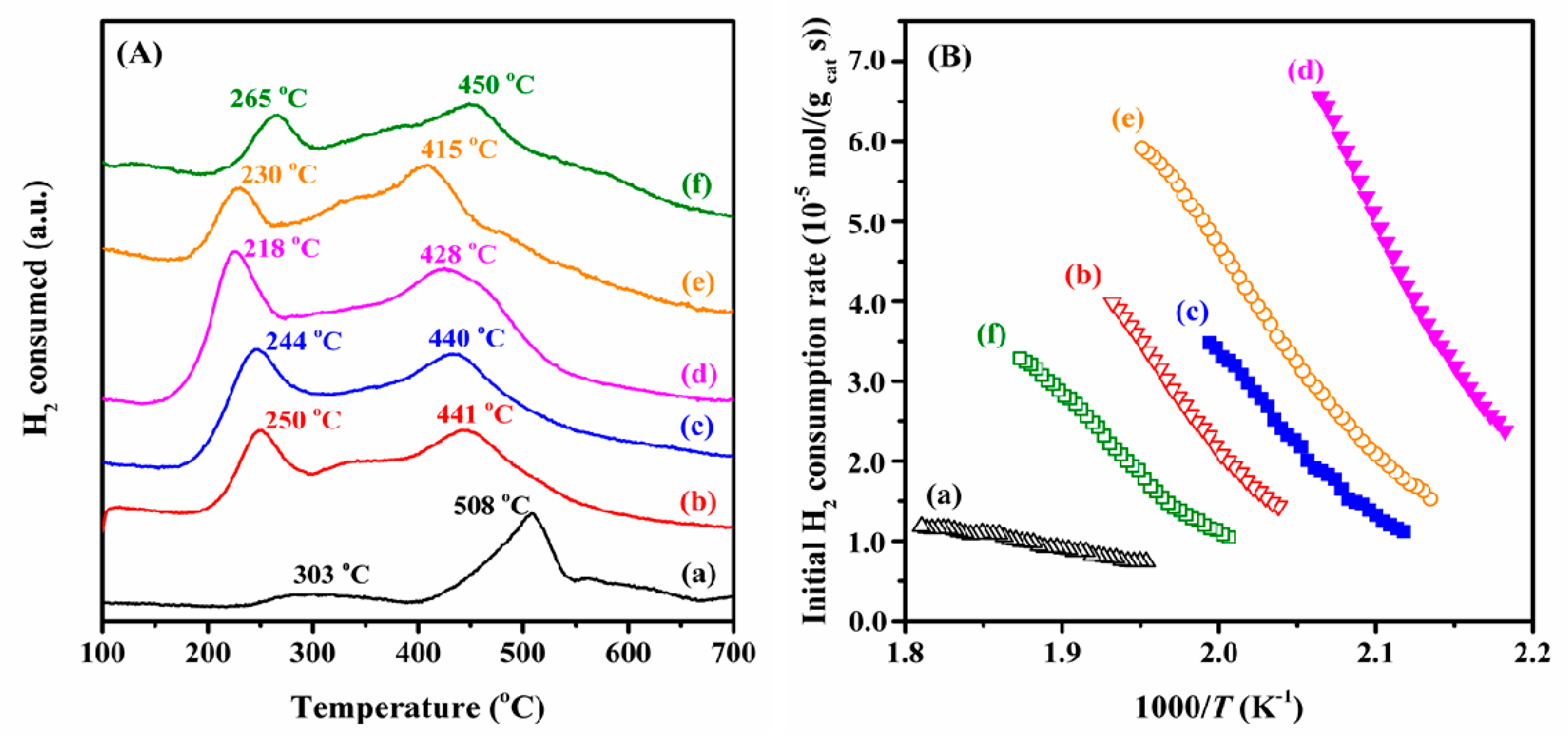
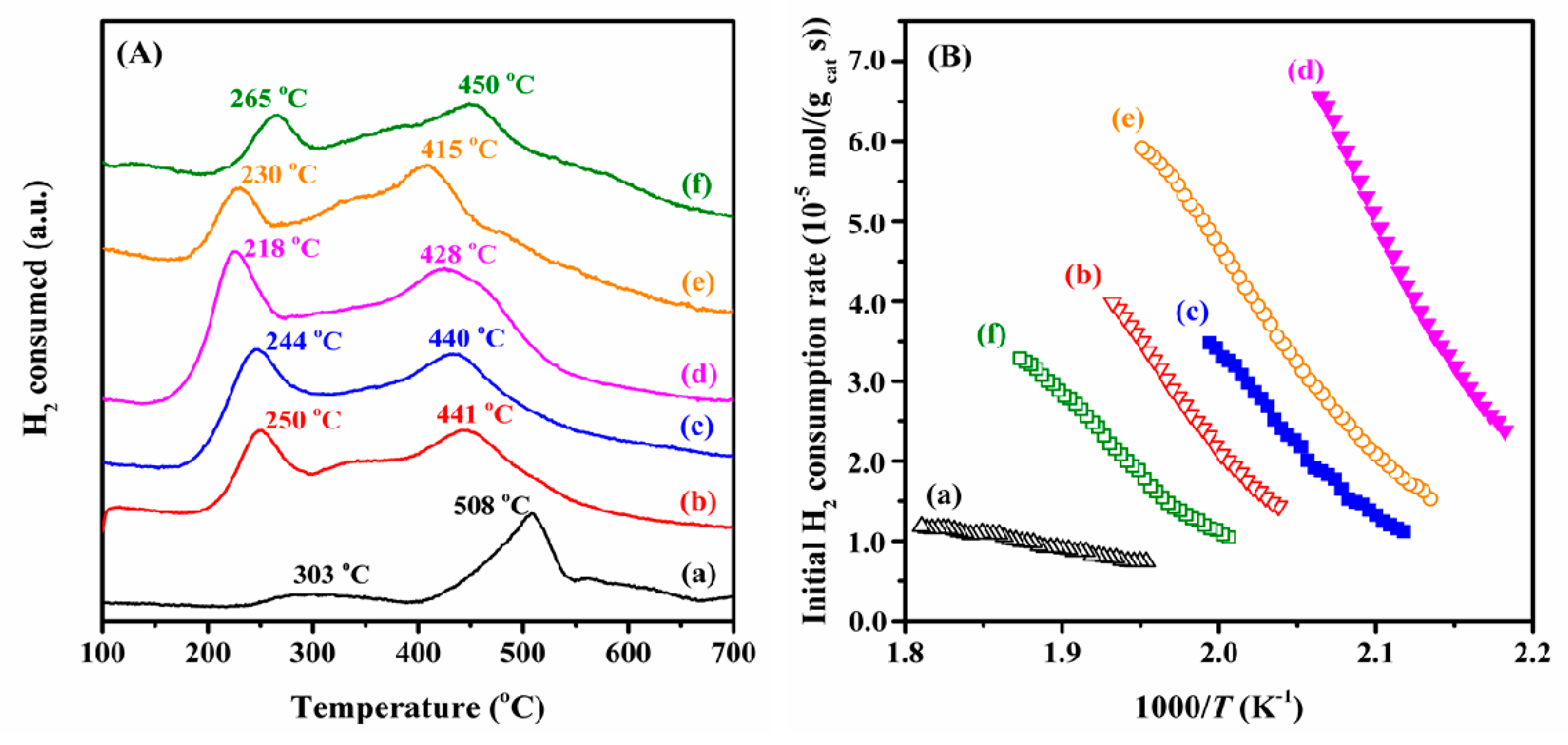
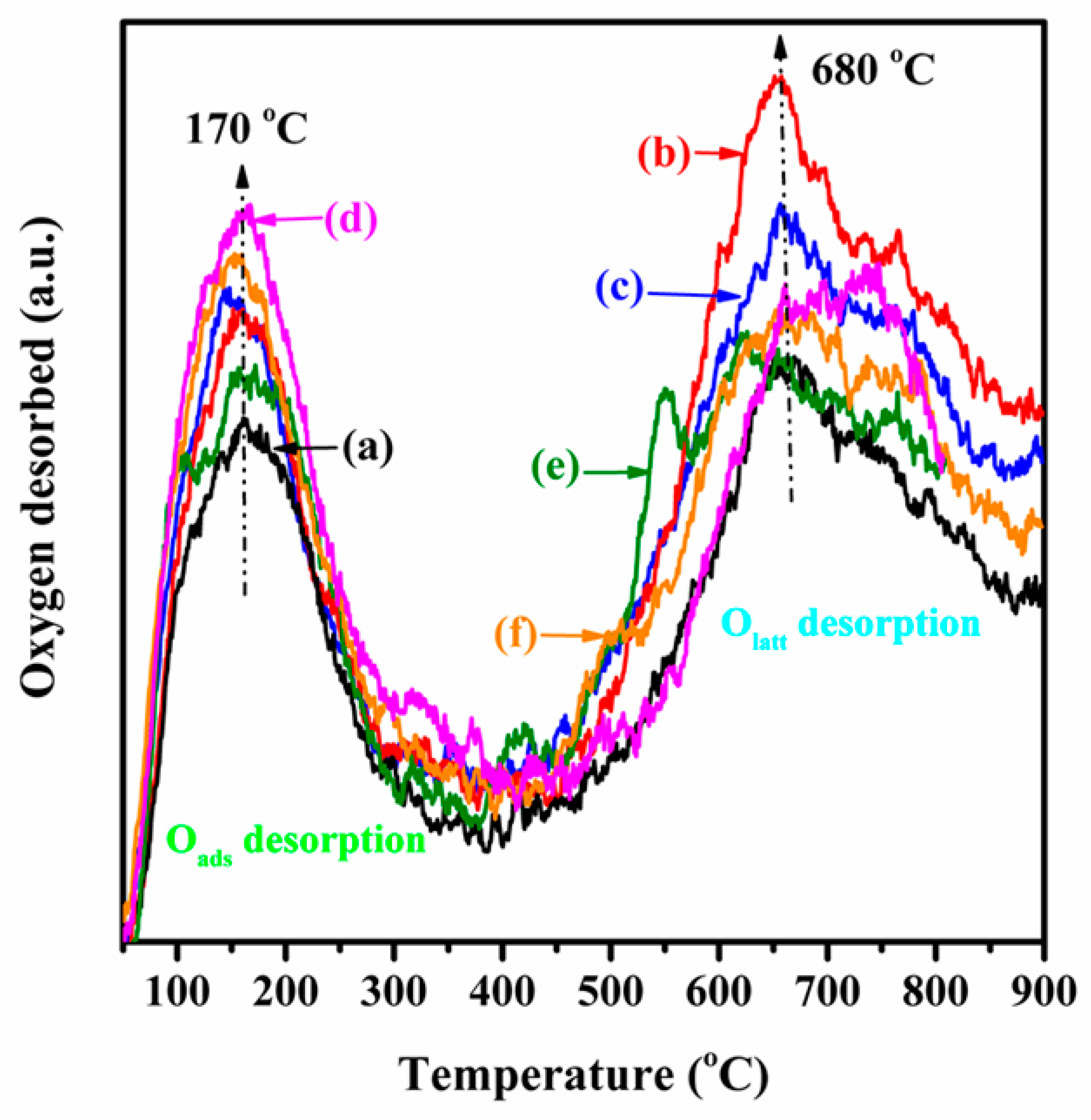
2.3. Low-Temperature Reducibility and Oxygen Species
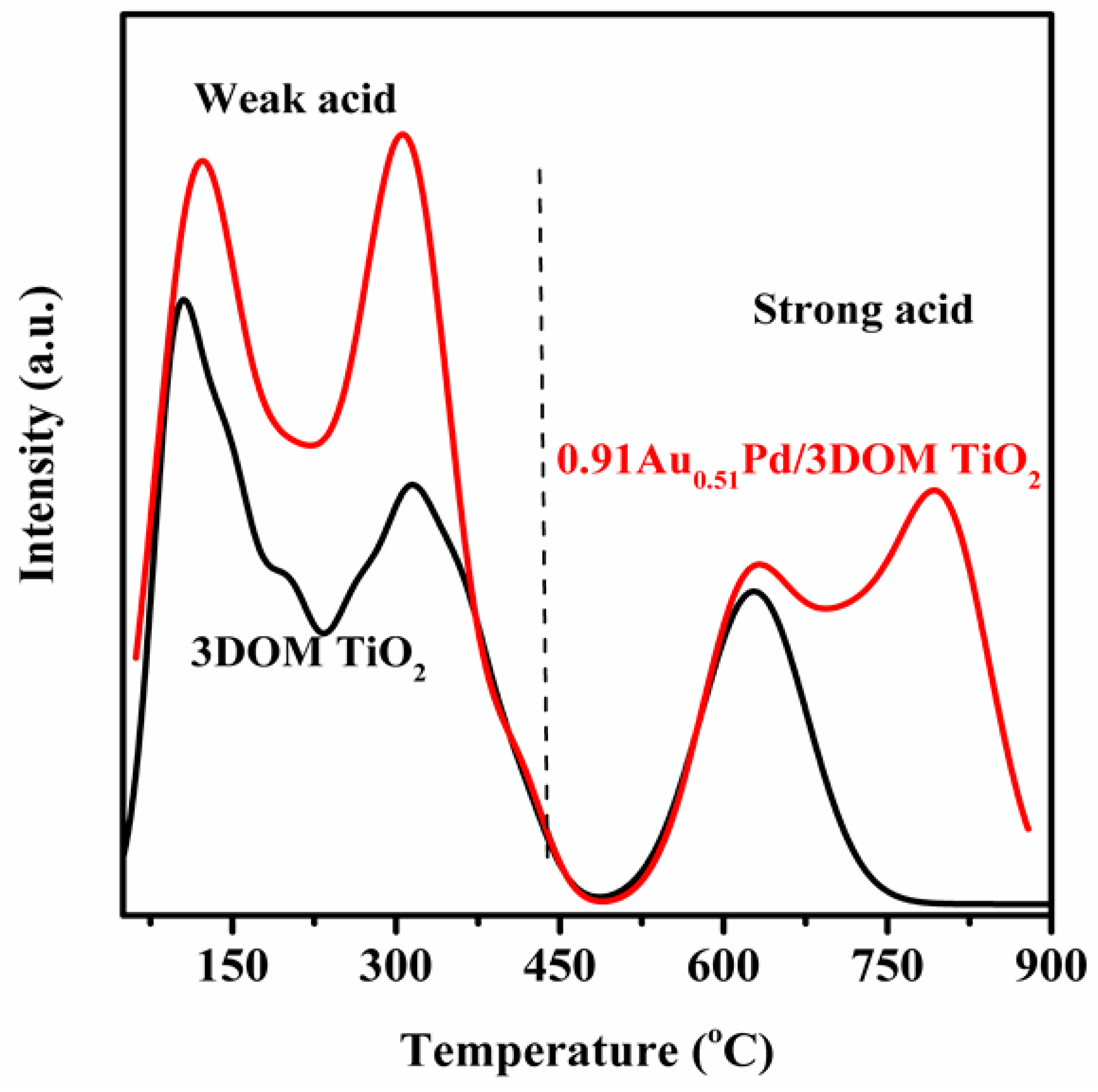
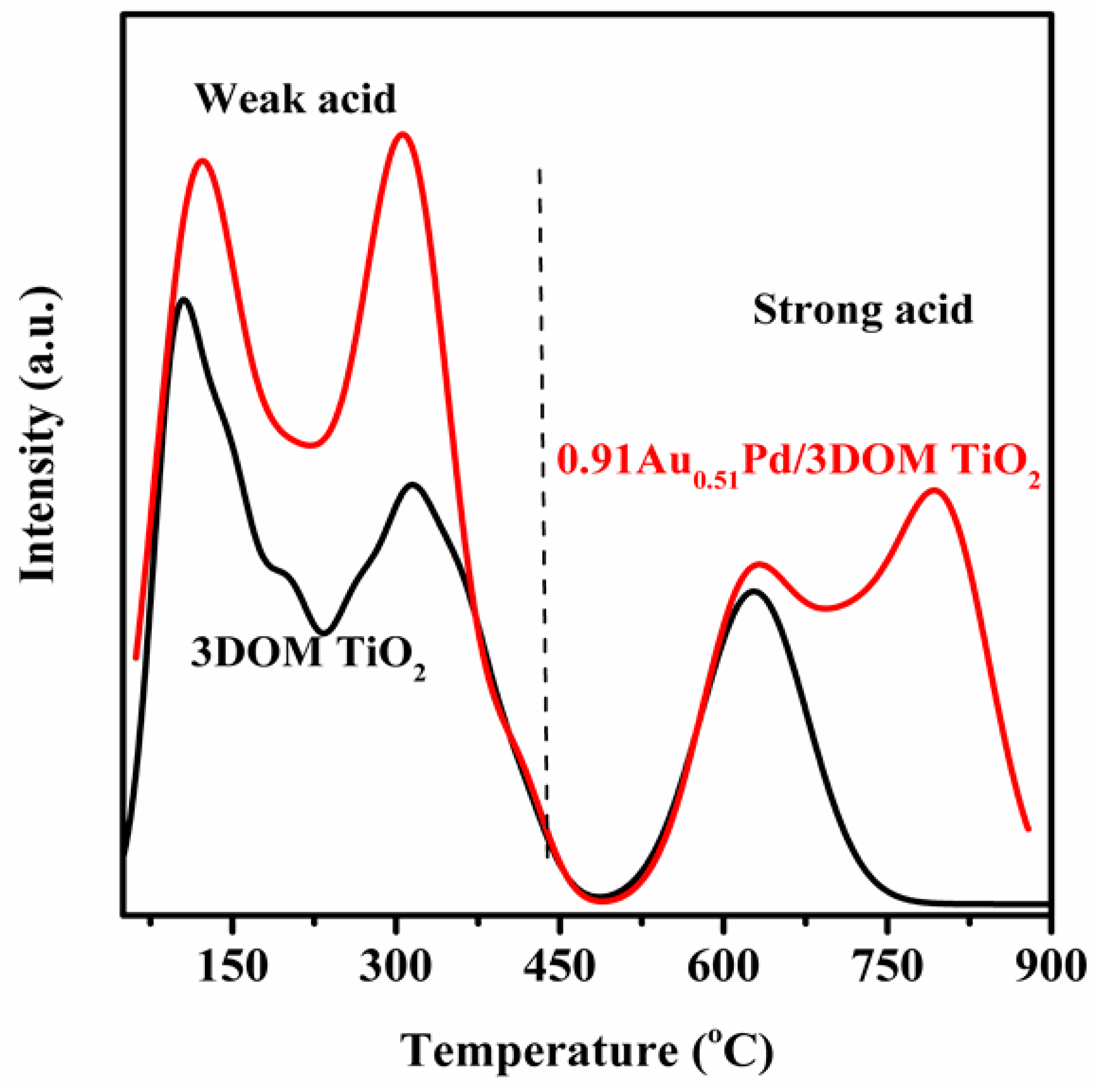
2.4. Surface Acid Property
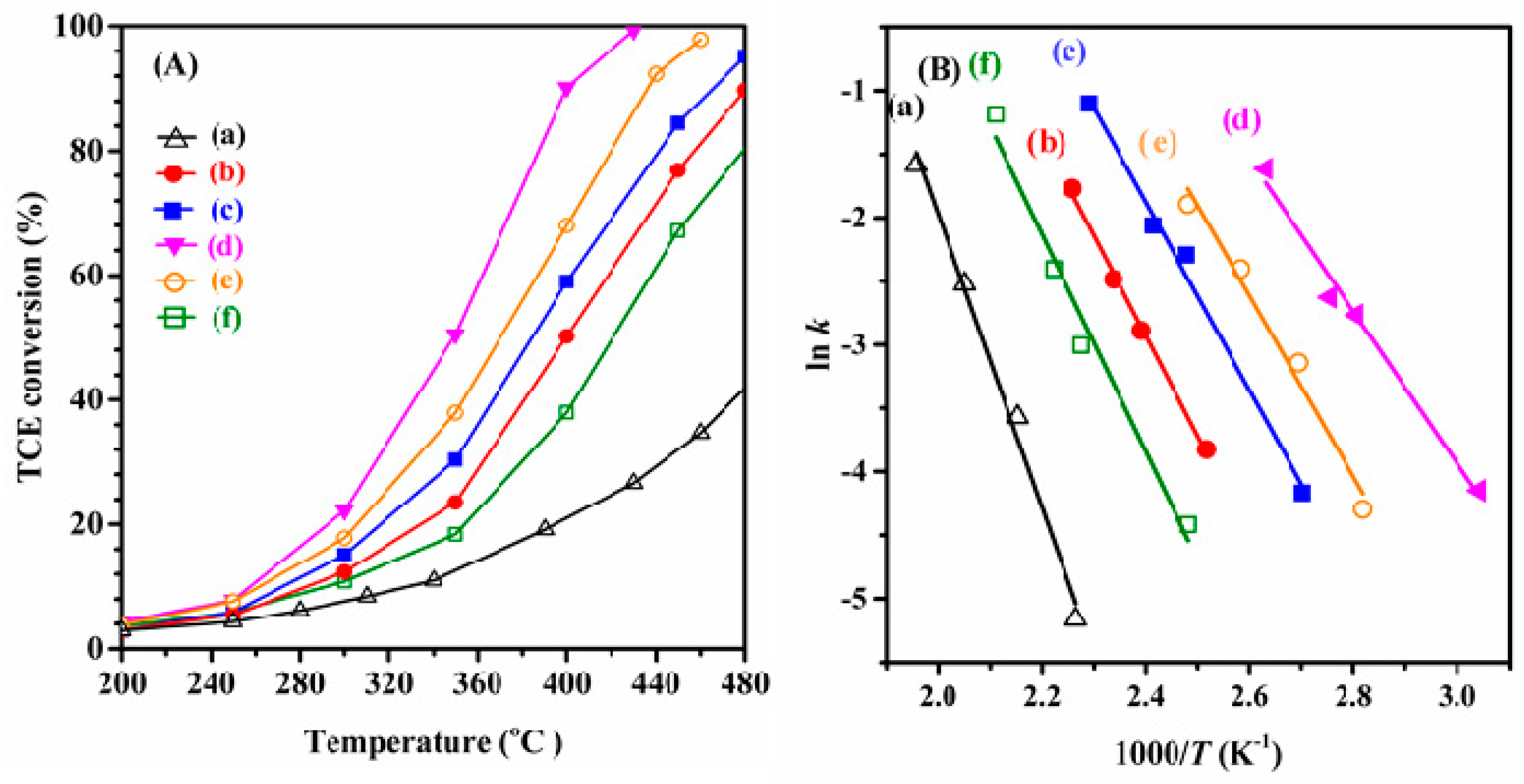
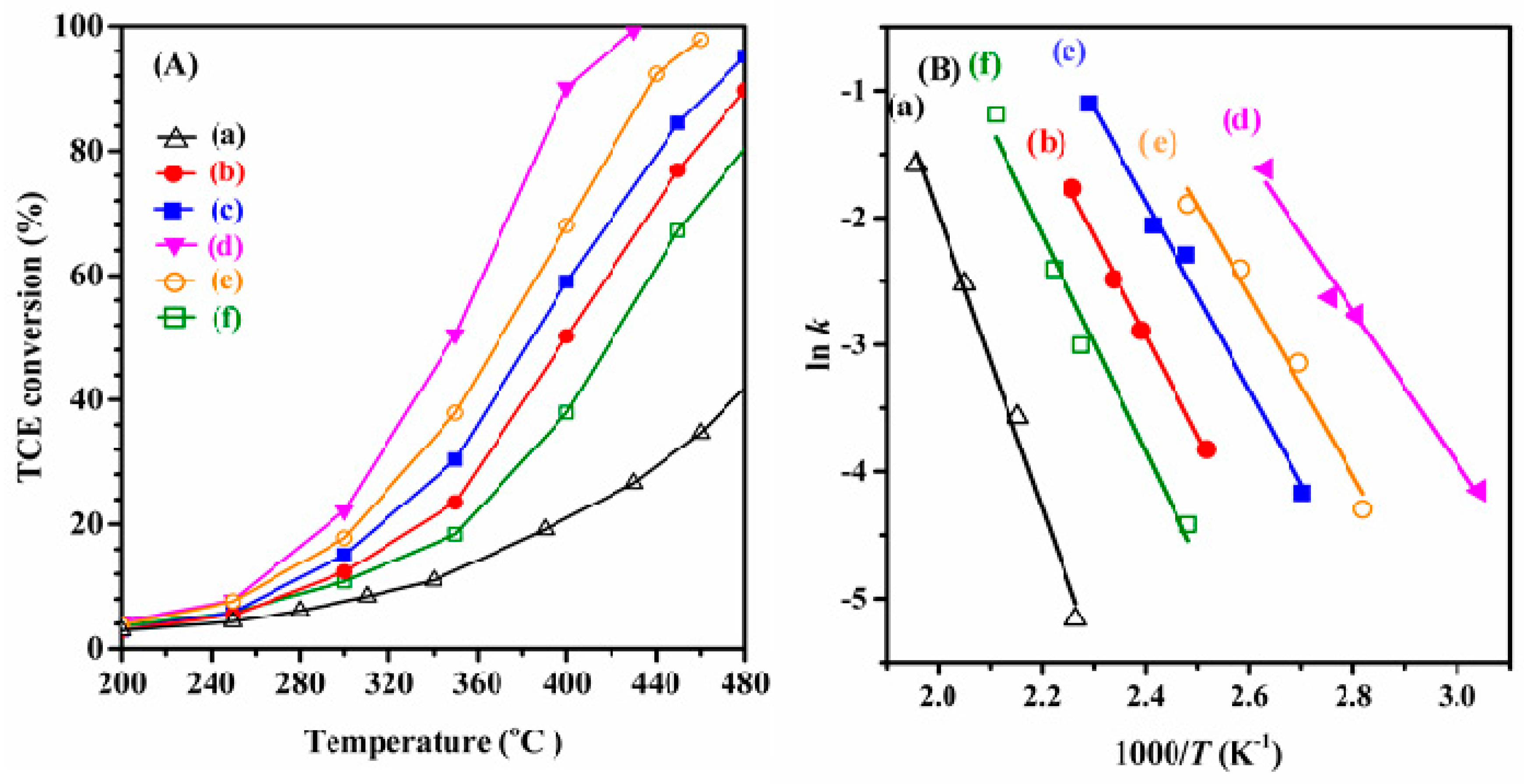
2.5. Catalytic Performance
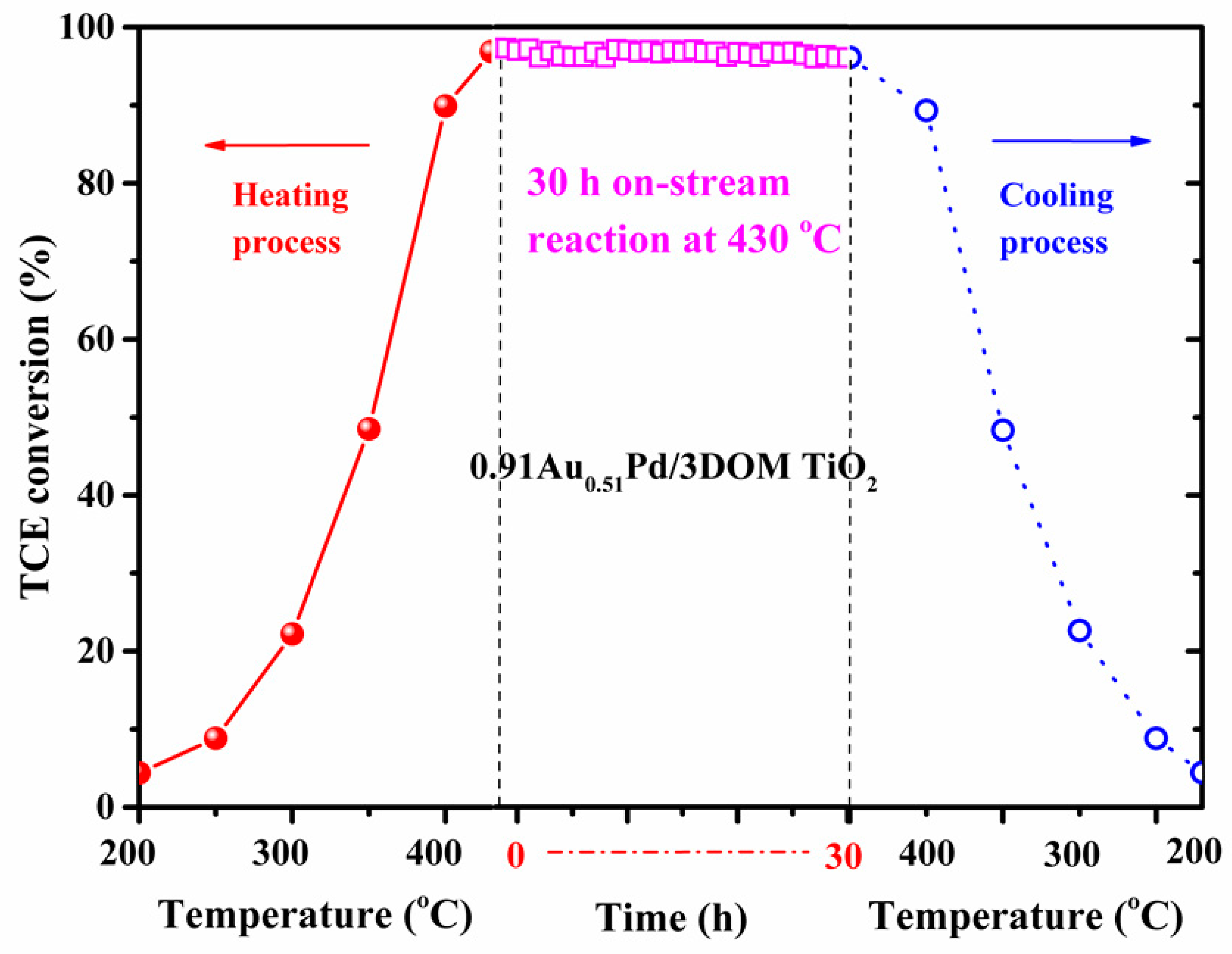
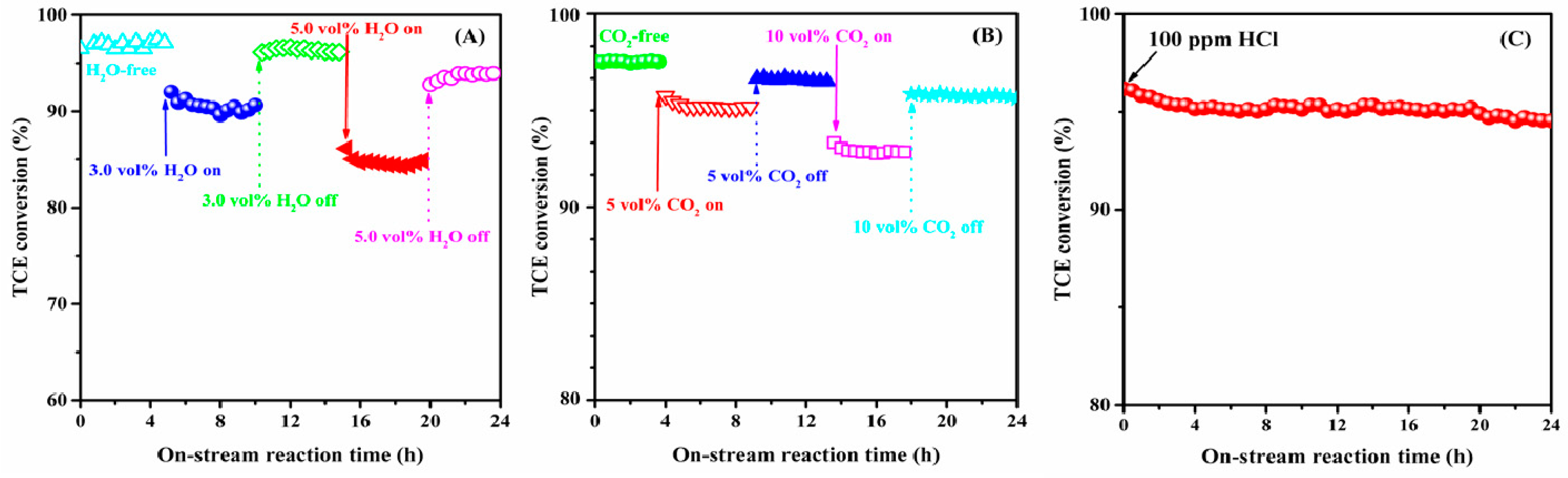
2.6. Catalytic Stability and Effects of H2O, CO2, and HCl on Catalytic Activity
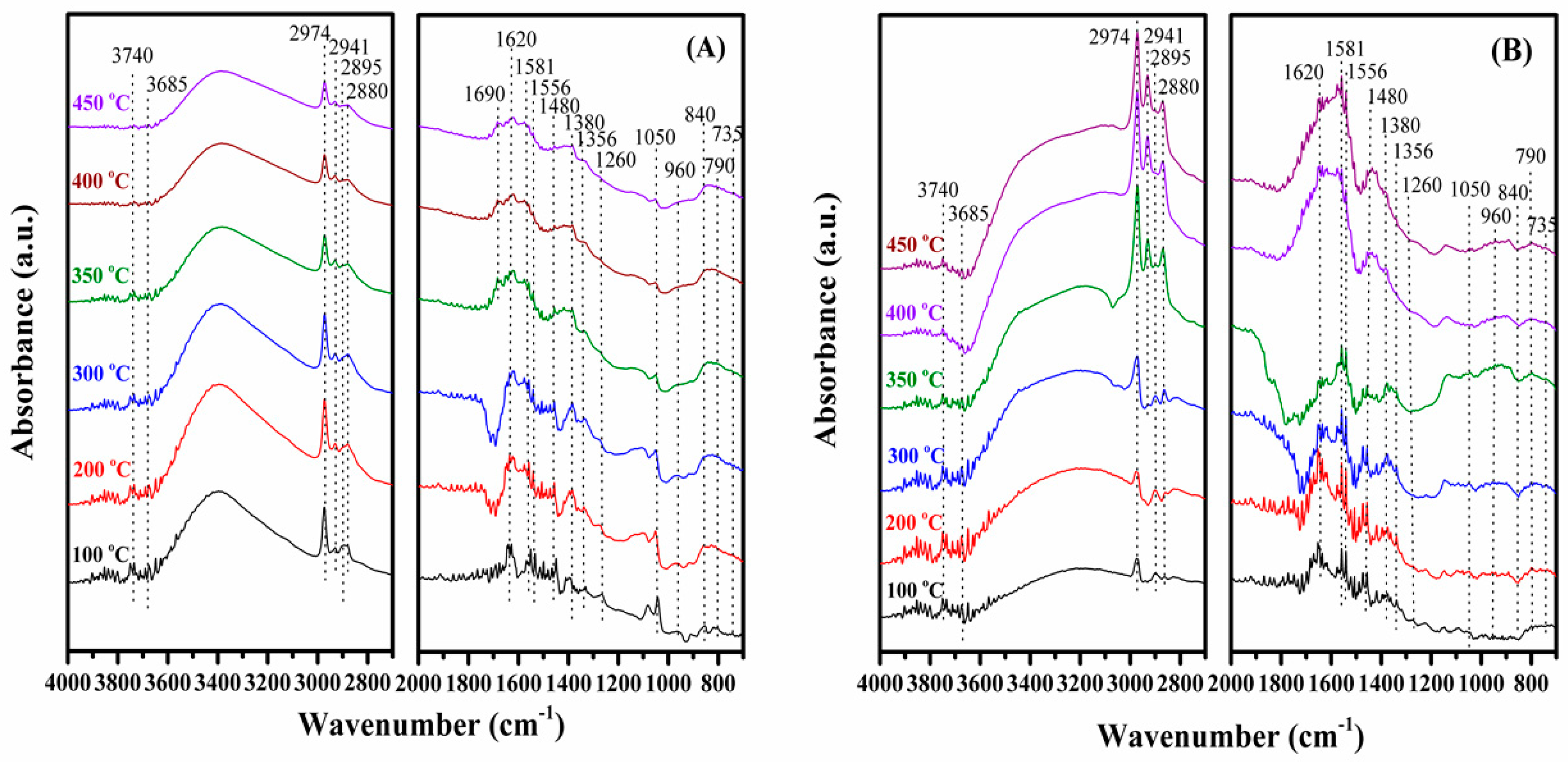
2.7. Possible Reaction Pathway
3. Materials and Methods
3.1. Preparation of the Catalysts
3.2. Catalytic Activity Evaluation
3.3. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pitkaaho, S.; Nevanpera, T.; Matejova, L.; Ojala, S.; Keiski, R.L. Oxidation of perchloroethylene over Pt, Pd, Rh, and V2O5 catalysis supported on Al2O3, Al2O3-TiO2, and Al2O3-CeO2. Appl. Catal. B 2013, 138–139, 33–42. [Google Scholar] [CrossRef]
- Miranda, B.; Diaz, E.; Ordonez, S.; Diez, F.V. Catalytic combustion of trichloroethene over Ru/AlO: Reaction mechanism and kinetic study. Catal. Commun. 2006, 7, 945–949. [Google Scholar] [CrossRef]
- Zhou, G.L.; Lan, H.; Song, R.Y.; Xie, H.M.; Du, Q.X. Effects of preparation method on CeCu oxide catalyst performance. RSC Adv. 2014, 4, 50840–50850. [Google Scholar] [CrossRef]
- Maupin, I.; Pinard, L.; Mijoin, J.; Magnoux, P. Bifunctional mechanism of dichloromethane oxidation over Pt/Al2O3: CH2Cl2 disproportionation over alumina and oxidation over platinum. J. Catal. 2012, 291, 104–109. [Google Scholar] [CrossRef]
- Rivas, B.; Lopez-Fonseca, R.; Jimenez-Gonzalez, C.; Gutierrez-Ortiz, J.I. Highly active behaviour of nanocrystalline Co3O4 from oxalate nanorods in the oxidation of chlorinated short chain alkanes. Chem. Eng. J. 2012, 184, 184–192. [Google Scholar] [CrossRef]
- Pitkäaho, S.; Matejova, L.; Ojala, S.; Gaalova, J.; Keiski, R.L. Catalytic abatement of trichloroethylene over Mo and/or W-based bronzes. Appl. Catal. B 2012, 111–114, 150–159. [Google Scholar] [CrossRef]
- Hutchings, G.J. Activated–carbon–supported gold–cesium(I) as highly effective catalysts for hydrochlorination of acetylene to vinyl chloride. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Liu, Y.X.; Dai, H.X.; Deng, J.G.; Li, X.W.; Wang, Y.; Arandiyan, H.; Guo, G.S. Au/3DOM La0.6Sr0.4MnO3: Highly active nanocatalysts for the oxidation of carbon monoxide and toluene. J. Catal. 2013, 305, 146–153. [Google Scholar] [CrossRef]
- Liu, Y.X.; Dai, H.X.; Deng, J.G.; Xie, S.H.; Yang, H.G.; Tan, W.; Han, W.; Jiang, Y.; Guo, G.S. Mesoporous Co3O4-supported gold nanocatalysts: Highly active for the oxidation of carbon monoxide, benzene, toluene, and o-xylene. J. Catal. 2014, 309, 408–418. [Google Scholar] [CrossRef]
- Saiman, M.I.B.; Brett, G.L.; Tiruvalam, R.; Forde, M.M.; Sharples, K.; Thetford, A.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Murphy, D.M.; et al. Involvement of surface-bound radicals in the oxidation of toluene using supported Au−Pd nanoparticles. Angew. Chem. 2012, 51, 5981–5985. [Google Scholar] [CrossRef] [PubMed]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Solsona-Espriu, B.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Bimetallic palladium–gold nanoparticl es synthesized in ionic liquid microemulsion. Science 2006, 311, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent advances in catalysts for methane combustion. Catal. Surv. 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Gao, T.T.; Andino, J.M.; Li, Y. Copper and iodine co-modified TiO2 nanoparticles for improved activity of CO2 photoreduction with water vapor. Appl. Catal. B 2012, 123–124, 257–264. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, K.; Wang, H.; Wang, Y.; Tian, D.; Wei, Y.; Zhu, X.; Zeng, C.; Luo, Y. Structure dependence and reaction mechanism of CO oxidation: A model study on macroporous CeO2 and CeO2–ZrO2 catalysts. J. Catal. 2016, 344, 365–377. [Google Scholar] [CrossRef]
- Xie, S.H.; Deng, J.G.; Zang, S.M.; Yang, H.G.; Guo, G.S.; Arandiyan, H.; Dai, H.X. Au–Pd/3DOM Co3O4: Highly active and stable nanocatalysts for toluene oxidation. J. Catal. 2015, 322, 38–48. [Google Scholar] [CrossRef]
- Xie, S.H.; Deng, J.G.; Liu, Y.X.; Zhang, Z.H.; Yang, H.G.; Jiang, Y.; Arandiyan, H.; Dai, H.X.; Au, C.T. Excellent catalytic performance, thermal stability, and water resistance of 3DOM Mn2O3-supported Au–Pd alloy nanoparticles for the complete oxidation of toluene. Appl. Catal. A 2015, 507, 82–90. [Google Scholar] [CrossRef]
- Wang, Y.; Arandiyan, H.; Scott, J.; Akia, M.; Dai, H.X.; Deng, J.G.; Amal, R. High performance Au−Pd supported on 3D hybrid strontium-substituted lanthanum manganite perovskite catalyst for methane combustion. ACS Catal. 2016, 6, 6935–6947. [Google Scholar] [CrossRef]
- Jiao, J.Q.; Wei, Y.C.; Zhao, Z.; Zhong, W.J.; Liu, J.; Li, J.M.; Jiang, G.Y. Synthesis of 3D ordered macroporous TiO2-supported Au nanoparticle photocatalysts and their photocatalytic performances for the reduction of CO2 to methane. Catal. Today 2015, 258, 319–326. [Google Scholar] [CrossRef]
- Arandiyan, H.; Dai, H.X.; Ji, K.M.; Sun, H.Y.; Li, J.H. Pt nanoparticles embedded in colloidal crystal template derived 3D ordered macroporous Ce0.6Zr0.3Y0.1O2: Highly efficient catalysts for methane combustion. ACS Catal. 2015, 5, 1781–1793. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, B.C.; Wang, Q.; Li, C.Y.; Hu, W.T.; Liu, Y.X.; Jing, P.; Zhao, W.Z.; Zhang, J. Three-dimensionally ordered macroporous Au/CeO2–Co3O4 catalysts with mesoporous walls for enhanced CO preferential oxidation in H2-rich gases. J. Catal. 2012, 296, 65–76. [Google Scholar] [CrossRef]
- Wei, Y.C.; Liu, J.; Zhao, Z.; Chen, Y.S.; Xu, C.M.; Duan, A.J.; Jiang, G.Y.; He, H. Highly active catalysts of gold nanoparticles supported on three-dimensionally ordered macroporous LaFeO3 for soot oxidation. Angew. Chem. Int. Ed. 2011, 50, 2326–2329. [Google Scholar] [CrossRef] [PubMed]
- Priolkar, K.R.; Bera, P.; Sarode, P.R.; Hegde, M.S.; Emura, S.; Kumashiro, R.; Lalla, N.P. In situ preparation and investigation of Pd/CeO2 catalysts for the low-temperature oxidation of CO. Chem. Mater. 2002, 14, 2120–2128. [Google Scholar] [CrossRef]
- Kesavan, L.; Tiruvalam, R.; Rahim, M.H.A.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; Kiely, C.J.; et al. Solvent free oxidation of primary carbon-hydrogen bonds in toluene using Au−Pd alloy nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Cao, Y.; Yao, X.; Ge, C.; Gao, F.; Deng, Y.; Tang, C.; Dong, L. Getting insight into the influence of SO2 on TiO2/CeO2 for the selective catalytic reduction of NO by NH3. Appl. Catal. B 2015, 165, 589–598. [Google Scholar] [CrossRef]
- Arandiyan, H.; Dai, H.X.; Deng, J.G.; Liu, Y.X.; Bai, B.Y.; Wang, Y.; Li, X.W.; Xie, S.H.; Li, J.H. Three-dimensionally ordered macroporous La0.6Sr0.4MnO3 with high surface areas: Active catalysts for the combustion of methane. J. Catal. 2013, 307, 327–339. [Google Scholar] [CrossRef]
- Batzill, M.; Diebold, U. Electronic properties of post-transition metal oxide semiconductor surfaces. Prog. Surf. Sci. 2005, 79, 47–154. [Google Scholar] [CrossRef]
- Xie, S.H.; Liu, Y.X.; Deng, J.G.; Zhao, X.T.; Yang, J.; Zhang, K.F.; Han, Z.; Arandiyan, H.; Dai, H.X. Effect of transition metal doping on the catalytic performance of Au–Pd/3DOM Mn2O3 for the oxidation of methane and o-xylene. Appl. Catal. B 2017, 206, 221–232. [Google Scholar] [CrossRef]
- Lucas, A.D.; Cañizares, P.; Durán, A. Improving deactivation behaviour of HZSM-5 catalysts. Appl. Catal. A 2001, 206, 87–93. [Google Scholar] [CrossRef]
- Steltenpohl, P.; Aranzabal, A.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I.; González-Velasco, J.R. Performance of zeolites and product selectivity in the gas-phase oxidation of 1,2-dichloroethane. Catal. Today 2000, 62, 367–377. [Google Scholar]
- López-Fonseca, R.; Cibrián, S.; Gutiérrez-Ortiz, J.I.; Gutiérrez-Ortiz, M.A.; González-Velasco, J.R. Oxidative destruction of dichloromethane over protonic zeolites. AIChE J. 2010, 49, 496–504. [Google Scholar] [CrossRef]
- Yang, P.; Zuo, S.F.; Shi, Z.N.; Tao, F.; Zhou, R.X. Accelerating effect of ZrO2 doping on catalytic performance and thermal stability of CeO2–CrOx mixed oxide for 1,2-dichloroethane elimination. Appl. Catal. B 2016, 191, 53–61. [Google Scholar] [CrossRef]
- Miranda, B.; Díaz, E.; Ordóñ ez, S.; Vega, A.; Díez, F.V. Performance of alumina-supported noble metal catalysts for the combustion of trichloroethene at dry and wet conditions. Appl. Catal. B 2006, 64, 262–271. [Google Scholar] [CrossRef]
- Maghsoodi, S.; TowFigurehi, J.; Khodadadi, A.; Mortazavi, Y. Highly active Fe2O3-doped TiO2 photocatalyst for degradation of trichloroethylene in air under UV and visible light irradiation: Experimental and computational studies. Chem. Eng. J. 2013, 215, 827–837. [Google Scholar] [CrossRef]
- Weng, X.L.; Weng, P.F.; Long, Y.; Meng, Q.J.; Wu, Z.B. Catalytic oxidation of chlorobenzene over MnxCe1−xO2/HZSM-5 catalysts: A study with practical implications. Environ. Sci. Technol. 2017, 51, 8057–8066. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, S.; Vega, A.; Miranda, B.; Díaz, E.; Díez, F.V. Oxidation of trichloroethene over metal oxide catalysts: Kinetic studies and correlation with adsorption properties. Chemosphere 2007, 66, 1706–1715. [Google Scholar]
- Li, H.F.; Lu, G.Z.; Dai, Q.G.; Wang, Y.Q.; Guo, Y.; Guo, Y.L. Efficient low-temperature catalytic combustion of trichloroethylene over flower-like mesoporous Mn-doped CeO2 microspheres. Appl. Catal. B 2011, 102, 475–483. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.L.; Zeng, J.L.; Zhu, T.Y. Catalytic oxidation of trichloroethylene over TiO2 supported ruthenium catalysts. Catal. Commun. 2016, 76, 13–18. [Google Scholar] [CrossRef]
- Rivas, B.; Sampedro, C.; García-Real, M.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I. Promoted activity of sulphated Ce/Zr mixed oxides for chlorinated VOC oxidative abatement. Appl. Catal. B 2013, 129, 225–235. [Google Scholar] [CrossRef]
- Yang, P.; Shi, Z.N.; Yang, S.S.; Zhou, R.X. High catalytic performances of CeO2–CrOx catalysts for chlorinated VOCs elimination. Chem. Eng. Sci. 2015, 126, 361–369. [Google Scholar] [CrossRef]
- Wu, M.; Dai, Q.G.; Wang, X.Y. Alumina hollow microspheres supported gold catalysts for low-temperature CO oxidation: Effect of the pretreatment atmospheres on the catalytic activity and stability. Catal. Commun. 2012, 18, 72–75. [Google Scholar] [CrossRef]
- Gutiérrez-Ortiz, J.I.; López-Fonseca, R.; Aurrekoetxea, U.; González-Velasco, J.R. Low-temperature deep oxidation of dichloromethane and trichloroethylene by H-ZSM-5-supported manganese oxide catalysts. J. Catal. 2003, 218, 148–154. [Google Scholar] [CrossRef]
- Feijen-Jeurissen, M.M.R.; Jornaa, J.J.; Nieuwenhuysa, B.E.; Sinquinb, G. Mechanism of catalytic destruction of 1,2-dichloroethane and trichloroethylene over γ-Al2O3 and γ-Al2O3 supported chromium and palladium catalysts. Catal. Today 1999, 54, 65–79. [Google Scholar] [CrossRef]
- Blanch-Raga, N.; Palomares, A.E.; Martínez-Triguero, J.; Puche, M.; Fetter, G.; Bosch, P. The oxidation of trichloroethylene over different mixed oxides derived from hydrotalcites. Appl. Catal. B 2014, 160–161, 129–134. [Google Scholar] [CrossRef]
- Burch, R.; Urbano, F.J.; Loader, P.K. Methane combustion over palladium catalysts: The effect of carbon dioxide and water on activity. Appl. Catal. A 1995, 123, 173–184. [Google Scholar] [CrossRef]
- Datka, J.; Sarbak, Z.; Eischens, R.P. Infrared study of coke on alumina and zeolite. J. Catal. 1994, 145, 544–550. [Google Scholar] [CrossRef]
- Kagel, R.O. Infrared investigation of the adsorption and surface reactions of the C1 through C4 normal alcohols on γ-alumina. J. Phys. Chem. 1967, 71, 844–850. [Google Scholar] [CrossRef]
- Eisenbach, D.; Gallei, E. Infrared spectroscopic investigations relating to coke formation on zeolites: I. Adsorption of hexene-1 and n-hexane on zeolites of type Y. J. Catal. 1979, 56, 377–389. [Google Scholar] [CrossRef]
- Trombetta, M.; Busca, G.; Rossini, S.A.; Piccoli, V.; Cornaro, U. FT-IR studies on light olefin skeletal isomerization catalysis.1. The interaction of C4 olefins and alcohols with pure gamma-alumina. J. Catal. 1997, 168, 334–348. [Google Scholar] [CrossRef]
- Phillips, L.A.; Raupp, G.B. Infrared spectroscopic investigation of gas–solid heterogeneous photocatalytic oxidation of trichloroethylene. J. Mol. Catal. 1992, 77, 297–311. [Google Scholar] [CrossRef]
- Halasz, J.; Imre, B.; Hannus, I. IR spectroscopic investigation of hydrodechlorination on Pt-containing zeolites. Appl. Catal. A 2004, 27, 47–53. [Google Scholar] [CrossRef]
- Schmal, M.; Mariana Souza, M.V.M.; Alegre, V.V.; Monica Silva, A.P.D.; Cesar, D.V.; Perez, C.A.C. Methane oxidation–effect of support, precursor and pretreatment conditions—In situ reaction XPS and DRIFT. Catal. Today 2006, 118, 392–401. [Google Scholar] [CrossRef]
- Aranzabal, A.; Ayastuy-Arizti, J.L.; González-Marcos, J.A.; González-Velasco, J.R. The reaction pathway and kinetic mechanism of the catalytic oxidation of gaseous lean TCE on Pd/alumina catalysts. J. Catal. 2003, 214, 130–135. [Google Scholar] [CrossRef]
- Li, H.N.; Zhang, L.; Dai, H.X.; He, H. Facile synthesis and unique physicochemical properties of three-dimensionally ordered macroporous magnesium oxide, gamma-alumina, and ceria-zirconia solid solutions with crystalline mesoporous walls. Inorg. Chem. 2009, 48, 4421–4434. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Xue, X.M.; Meng, Z.H.; Zhou, R.X. Enhanced catalytic activity and stability of Ce doping on Cr supported HZSM-5 catalysts for deep oxidation of chlorinated volatile organic compounds. Chem. Eng. J. 2013, 234, 203–210. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Actual Au Content a (wt%) | Actual Pd Content a (wt%) | Actual Au/Pd Molar Ratio a (mol/mol) | Db (nm) | Noble Metal Particle Size c (nm) | Mesopores Diameter d (nm) | Macropores Diameter e (nm) | BET Surface Area e (m2/g) | Pore Volume e (cm3/g) |
|---|---|---|---|---|---|---|---|---|---|
| 3DOM TiO2 | – | – | – | 20.1 | - | 180–200 | 3.1–4.2 | 53.3 | 0.209 |
| 0.89Au1.86Pd/3DOM TiO2 | 0.69 | 0.20 | 1.86 | 19.6 | 3.4 | 175–194 | 2.6–4.1 | 50.2 | 0.205 |
| 0.87Au0.95Pd/3DOM TiO2 | 0.56 | 0.31 | 0.95 | 20.4 | 3.2 | 186–205 | 2.9–4.4 | 49.5 | 0.208 |
| 0.91Au0.51Pd/3DOM TiO2 | 0.44 | 0.47 | 0.51 | 19.8 | 2.9 | 180–201 | 2.8–4.3 | 51.2 | 0.210 |
| 0.93Au/3DOM TiO2 | 0.93 | – | – | 20.3 | 3.2 | 176–195 | 2.7–4.2 | 52.1 | 0.196 |
| 0.89Pd/3DOM TiO2 | – | 0.89 | – | 19.5 | 3.5 | 178–194 | 3.0–4.3 | 50.6 | 0.190 |
| Sample | Surface Element Composition a | O2 Desorption b (mmol/g) | H2 Consumption c (mmol/g) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Auδ+/Au0 Molar Ratio | Pd2+/Pd0 Molar Ratio | Oads/Olatt Molar Ratio | 50–450 °C | 450–800 °C | Total | <350 °C | ≥350 °C | Total | |
| 3DOM TiO2 | – | – | 0.31 | 0.46 | 1.92 | 2.38 | 2.8 | 11.4 | 14.2 |
| 0.89Au1.86Pd/3DOM TiO2 | 0.29 | 0.65 | 0.45 | 0.98 | 2.21 | 3.19 | 10.2 | 5.4 | 15.6 |
| 0.87Au0.95Pd/3DOM TiO2 | 0.31 | 0.68 | 0.47 | 1.03 | 2.18 | 3.21 | 10.8 | 5.3 | 16.1 |
| 0.91Au0.51Pd/3DOM TiO2 | 0.36 | 0.77 | 0.56 | 1.16 | 2.31 | 3.47 | 12.9 | 4.3 | 17.2 |
| 0.93Au/3DOM TiO2 | 0.21 | – | 0.43 | 0.92 | 2.13 | 3.05 | 9.7 | 5.7 | 15.4 |
| 0.89Pd/3DOM TiO2 | – | 0.63 | 0.49 | 1.08 | 2.20 | 3.28 | 11.2 | 5.1 | 16.3 |
| Catalyst | Catalytic Activity | TCE oxidation at 300 °C | Ea (kJ/mol) | ||||
|---|---|---|---|---|---|---|---|
| T10% (°C) | T50% (°C) | T90% (°C) | SHCla (%) | Reaction Rate (μmol/(gNoble metal s)) | TOFNoble metal (×10−3 s−1) | ||
| 3DOM TiO2 | 320 | – | – | 32.3 | – | – | 97.6 |
| 0.89Au1.86Pd/3DOM TiO2 | 283 | 401 | 479 | 85.1 | 28.1 | 0.54 | 66.2 |
| 0.87Au0.95Pd/3DOM TiO2 | 270 | 386 | 462 | 88.4 | 36.5 | 0.71 | 63.5 |
| 0.91Au0.51Pd/3DOM TiO2 | 255 | 354 | 400 | 94.2 | 50.4 | 0.96 | 51.7 |
| 0.93Au/3DOM TiO2 | 294 | 419 | – | 82.9 | 24.6 | 0.47 | 71.4 |
| 0.89Pd/3DOM TiO2 | 261 | 371 | 435 | 91.6 | 45.1 | 0.85 | 59.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, Y.; Deng, J.; Zhang, K.; Yang, J.; Han, Z.; Dai, H. AuPd/3DOM TiO2 Catalysts: Good Activity and Stability for the Oxidation of Trichloroethylene. Catalysts 2018, 8, 666. https://doi.org/10.3390/catal8120666
Zhang X, Liu Y, Deng J, Zhang K, Yang J, Han Z, Dai H. AuPd/3DOM TiO2 Catalysts: Good Activity and Stability for the Oxidation of Trichloroethylene. Catalysts. 2018; 8(12):666. https://doi.org/10.3390/catal8120666
Chicago/Turabian StyleZhang, Xing, Yuxi Liu, Jiguang Deng, Kunfeng Zhang, Jun Yang, Zhuo Han, and Hongxing Dai. 2018. "AuPd/3DOM TiO2 Catalysts: Good Activity and Stability for the Oxidation of Trichloroethylene" Catalysts 8, no. 12: 666. https://doi.org/10.3390/catal8120666
APA StyleZhang, X., Liu, Y., Deng, J., Zhang, K., Yang, J., Han, Z., & Dai, H. (2018). AuPd/3DOM TiO2 Catalysts: Good Activity and Stability for the Oxidation of Trichloroethylene. Catalysts, 8(12), 666. https://doi.org/10.3390/catal8120666