Catalytic Pyrolysis of Biomass and Polymer Wastes


Abstract
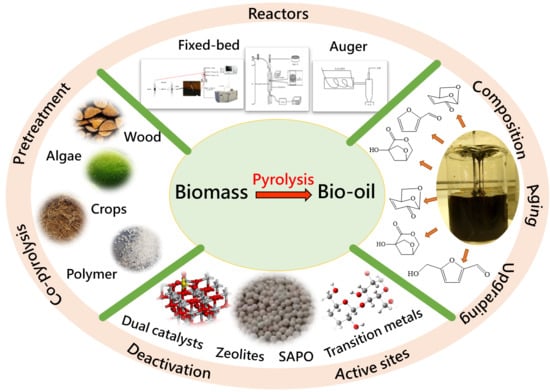
1. Introduction
1.1. Drive for Sustainable Energy
1.2. Catalytic Fast Pyrolysis of Biomass and Waste Polymers
2. Fundamentals of Catalytic Fast Pyrolysis (CFP)
2.1. Feedstock
2.1.1. Plant Biomass and Its Pretreatment
Effect of Biomass Composition on Bio-Oil Properties
Effect of Feedstock Elemental Composition
Effect of Biomass Pretreatment
Effect of Indigenous Catalysts/Alkali and Alkaline Earth Metals
2.1.2. Waste Polymers
2.1.3. Polymers and Co-Pyrolysis of Polymers with Biomass
Mechanism of Catalytic Co-Pyrolysis
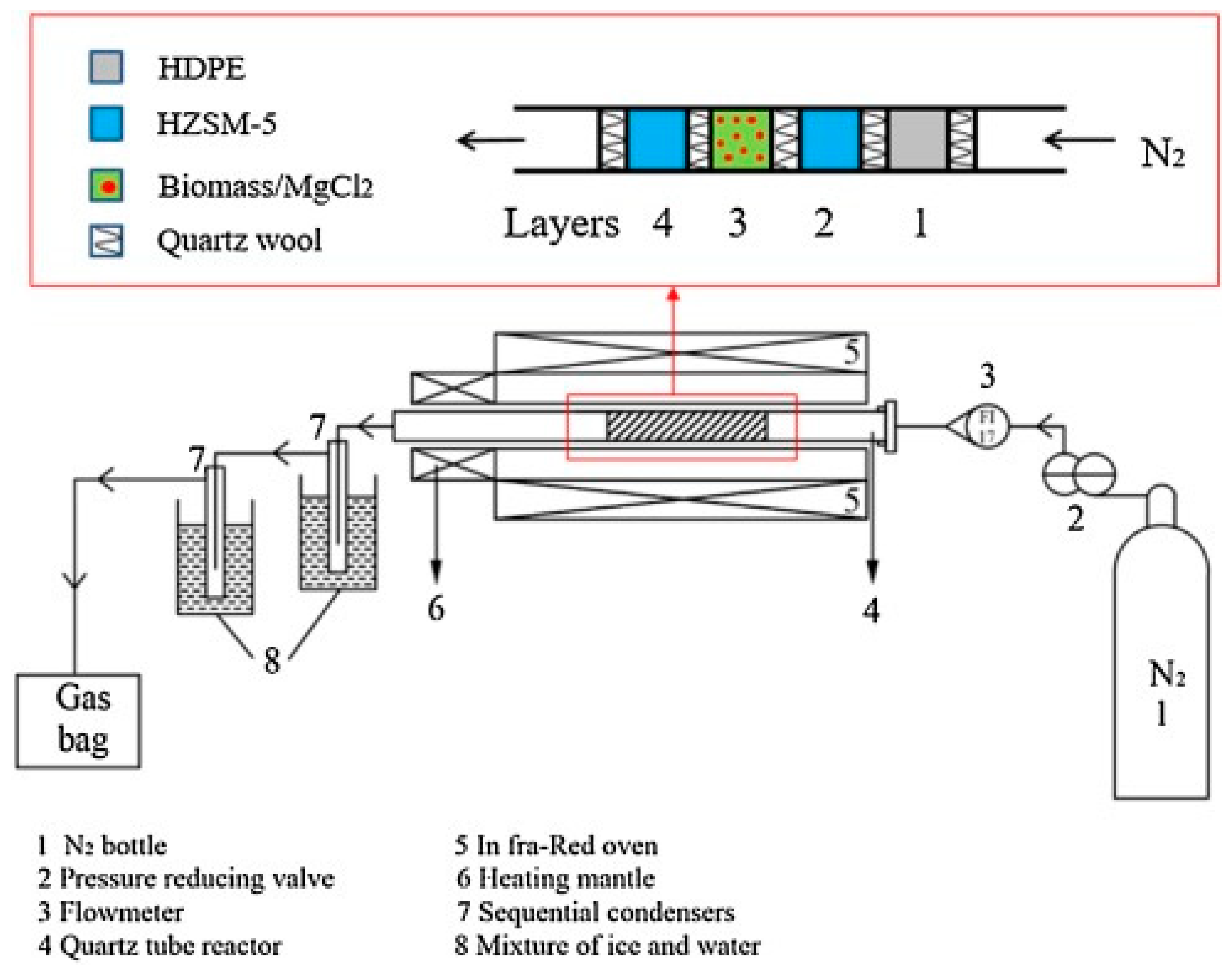
2.2. Reactors
2.3. Vapor Cleaning and Quenching
2.4. Optimization of Bio-Oil Yield Using Statistical Method
2.5. Pyrolysis Oil Stability
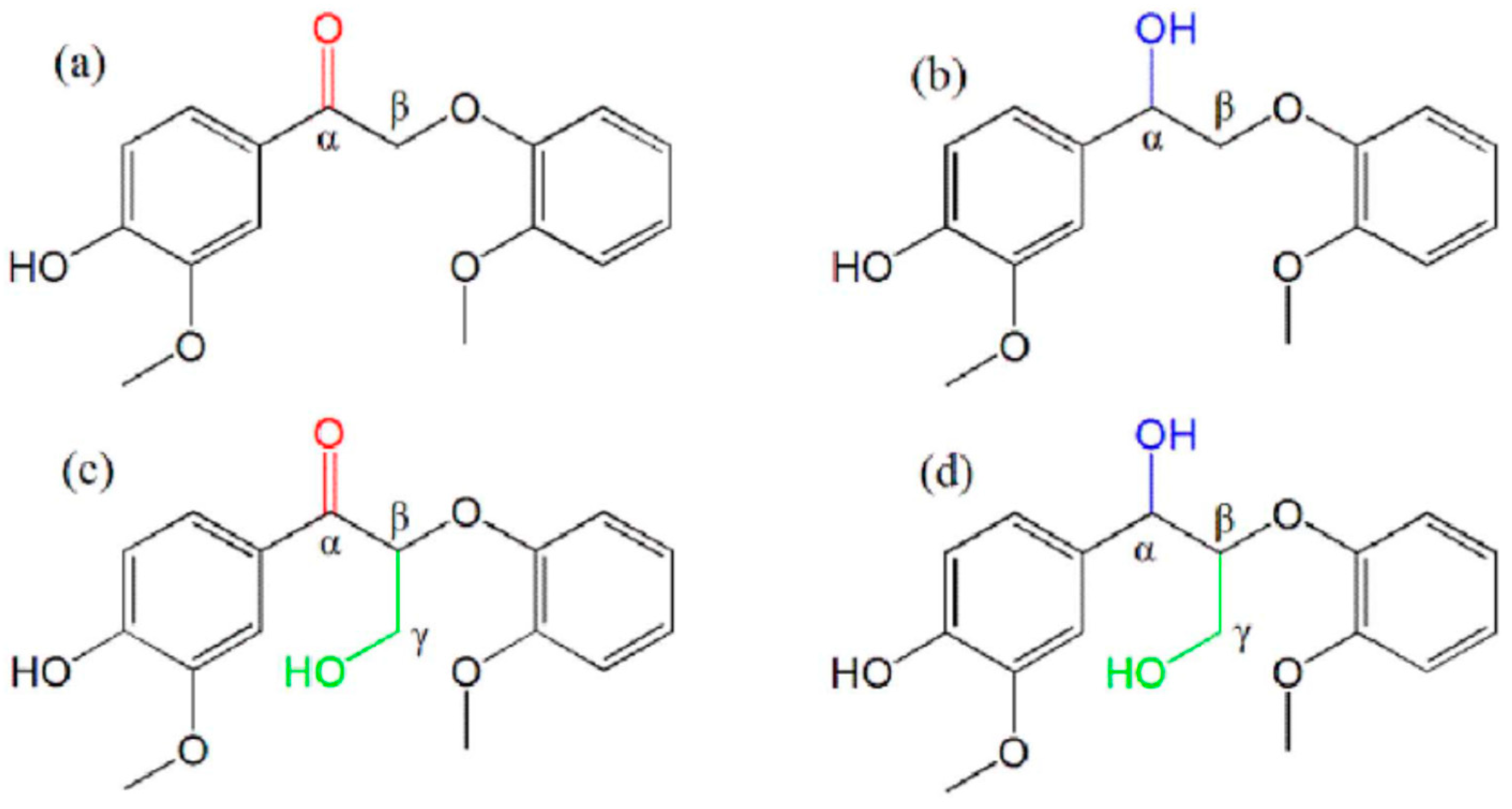
2.5.1. Pyrolysis Oil Composition
- (1)
- feedstock (including particle size, dirt, moisture, and protein content);
- (2)
- heat transfer rate;
- (3)
- extent of vapor dilution, residence time and temperature of vapors in the reactor;
- (4)
- efficiency of the condensation equipment;
- (5)
- char temperature during pyrolysis;
- (6)
- efficiency of the char removal system;
- (7)
- storage time and temperature; and
- (8)
- extent of contamination during pyrolysis and storage.
Organic Compounds
Inorganic Compounds
2.5.2. Accelerated Aging
2.5.3. Additives to Stabilize Pyrolysis Oil
2.6. Pyrolysis Oil Upgrading
3. Catalyst Design and Product Distribution Optimization
3.1. Catalysts for CFP
3.1.1. Zeolites
ZSM-5
HZSM-5
MCM-22
Zeolite Y
Metal Doping of Zeolites for In-Bed CFP Processes
3.1.2. SAPO
3.1.3. Alkali and Alkaline Earth Metallic (AAEM) Species
3.1.4. Transition Metal and Other Metal Based Catalysts
3.1.5. Dual Catalyst Systems
3.2. Catalyst Deactivation and Regeneration
3.2.1. Catalyst Deactivation in Biomass Pyrolysis
3.2.2. Catalyst Deactivation in Polymer Pyrolysis
3.2.3. Catalyst Regeneration
4. Summary and Outlook
Funding
Conflicts of Interest
References
- EIA. International Energy Annual 2001 Edition; U.S. Department of Energy: Washington, DC, USA, 2003.
- Wessner, C. What is the best guess as to when the world’s oil reserves will run out? Popul. Sci. 1999. [Google Scholar]
- Appenzeller, T. The End of Cheap Oil, National Geographic. Available online: https://www.nationalgeographic.com/environment/global-warming/end-cheap-oil/ (accessed on 15 July 2018).
- EIA. Annual Energy Review 2010; Energy Information Administration: Washington, DC, USA, 2011.
- Chow, J.; Kopp, R.J.; Portney, P.R. Energy resources and global development. Science 2003, 302, 1528–1531. [Google Scholar] [CrossRef] [PubMed]
- EIA. Annual Energy Outlook 2014 with Projections to 2040; Enery Information Adminstration: Washington, DC, USA, 2014.
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, C.; Reyes-Sosa, F.M.; Díez, B. Enzymatic hydrolysis of biomass from wood. Microb. Biotechnol. 2016, 9, 149–156. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Li, X.; Chen, G.; Liu, C.; Ma, W.; Yan, B.; Zhang, J. Hydrodeoxygenation of lignin-derived bio-oil using molecular sieves supported metal catalysts: A critical review. Renew. Sustain. Energy Rev. 2017, 71, 296–308. [Google Scholar] [CrossRef]
- Li, X.; Luo, X.; Jin, Y.; Li, J.; Zhang, H.; Zhang, A.; Xie, J. Heterogeneous sulfur-free hydrodeoxygenation catalysts for selectively upgrading the renewable bio-oils to second generation biofuels. Renew. Sustain. Energy Rev. 2018, 82, 3762–3797. [Google Scholar] [CrossRef]
- Filiciotto, L.; Balu, A.M.; Van der Waal, J.C.; Luque, R. Catalytic insights into the production of biomass-derived side products methyl levulinate, furfural and humins. Catal. Today 2018, 302, 2–15. [Google Scholar] [CrossRef]
- Morgan, H.M.; Bu, Q.; Liang, J.; Liu, Y.; Mao, H.; Shi, A.; Lei, H.; Ruan, R. A review of catalytic microwave pyrolysis of lignocellulosic biomass for value-added fuel and chemicals. Bioresour. Technol. 2017, 230, 112–121. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, Y.; Liu, S.; Zhou, N.; Chen, P.; Cheng, Y.; Addy, M.; Lu, Q.; Omar, M.M.; Liu, Y.; et al. Bio-oil from fast pyrolysis of lignin: Effects of process and upgrading parameters. Bioresour. Technol. 2017, 241, 1118–1126. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Chen, S.; Wu, J. Catalytic co-pyrolysis of lignocellulosic biomass with polymers: A critical review. Green Chem. 2016, 18, 4145–4169. [Google Scholar] [CrossRef]
- Lin, F.; Waters, C.L.; Mallinson, R.G.; Lobban, L.L.; Bartley, L.E. Relationships between biomass composition and liquid products formed via pyrolysis. Front. Energy Res. 2015, 3, 45. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Garrett, D.E.; Mallan, G.M. Pyrolysis Process for Solid Wastes. U.S. Patent 4,153,514, 8 May 1979. [Google Scholar]
- Mao, J.-D.; Johnson, R.; Lehmann, J.; Olk, D.; Neves, E.G.; Thompson, M.; Schmidt-Rohr, K. Abundant and stable char residues in soils: Implications for soil fertility and carbon sequestration. Environ. Sci. Technol. 2012, 46, 9571–9576. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cao, X.; Zhao, L.; Wang, H.; Yu, H.; Gao, B. Removal of Cu, Zn, and Cd from aqueous solutions by the dairy manure-derived biochar. Environ. Sci. Pollut. Res. 2013, 20, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wawra, A.; Friesl-Hanl, W.; Jäger, A.; Puschenreiter, M.; Soja, G.; Reichenauer, T.; Watzinger, A. Investigations of microbial degradation of polycyclic aromatic hydrocarbons based on 13 c-labeled phenanthrene in a soil co-contaminated with trace elements using a plant assisted approach. Environ. Sci. Pollut. Res. 2018, 25, 6364–6377. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yi, Y.; Fang, Z.; Tsang, E.P. Effects of biochar on phytotoxicity and translocation of polybrominated diphenyl ethers in ni/fe bimetallic nanoparticle-treated soil. Environ. Sci. Pollut. Res. 2018, 25, 2570–2579. [Google Scholar] [CrossRef]
- Fellmann, T.; Witzke, P.; Weiss, F.; Van Doorslaer, B.; Drabik, D.; Huck, I.; Salputra, G.; Jansson, T.; Leip, A. Major challenges of integrating agriculture into climate change mitigation policy frameworks. Mitig. Adapt. Strategy Glob. Chang. 2018, 23, 451–468. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Zeng, G.; Wang, X.; Hu, X.; Gu, Y.; Yang, Z. Application of biochar for the removal of pollutants from aqueous solutions. Chemosphere 2015, 125, 70–85. [Google Scholar] [CrossRef]
- Jaiswal, A.K.; Elad, Y.; Paudel, I.; Graber, E.R.; Cytryn, E.; Frenkel, O. Linking the belowground microbial composition, diversity and activity to soilborne disease suppression and growth promotion of tomato amended with biochar. Sci. Rep. 2017, 7, 44382. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Dickerson, T.; Soria, J. Catalytic fast pyrolysis: A review. Energies 2013, 6, 514–538. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Kaiqi, S.; Shuangxi, S.; Qiang, H.; Xuwen, L.; Lan, J.; Ya, L. Review of catalytic pyrolysis of biomass for bio-oil. In Proceedings of the 2011 International Conference on Materials for Renewable Energy. Environment, Shanghai, China, 20–22 May 2011; pp. 317–321. [Google Scholar] [CrossRef]
- Rosendahl, L. Biomass Combustion Science, Technology and Engineering; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Carpenter, D.; Westover, T.L.; Czernik, S.; Jablonski, W. Biomass feedstocks for renewable fuel production: A review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors. Green Chem. 2014, 16, 384–406. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Wang, K.; Mante, O.D.; Peters, J.E.; Dayton, D.C. Influence of the feedstock on catalytic fast pyrolysis with a solid acid catalyst. Energy Technol. 2017, 5, 183–188. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Su, L.; Zhang, Y.; Wang, Y.; Zhang, H. The role of shape selectivity in catalytic fast pyrolysis of lignin with zeolite catalysts. Appl. Catal. A 2012, 447–448, 115–123. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Zhang, J.; Choi, Y.S.; Yoo, C.G.; Kim, T.H.; Brown, R.C.; Shanks, B.H. Cellulose-hemicellulose and cellulose-lignin interactions during fast pyrolysis. ACS Sustain. Chem. Eng. 2015, 3, 293–301. [Google Scholar] [CrossRef]
- Wu, S.; Shen, D.; Hu, J.; Zhang, H.; Xiao, R. Cellulose-lignin interactions during fast pyrolysis with different temperatures and mixing methods. Biomass Bioenergy 2016, 90, 209–217. [Google Scholar] [CrossRef]
- Mukkamala, S.; Wheeler, M.C.; van Heiningen, A.R.P.; DeSisto, W.J. Formate-assisted fast pyrolysis of lignin. Energy Fuels 2012, 26, 1380–1384. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xue, Y.; Sharma, A.; Bai, X. Lignin valorization through thermochemical conversion: Comparison of hardwood, softwood and herbaceous lignin. ACS Sustain. Chem. Eng. 2016, 4, 6608–6617. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, S.; Lucia, L.A.; Chu, J. A comparison of the pyrolysis behavior of selected β-o-4 type lignin model compounds. J. Anal. Appl. Pyrolysis 2017, 125, 185–192. [Google Scholar] [CrossRef]
- Guo, D.; Wu, S.; Lyu, G.; Guo, H. Effect of molecular weight on the pyrolysis characteristics of alkali lignin. Fuel 2017, 193, 45–53. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Y.-T.; Vispute, T.P.; Xiao, R.; Huber, G.W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with zsm-5: The hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4, 2297–2307. [Google Scholar] [CrossRef]
- Horne, P.A.; Nugranad, N.; Williams, P.T. Catalytic coprocessing of biomass-derived pyrolysis vapours and methanol. J. Anal. Appl. Pyrolysis 1995, 34, 87–108. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, S.; Bai, X. Role of hydrogen transfer during catalytic copyrolysis of lignin and tetralin over hzsm-5 and hy zeolite catalysts. ACS Sustain. Chem. Eng. 2016, 4, 4237–4250. [Google Scholar] [CrossRef]
- Cheng, Y.-T.; Huber, G.W. Production of targeted aromatics by using diels-alder classes of reactions with furans and olefins over zsm-5. Green Chem. 2012, 14, 3114–3125. [Google Scholar] [CrossRef]
- Zhang, H.; Carlson, T.R.; Xiao, R.; Huber, G.W. Catalytic fast pyrolysis of wood and alcohol mixtures in a fluidized bed reactor. Green Chem. 2012, 14, 98–110. [Google Scholar] [CrossRef]
- Zhang, B.; Zhong, Z.; Min, M.; Ding, K.; Xie, Q.; Ruan, R. Catalytic fast co-pyrolysis of biomass and food waste to produce aromatics: Analytical Py-GC/MS study. Bioresour. Technol. 2015, 189, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Badger, P.C. Processing cost analysis for biomass feedstocks, ORNL/TM-2002/199; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1996.
- Westover, T.L.; Phanphanich, M.; Clark, M.L.; Rowe, S.R.; Egan, S.E.; Zacher, A.H.; Santosa, D. Impact of thermal pretreatment on the fast pyrolysis conversion of southern pine. Biofuels 2013, 4, 45–61. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Peacocke, G.V.C. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Uzun, B.B.; Pütün, A.E.; Pütün, E. Rapid pyrolysis of olive residue. 1. Effect of heat and mass transfer limitations on product yields and bio-oil compositions. Energy Fuels 2007, 21, 1768–1776. [Google Scholar] [CrossRef]
- Chen, D.; Li, Y.; Deng, M.; Wang, J.; Chen, M.; Yan, B.; Yuan, Q. Effect of torrefaction pretreatment and catalytic pyrolysis on the pyrolysis poly-generation of pine wood. Bioresour. Technol. 2016, 214, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Zhao, Z.; Huang, Z.; Zhao, K.; Wei, G.; Wang, X.; He, F.; Li, H. Catalytic fast pyrolysis of biomass pretreated by torrefaction with varying severity. Energy Fuels 2014, 28, 5804–5811. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Jiang, Y.; Vitidsant, T.; Reubroycharoen, P.; Xiao, R. Improving hydrocarbon yield by two-step pyrolysis of pinewood in a fluidized-bed reactor. Fuel Process. Technol. 2017, 159, 19–26. [Google Scholar] [CrossRef]
- Zheng, A.; Jiang, L.; Zhao, Z.; Huang, Z.; Zhao, K.; Wei, G.; Wang, X.; He, F.; Li, H. Impact of torrefaction on the chemical structure and catalytic fast pyrolysis behavior of hemicellulose, lignin, and cellulose. Energy Fuels 2015, 29, 8027–8034. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Besler, S. Inorganic compounds in biomass feedstocks. 1. Effect on the quality of fast pyrolysis oils. Energy Fuels 1996, 10, 293–298. [Google Scholar] [CrossRef]
- Zhou, L.; Jia, Y.; Nguyen, T.-H.; Adesina, A.A.; Liu, Z. Hydropyrolysis characteristics and kinetics of potassium-impregnated pine wood. Fuel Process. Technol. 2013, 116, 149–157. [Google Scholar] [CrossRef]
- Hu, S.; Jiang, L.; Wang, Y.; Su, S.; Sun, L.; Xu, B.; He, L.; Xiang, J. Effects of inherent alkali and alkaline earth metallic species on biomass pyrolysis at different temperatures. Bioresour. Technol. 2015, 192, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zhang, Z.B.; Yang, X.C.; Dong, C.Q.; Zhu, X.F. Catalytic fast pyrolysis of biomass impregnated with k3po4 to produce phenolic compounds: Analytical py-gc/ms study. J. Anal. Appl. Pyrolysis 2013, 104, 139–145. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Lu, Q.; Ye, X.N.; Li, W.T.; Hu, B.; Dong, C.Q. Production of phenolic-rich bio-oil from catalytic fast pyrolysis of biomass using magnetic solid base catalyst. Energy Convers. Manag. 2015, 106, 1309–1317. [Google Scholar] [CrossRef]
- Fermoso, J.; Hernando, H.; Jiménez-Sánchez, S.; Lappas, A.A.; Heracleous, E.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Bio-oil production by lignocellulose fast-pyrolysis: Isolating and comparing the effects of indigenous versus external catalysts. Fuel Process. Technol. 2017, 167, 563–574. [Google Scholar] [CrossRef]
- Mourant, D.; Wang, Z.; He, M.; Wang, X.S.; Garcia-Perez, M.; Ling, K.; Li, C.-Z. Mallee wood fast pyrolysis: Effects of alkali and alkaline earth metallic species on the yield and composition of bio-oil. Fuel 2011, 90, 2915–2922. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Satrio, J.A.; Brown, R.C.; Shanks, B.H. Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresour. Technol. 2010, 101, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Trendewicz, A.; Evans, R.; Dutta, A.; Sykes, R.; Carpenter, D.; Braun, R. Evaluating the effect of potassium on cellulose pyrolysis reaction kinetics. Biomass Bioenergy 2015, 74, 15–25. [Google Scholar] [CrossRef]
- Larsen, M.B.; Schultz, L.; Glarborg, P.; Skaarup-Jensen, L.; Dam-Johansen, K.; Frandsen, F.; Henriksen, U. Devolatilization characteristics of large particles of tyre rubber under combustion conditions. Fuel 2006, 85, 1335–1345. [Google Scholar] [CrossRef]
- Leung, D.; Yin, X.; Zhao, Z.; Xu, B.; Chen, Y. Pyrolysis of tire powder: Influence of operation variables on the composition and yields of gaseous product. Fuel Process. Technol. 2002, 79, 141–155. [Google Scholar] [CrossRef]
- Isoda, T.; Nakahara, T.; Kusakabe, K.; Morooka, S. Catalytic cracking of polyethylene-liquefied oil over amorphous aluminosilicate catalysts. Energy. Fuels 1998, 12, 1161–1167. [Google Scholar] [CrossRef]
- Mastral, A.; Murillo, R.; Garcıa, T.; Navarro, M.; Callen, M.; López, J. Study of the viability of the process for hydrogen recovery from old tyre oils. Fuel Process. Technol. 2002, 75, 185–199. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Q.; Zhou, Z.; Yang, J.; Qi, F.; Pan, Y. Online study on the pyrolysis of polypropylene over the hzsm-5 zeolite with photoionization time-of-flight mass spectrometry. Energy Fuels 2015, 29, 1090–1098. [Google Scholar] [CrossRef]
- Palza, H.; Aravena, C.; Colet, M. Role of the catalyst in the pyrolysis of polyolefin mixtures and used tires. Energy Fuels 2017, 31, 3111–3120. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Kim, C.; Kim, R.; Pak, D. Thermogravimetric characteristics and kinetic study of biomass co-pyrolysis with plastics. Korean J. Chem. Eng. 2009, 25, 1047. [Google Scholar] [CrossRef]
- Paradela, F.; Pinto, F.; Gulyurtlu, I.; Cabrita, I.; Lapa, N. Study of the co-pyrolysis of biomass and plastic wastes. Clean Technol. Environ. Policy 2009, 11, 115–122. [Google Scholar] [CrossRef]
- Mushtaq, F.; Mat, R.; Ani, F.N. A review on microwave assisted pyrolysis of coal and biomass for fuel production. Renew. Sustain. Energy Rev. 2014, 39, 555–574. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Xia, X.; Lin, C.-X.; Tong, D.-S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, P.; Wang, J.; Jiang, P.; Wu, X.; Xue, H.; Liu, J.; Zhou, X.; Li, Q. Production of jet and diesel biofuels from renewable lignocellulosic biomass. Appl. Energy 2015, 150, 128–137. [Google Scholar] [CrossRef]
- Martínez, J.D.; Puy, N.; Murillo, R.; García, T.; Navarro, M.V.; Mastral, A.M. Waste tyre pyrolysis—A review. Renew. Sustain. Energy Rev. 2013, 23, 179–213. [Google Scholar] [CrossRef]
- Cho, M.-H.; Jung, S.-H.; Kim, J.-S. Pyrolysis of mixed plastic wastes for the recovery of benzene, toluene, and xylene (btx) aromatics in a fluidized bed and chlorine removal by applying various additives. Energy Fuels 2010, 24, 1389–1395. [Google Scholar] [CrossRef]
- Xue, Y.; Kelkar, A.; Bai, X. Catalytic co-pyrolysis of biomass and polyethylene in a tandem micropyrolyzer. Fuel 2016, 166, 227–236. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; Li, J.; Su, L.; Zuo, J.; Komarneni, S.; Wang, Y. Improving the aromatic production in catalytic fast pyrolysis of cellulose by co-feeding low-density polyethylene. Appl. Catal. A 2013, 455, 114–121. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, J.; Xiao, R.; Shen, D.; Jin, B.; Xiao, G.; Chen, R. Co-catalytic pyrolysis of biomass and waste triglyceride seed oil in a novel fluidized bed reactor to produce olefins and aromatics integrated with self-heating and catalyst regeneration processes. RSC Adv. 2013, 3, 5769–5774. [Google Scholar] [CrossRef]
- Dorado, C.; Mullen, C.A.; Boateng, A.A. H-zsm5 catalyzed co-pyrolysis of biomass and plastics. ACS Sustain. Chem. Eng. 2014, 2, 301–311. [Google Scholar] [CrossRef]
- Artetxe, M.; Lopez, G.; Amutio, M.; Elordi, G.; Bilbao, J.; Olazar, M. Light olefins from hdpe cracking in a two-step thermal and catalytic process. Chem. Eng. J. 2012, 207–208, 27–34. [Google Scholar] [CrossRef]
- Serrano, D.P.; Aguado, J.; Escola, J.M.; Rodríguez, J.M.; San Miguel, G. An investigation into the catalytic cracking of ldpe using py-gc/ms. J. Anal. Appl. Pyrolysis 2005, 74, 370–378. [Google Scholar] [CrossRef]
- Brassard, P.; Godbout, S.; Raghavan, V. Pyrolysis in auger reactors for biochar and bio-oil production: A review. Biosyst. Eng. 2017, 161, 80–92. [Google Scholar] [CrossRef]
- Diebold, J.P.; Bridgwater, A.V. Overview of fast pyrolysis of biomass for the production of liquid fuels. In Developments in Thermochemical Biomass Conversion; Bridgwater, A.V., Boocock, D.G.B., Eds.; Springer: The Netherlands, 1997; pp. 5–23. [Google Scholar]
- Boateng, A.A.; Daugaard, D.E.; Goldberg, N.M.; Hicks, K.B. Bench-scale fluidized-bed pyrolysis of switchgrass for bio-oil production. Ind. Eng. Chem. Res. 2007, 46, 1891–1897. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of biomass in a fluidized bed reactor: Influence of the acidity of h-beta zeolite. Process. Saf. Environ. Prot. 2007, 85, 473–480. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, R.; Wang, D.; He, G.; Shao, S.; Zhang, J.; Zhong, Z. Biomass fast pyrolysis in a fluidized bed reactor under n2, co2, co, ch4 and h2 atmospheres. Bioresour. Technol. 2011, 102, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, E.; Hogendoorn, K.J.A.; Wang, X.; Westerhof, R.J.M.; Kersten, S.R.A.; van Swaaij, W.P.M.; Groeneveld, M.J. Fast pyrolysis of biomass in a fluidized bed reactor: In situ filtering of the vapors. Ind. Eng. Chem. Res. 2009, 48, 4744–4756. [Google Scholar] [CrossRef]
- Lee, K.-H.; Kang, B.-S.; Park, Y.-K.; Kim, J.-S. Influence of reaction temperature, pretreatment, and a char removal system on the production of bio-oil from rice straw by fast pyrolysis, using a fluidized bed. Energy Fuels 2005, 19, 2179–2184. [Google Scholar] [CrossRef]
- Kang, B.-S.; Lee, K.H.; Park, H.J.; Park, Y.-K.; Kim, J.-S. Fast pyrolysis of radiata pine in a bench scale plant with a fluidized bed: Influence of a char separation system and reaction conditions on the production of bio-oil. J. Anal. Appl. Pyrolysis 2006, 76, 32–37. [Google Scholar] [CrossRef]
- Rüdisüli, M.; Schildhauer, T.J.; Biollaz, S.M.A.; van Ommen, J.R. Scale-up of bubbling fluidized bed reactors—A review. Powder Technol. 2012, 217, 21–38. [Google Scholar] [CrossRef]
- Fernandez-Akarregi, A.R.; Makibar, J.; Lopez, G.; Amutio, M.; Olazar, M. Design and operation of a conical spouted bed reactor pilot plant (25kg/h) for biomass fast pyrolysis. Fuel Process. Technol. 2013, 112, 48–56. [Google Scholar] [CrossRef]
- Aguado, R.; Olazar, M.; San José, M.J.; Aguirre, G.; Bilbao, J. Pyrolysis of sawdust in a conical spouted bed reactor. Yields and product composition. Ind. Eng. Chem. Res. 2000, 39, 1925–1933. [Google Scholar] [CrossRef]
- Amutio, M.; Lopez, G.; Alvarez, J.; Olazar, M.; Bilbao, J. Fast pyrolysis of eucalyptus waste in a conical spouted bed reactor. Bioresour. Technol. 2015, 194, 225–232. [Google Scholar] [CrossRef]
- Makibar, J.; Fernandez-Akarregi, A.R.; Díaz, L.; Lopez, G.; Olazar, M. Pilot scale conical spouted bed pyrolysis reactor: Draft tube selection and hydrodynamic performance. Powder Technol. 2012, 219, 49–58. [Google Scholar] [CrossRef]
- López, G.; Olazar, M.; Aguado, R.; Bilbao, J. Continuous pyrolysis of waste tyres in a conical spouted bed reactor. Fuel 2010, 89, 1946–1952. [Google Scholar] [CrossRef]
- Garcìa-Pérez, M.; Chaala, A.; Pakdel, H.; Kretschmer, D.; Roy, C. Vacuum pyrolysis of softwood and hardwood biomass: Comparison between product yields and bio-oil properties. J. Anal. Appl. Pyrolysis 2007, 78, 104–116. [Google Scholar] [CrossRef]
- Roy, C.; Chaala, A.; Darmstadt, H. The vacuum pyrolysis of used tires: End-uses for oil and carbon black products. J. Anal. Appl. Pyrolysis 1999, 51, 201–221. [Google Scholar] [CrossRef]
- Di Blasi, C. Heat transfer mechanisms and multi-step kinetics in the ablative pyrolysis of cellulose. Chem. Eng. Sci. 1996, 51, 2211–2220. [Google Scholar] [CrossRef]
- Helleur, R.; Popovic, N.; Ikura, M.; Stanciulescu, M.; Liu, D. Characterization and potential applications of pyrolytic char from ablative pyrolysis of used tires. J. Anal. Appl. Pyrolysis 2001, 58–59, 813–824. [Google Scholar] [CrossRef]
- Peacocke, G.V.C.; Bridgwater, A.V. Ablative plate pyrolysis of biomass for liquids. Biomass Bioenergy 1994, 7, 147–154. [Google Scholar] [CrossRef]
- Daugaard, D.; Jones, S.T.; Dalluge, D.L.; Brown, R.C. Selective Temperature Quench and Electrostatic Recovery of Bio-Oil Fractions. U.S. Patent 8,992,736, 31 March 2015. [Google Scholar]
- Chen, Y.-C.; Pan, Y.-N.; Hsieh, K.-H. Process optimization of fast pyrolysis reactor for converting forestry wastes into bio-oil with the taguchi method. Procedia Environ. Sci. 2011, 10, 1719–1725. [Google Scholar] [CrossRef]
- Brown, J.N.; Brown, R.C. Process optimization of an auger pyrolyzer with heat carrier using response surface methodology. Bioresour. Technol. 2012, 103, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Diebold, J.P. A Review of the Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-Oils; National Renewable Energy Laboratory: Golden, CO, USA, 2000.
- Piskorz, J.; Scott, D.S.; Radlein, D. Composition of oils obtained by fast pyrolysis of different woods. In Pyrolysis Oils from Biomass; American Chemical Society: Washington, DC, USA, 1988; Volume 376, pp. 167–178. [Google Scholar]
- Ed, J.S.; Thomas, A.M. Pyrolysis Oils from Biomass; American Chemical Society: Washington, DC, USA, 1988; Volume 376, p. 372. [Google Scholar]
- Garcìa-Pérez, M.; Chaala, A.; Pakdel, H.; Kretschmer, D.; Rodrigue, D.; Roy, C. Multiphase structure of bio-oils. Energy Fuels 2005, 20, 364–375. [Google Scholar] [CrossRef]
- Milne, T.A.A.; Davis, M.; Deutch, S.; Johnson, D. A Review of the Chemical Composition of Fast Pyrolysis Oils, in Developments in Thermal Biomass Conversion; Blackie Academic and Professional: London, UK, 1997. [Google Scholar]
- Diebold, J.P. A Review of the Toxicity of Biomass Pyrolysis Liquids Formed at Low Temperatures; National Renewable Energy Laboratory: Golden, CO, USA, 1997; p. 35. [Google Scholar]
- Branca, C.; Giudicianni, P.; Di Blasi, C. Gc/ms characterization of liquids generated from low-temperature pyrolysis of wood. Ind. Eng. Chem. Res. 2003, 42, 3190–3202. [Google Scholar] [CrossRef]
- Saiz-Jimenez, C.; De Leeuw, J.W. Lignin pyrolysis products: Their structures and their significance as biomarkers. Org. Geochem. 1986, 10, 869–876. [Google Scholar] [CrossRef]
- Kawamoto, H.; Murayama, M.; Saka, S. Pyrolysis behavior of levoglucosan as an intermediate in cellulose pyrolysis: Polymerization into polysaccharide as a key reaction to carbonized product formation. J. Wood Sci. 2003, 49, 469–473. [Google Scholar] [CrossRef]
- Shafizadeh, F.; Furneaux, R.H.; Cochran, T.G.; Scholl, J.P.; Sakai, Y. Production of levoglucosan and glucose from pyrolysis of cellulosic materials. J. Appl. Polym. Sci. 1979, 23, 3525–3539. [Google Scholar] [CrossRef]
- Piskorz, J.; Radlein, D.; Scott, D.S. On the mechanism of the rapid pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 1986, 9, 121–137. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Besler, S.; Evans, R.J. Inorganic Compounds in Biomass Feedstocks: Their Role in Char Formation and Effect on the Quality of Fast Pyrolysis Oils; National Renewable Energy Laboratory: Golden, CO, USA, 1994. [Google Scholar]
- French, R.J.; Milne, T.A. Vapor phase release of alkali species in the combustion of biomass pyrolysis oils. Biomass Bioenergy 1994, 7, 315–325. [Google Scholar] [CrossRef]
- Dayton, D. The fate of alkali metal during biomass thermochemical conversion. In Developments in Thermochemical Biomass Conversion; Bridgwater, A.V., Boocock, D.G.B., Eds.; Springer: The Netherlands, 1997; pp. 1263–1277. [Google Scholar]
- French, R.J.; Dayton, D.C.; Milne, T.A. The Direct Observation of Alkali Vapor Species in Biomass Combustion and Gasification; National Renewable Energy Laboratory: Golden, CO, USA, 1994. [Google Scholar]
- Lu, Q.; Yang, X.-L.; Zhu, X.-F. Analysis on chemical and physical properties of bio-oil pyrolyzed from rice husk. J. Anal. Appl. Pyrolysis 2008, 82, 191–198. [Google Scholar] [CrossRef]
- Diebold, J.P.; Czernik, S. Additives to lower and stabilize the viscosity of pyrolysis oils during storage. Energy Fuels 1997, 11, 1081–1091. [Google Scholar] [CrossRef]
- Czernik, S.; Johnson, D.K.; Black, S. Stability of wood fast pyrolysis oil. Biomass Bioenergy 1994, 7, 187–192. [Google Scholar] [CrossRef]
- Fratini, E.; Bonini, M.; Oasmaa, A.; Solantausta, Y.; Teixeira, J.; Baglioni, P. Sans analysis of the microstructural evolution during the aging of pyrolysis oils from biomass. Langmuir 2005, 22, 306–312. [Google Scholar] [CrossRef]
- Boucher, M.E.; Chaala, A.; Pakdel, H.; Roy, C. Bio-oils obtained by vacuum pyrolysis of softwood bark as a liquid fuel for gas turbines. Part ii: Stability and ageing of bio-oil and its blends with methanol and a pyrolytic aqueous phase. Biomass Bioenergy 2000, 19, 351–361. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E. Fast pyrolysis of forestry residue. 3. Storage stability of liquid fuel. Energy Fuels 2003, 17, 1075–1084. [Google Scholar] [CrossRef]
- Hilten, R.N.; Das, K.C. Comparison of three accelerated aging procedures to assess bio-oil stability. Fuel 2010, 89, 2741–2749. [Google Scholar] [CrossRef]
- Ortega, J.V.; Renehan, A.M.; Liberatore, M.W.; Herring, A.M. Physical and chemical characteristics of aging pyrolysis oils produced from hardwood and softwood feedstocks. J. Anal. Appl. Pyrolysis 2011, 91, 190–198. [Google Scholar] [CrossRef]
- Ben, H.; Ragauskas, A.J. In situ nmr characterization of pyrolysis oil during accelerated aging. ChemSusChem 2012, 5, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Y.; Mourant, D.; Gunawan, R.; Lievens, C.; Chaiwat, W.; Gholizadeh, M.; Wu, L.; Li, X.; Li, C.-Z. Polymerization on heating up of bio-oil: A model compound study. AlChE J. 2013, 59, 888–900. [Google Scholar] [CrossRef]
- Alsbou, E.; Helleur, B. Accelerated aging of bio-oil from fast pyrolysis of hardwood. Energy Fuels 2014, 28, 3224–3235. [Google Scholar] [CrossRef]
- Zhang, L.; Chaparro Sosa, A.; Walters, K.B. Impacts of thermal processing on the physical and chemical properties of pyrolysis oil produced by a modified fluid catalytic cracking pyrolysis process. Energy Fuels 2016, 30, 7367–7378. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A. Storage stability of biocrude oils from fast pyrolysis of poultry litter. Waste Manag. 2012, 32, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Oasmaa, A.; Kuoppala, E.; Selin, J.-F.; Gust, S.; Solantausta, Y. Fast pyrolysis of forestry residue and pine. 4. Improvement of the product quality by solvent addition. Energy Fuels 2004, 18, 1578–1583. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Sharma, R.K.; Bakhshi, N.N. Characterization and stability analysis of wood-derived bio-oil. Fuel Process. Technol. 1992, 31, 241–256. [Google Scholar] [CrossRef]
- Meng, J.; Smirnova, T.I.; Song, X.; Moore, A.; Ren, X.; Kelley, S.; Park, S.; Tilotta, D. Identification of free radicals in pyrolysis oil and their impact on bio-oil stability. RSC Adv. 2014, 4, 29840–29846. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, Y.; Wijayapala, R.; Walters, K.B. Alcohol stabilization of low water content pyrolysis oil during high temperature treatment. Energy Fuels 2017, 31, 13666–13674. [Google Scholar] [CrossRef]
- Vitolo, S.; Seggiani, M.; Frediani, P.; Ambrosini, G.; Politi, L. Catalytic upgrading of pyrolytic oils to fuel over different zeolites. Fuel 1999, 78, 1147–1159. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Upgrading bio-oil over different solid catalysts. Energy Fuels 2006, 20, 2717–2720. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeres, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Rioche, C.; Kulkarni, S.; Meunier, F.C.; Breen, J.P.; Burch, R. Steam reforming of model compounds and fast pyrolysis bio-oil on supported noble metal catalysts. Appl. Catal. B Environ. 2005, 61, 130–139. [Google Scholar] [CrossRef]
- Takanabe, K.; Aika, K.-I.; Seshan, K.; Lefferts, L. Sustainable hydrogen from bio-oil—Steam reforming of acetic acid as a model oxygenate. J. Catal. 2004, 227, 101–108. [Google Scholar] [CrossRef]
- Chiaramonti, D.; Bonini, M.; Fratini, E.; Tondi, G.; Gartner, K.; Bridgwater, A.V.; Grimm, H.P.; Soldaini, I.; Webster, A.; Baglioni, P. Development of emulsions from biomass pyrolysis liquid and diesel and their use in engines—Part 1: Emulsion production. Biomass Bioenergy 2003, 25, 85–99. [Google Scholar] [CrossRef]
- Javaid, A.; Ryan, T.; Berg, G.; Pan, X.; Vispute, T.; Bhatia, S.R.; Huber, G.W.; Ford, D.M. Removal of char particles from fast pyrolysis bio-oil by microfiltration. J. Membr. Sci. 2010, 363, 120–127. [Google Scholar] [CrossRef]
- Naske, C.D.; Polk, P.; Wynne, P.Z.; Speed, J.; Holmes, W.E.; Walters, K.B. Postcondensation filtration of pine and cottonwood pyrolysis oil and impacts on accelerated aging reactions. Energy Fuels 2011, 26, 1284–1297. [Google Scholar] [CrossRef]
- McCall, M.J.; Brandvold, T.A. Fuel and Fuel Blending Components from Biomass Derived Pyrolysis Oil. U.S. Patent 8,329,969, 11 December 2012. [Google Scholar]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Gagnon, J.; Kaliaguine, S. Catalytic hydrotreatment of vacuum pyrolysis oils from wood. Ind. Eng. Chem. Res. 1988, 27, 1783–1788. [Google Scholar] [CrossRef]
- Pham, T.N.; Sooknoi, T.; Crossley, S.P.; Resasco, D.E. Ketonization of carboxylic acids: Mechanisms, catalysts, and implications for biomass conversion. Acs Catal. 2013, 3, 2456–2473. [Google Scholar] [CrossRef]
- Lilga, M.A.; Padmaperuma, A.B.; Auberry, D.L.; Job, H.M.; Swita, M.S. Ketonization of levulinic acid and γ-valerolactone to hydrocarbon fuel precursors. Catal. Today 2018, 302, 80–86. [Google Scholar] [CrossRef]
- Gliński, M.; Kijeński, J.; Jakubowski, A. Ketones from monocarboxylic acids: Catalytic ketonization over oxide systems. Appl. Catal. A Gen. 1995, 128, 209–217. [Google Scholar] [CrossRef]
- Weber, J.; Thompson, A.; Wilmoth, J.; Batra, V.S.; Janulaitis, N.; Kastner, J.R. Effect of metal oxide redox state in red mud catalysts on ketonization of fast pyrolysis oil derived oxygenates. Appl. Catal. B Environ. 2019, 241, 430–441. [Google Scholar] [CrossRef]
- Fan, L.; Chen, P.; Zhou, N.; Liu, S.; Zhang, Y.; Liu, Y.; Wang, Y.; Omar, M.M.; Peng, P.; Addy, M.; et al. In-situ and ex-situ catalytic upgrading of vapors from microwave-assisted pyrolysis of lignin. Bioresour. Technol. 2018, 247, 851–858. [Google Scholar] [CrossRef]
- Gamliel, D.P.; Du, S.; Bollas, G.M.; Valla, J.A. Investigation of in situ and ex situ catalytic pyrolysis of miscanthus × giganteus using a pygc-ms microsystem and comparison with a bench-scale spouted-bed reactor. Bioresour. Technol. 2015, 191, 187–196. [Google Scholar] [CrossRef]
- Luo, G.; Resende, F.L.P. In-situ and ex-situ upgrading of pyrolysis vapors from beetle-killed trees. Fuel 2016, 166, 367–375. [Google Scholar] [CrossRef]
- Wang, K.; Johnston, P.A.; Brown, R.C. Comparison of in-situ and ex-situ catalytic pyrolysis in a micro-reactor system. Bioresour. Technol. 2014, 173, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xiao, R.; Zhang, H. Ex-situ catalytic fast pyrolysis of biomass over hzsm-5 in a two-stage fluidized-bed/fixed-bed combination reactor. Bioresour. Technol. 2017, 243, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, H.; Karim, A.M.; Sun, J.; Wang, Y. Catalytic fast pyrolysis of lignocellulosic biomass. Chem. Soc. Rev. 2014, 43, 7594–7623. [Google Scholar] [CrossRef] [PubMed]
- Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B.F. Potential and challenges of zeolite chemistry in the catalytic conversion of biomass. Chem. Soc. Rev. 2016, 45, 584–611. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Lim, J.K.; Hameed, B.H. Recent progress on biomass co-pyrolysis conversion into high-quality bio-oil. Bioresour. Technol. 2016, 221, 645–655. [Google Scholar] [CrossRef]
- Venderbosch, R.H. A critical view on catalytic pyrolysis of biomass. ChemSusChem 2015, 8, 1306–1316. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. In situ fast pyrolysis of biomass with zeolite catalysts for bioaromatics/gasoline production: A review. Energy Convers. Manag. 2015, 105, 338–354. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- Imran, A.; Bramer, E.; Seshan, K.; Brem, G. Catalytic flash pyrolysis of biomass using different types of zeolite and online vapor fractionation. Energies 2016, 9, 187. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of woody biomass in a fluidized bed reactor: Influence of the zeolite structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Carlson, T.R.; Jae, J.; Lin, Y.-C.; Tompsett, G.A.; Huber, G.W. Catalytic fast pyrolysis of glucose with hzsm-5: The combined homogeneous and heterogeneous reactions. J. Catal. 2010, 270, 110–124. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Kurnia, I.; Karnjanakom, S.; Bayu, A.; Yoshida, A.; Rizkiana, J.; Prakoso, T.; Abudula, A.; Guan, G. In-situ catalytic upgrading of bio-oil derived from fast pyrolysis of lignin over high aluminum zeolites. Fuel Process. Technol. 2017, 167, 730–737. [Google Scholar] [CrossRef]
- Foster, A.J.; Jae, J.; Cheng, Y.-T.; Huber, G.W.; Lobo, R.F. Optimizing the aromatic yield and distribution from catalytic fast pyrolysis of biomass over zsm-5. Appl. Catal. A 2012, 423–424, 154–161. [Google Scholar] [CrossRef]
- Hoff, T.C.; Gardner, D.W.; Thilakaratne, R.; Wang, K.; Hansen, T.W.; Brown, R.C.; Tessonnier, J.P. Tailoring zsm-5 zeolites for the fast pyrolysis of biomass to aromatic hydrocarbons. ChemSusChem 2016, 9, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shao, S.; Luo, M.; Xiao, R. The comparison of chemical liquid deposition and acid dealumination modified zsm-5 for catalytic pyrolysis of pinewood using pyrolysis-gas chromatography/mass spectrometry. Bioresour. Technol. 2017, 244, 726–732. [Google Scholar] [CrossRef]
- Gou, J.; Wang, Z.; Li, C.; Qi, X.; Vattipalli, V.; Cheng, Y.-T.; Huber, G.; Conner, W.C.; Dauenhauer, P.J.; Mountziaris, T.J.; et al. The effects of zsm-5 mesoporosity and morphology on the catalytic fast pyrolysis of furan. Green Chem. 2017, 19, 3549–3557. [Google Scholar] [CrossRef]
- Hoff, T.C.; Gardner, D.W.; Thilakaratne, R.; Proano-Aviles, J.; Brown, R.C.; Tessonnier, J.-P. Elucidating the effect of desilication on aluminum-rich zsm-5 zeolite and its consequences on biomass catalytic fast pyrolysis. Appl. Catal. A 2017, 529, 68–78. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, Z.; Sun, J.; Wang, Q. Recent progress of catalytic pyrolysis of biomass by hzsm-5. Chin. J. Catal. 2013, 34, 641–650. [Google Scholar] [CrossRef]
- Jackson, M.A.; Compton, D.L.; Boateng, A.A. Screening heterogeneous catalysts for the pyrolysis of lignin. J. Anal. Appl. Pyrolysis 2009, 85, 226–230. [Google Scholar] [CrossRef]
- Mihalcik, D.J.; Mullen, C.A.; Boateng, A.A. Screening acidic zeolites for catalytic fast pyrolysis of biomass and its components. J. Anal. Appl. Pyrolysis 2011, 92, 224–232. [Google Scholar] [CrossRef]
- Ma, Z.; Troussard, E.; van Bokhoven, J.A. Controlling the selectivity to chemicals from lignin via catalytic fast pyrolysis. Appl. Catal. A 2012, 423–424, 130–136. [Google Scholar] [CrossRef]
- Shen, D.; Zhao, J.; Xiao, R.; Gu, S. Production of aromatic monomers from catalytic pyrolysis of black-liquor lignin. J. Anal. Appl. Pyrolysis 2015, 111, 47–54. [Google Scholar] [CrossRef]
- Jia, L.Y.; Raad, M.; Hamieh, S.; Toufaily, J.; Hamieh, T.; Bettahar, M.M.; Mauviel, G.; Tarrighi, M.; Pinard, L.; Dufour, A. Catalytic fast pyrolysis of biomass: Superior selectivity of hierarchical zeolites to aromatics. Green Chem. 2017, 19, 5442–5459. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Zhou, G.; Wang, W.; Wang, C.; Komarneni, S.; Wang, Y. Catalytic fast pyrolysis of biomass with mesoporous zsm-5 zeolites prepared by desilication with naoh solutions. Appl. Catal. A 2014, 470, 115–122. [Google Scholar] [CrossRef]
- Gamliel, D.P.; Cho, H.J.; Fan, W.; Valla, J.A. On the effectiveness of tailored mesoporous mfi zeolites for biomass catalytic fast pyrolysis. Appl. Catal. A 2016, 522, 109–119. [Google Scholar] [CrossRef]
- Leonowicz, M.E.; Lawton, J.A.; Lawton, S.L.; Rubin, M.K. Mcm-22: A molecular sieve with two independent multidimensional channel systems. Science 1994, 264, 1910–1913. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.J.; Kresge, C.T.; Vartuli, J.C.; Leonowicz, M.E.; Fung, A.S.; McCullen, S.B. Mcm-36: The first pillared molecular sieve with zeoliteproperties. Stud. Surf. Sci. Catal. 1995, 94, 301–308. [Google Scholar] [CrossRef]
- Hernando, H.; Fermoso, J.; Ochoa-Hernández, C.; Opanasenko, M.; Pizarro, P.; Coronado, J.M.; Čejka, J.; Serrano, D.P. Performance of mcm-22 zeolite for the catalytic fast-pyrolysis of acid-washed wheat straw. Catal. Today 2018, 304, 30–38. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kim, J.-Y.; Kim, J.; Ahn, W.-S. CO2 capture over amine-functionalized mcm-22, mcm-36 and itq-2. Fuel 2012, 97, 435–442. [Google Scholar] [CrossRef]
- Käldström, M.; Kumar, N.; Heikkilä, T.; Murzin, D.Y. Pillared h-mcm-36 mesoporous and h-mcm-22 microporous materials for conversion of levoglucosan: Influence of varying acidity. Appl. Catal. A 2011, 397, 13–21. [Google Scholar] [CrossRef]
- Naqvi, S.R.; Uemura, Y.; Yusup, S.; Sugiura, Y.; Nishiyama, N. In situ catalytic fast pyrolysis of paddy husk pyrolysis vapors over mcm-22 and itq-2 zeolites. J. Anal. Appl. Pyrolysis 2015, 114, 32–39. [Google Scholar] [CrossRef]
- Wang, W.; Shi, Y.; Cui, Y.; Li, X. Catalytic fast pyrolysis of cellulose for increasing contents of furans and aromatics in biofuel production. J. Anal. Appl. Pyrolysis 2018, 131, 93–100. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, R.; Song, W. Influence of hsapo-34, hzsm-5, and nay on pyrolysis of corn straw fermentation residue via py-gc/ms. J. Anal. Appl. Pyrolysis 2016, 122, 183–190. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, Y.-M.; Lee, B.; Kim, S.; Jae, J.; Jung, S.-C.; Kim, T.-W.; Park, Y.-K. Catalytic copyrolysis of torrefied cork oak and high density polyethylene over a mesoporous hy catalyst. Catal. Today 2018, 307, 301–307. [Google Scholar] [CrossRef]
- Ma, Z.; Ghosh, A.; Asthana, N.; van Bokhoven, J. Visualization of structural changes during deactivation and regeneration of fau zeolite for catalytic fast pyrolysis of lignin using nmr and electron microscopy techniques. ChemCatChem 2018, 10, 4431–4437. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Chen, L.; Zhao, B.; Yang, S.; Xie, X. Comparision of catalytic fast pyrolysis of biomass to aromatic hydrocarbons over zsm-5 and fe/zsm-5 catalysts. J. Anal. Appl. Pyrolysis 2016, 121, 342–346. [Google Scholar] [CrossRef]
- Saraçoğlu, E.; Uzun, B.B.; Apaydın-Varol, E. Upgrading of fast pyrolysis bio-oil over fe modified zsm-5 catalyst to enhance the formation of phenolic compounds. Int. J. Hydrogen Energy 2017, 42, 21476–21486. [Google Scholar] [CrossRef]
- Fermoso, J.; Hernando, H.; Jana, P.; Moreno, I.; Přech, J.; Ochoa-Hernández, C.; Pizarro, P.; Coronado, J.M.; Čejka, J.; Serrano, D.P. Lamellar and pillared zsm-5 zeolites modified with mgo and zno for catalytic fast-pyrolysis of eucalyptus woodchips. Catal. Today 2016, 277, 171–181. [Google Scholar] [CrossRef]
- Schultz, E.L.; Mullen, C.A.; Boateng, A.A. Aromatic hydrocarbon production from eucalyptus urophylla pyrolysis over several metal-modified zsm-5 catalysts. Energy Technol. 2017, 5, 196–204. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, F.; Yang, X.; Huang, Y.; Liu, C.; Zheng, Z.; Gu, J. Study on aromatics production via the catalytic pyrolysis vapor upgrading of biomass using metal-loaded modified h-zsm-5. J. Anal. Appl. Pyrolysis 2017, 126, 169–179. [Google Scholar] [CrossRef]
- Liu, T.-L.; Cao, J.-P.; Zhao, X.-Y.; Wang, J.-X.; Ren, X.-Y.; Fan, X.; Zhao, Y.-P.; Wei, X.-Y. In situ upgrading of shengli lignite pyrolysis vapors over metal-loaded hzsm-5 catalyst. Fuel Process. Technol. 2017, 160, 19–26. [Google Scholar] [CrossRef]
- Mullen, C.A.; Tarves, P.C.; Boateng, A.A. Role of potassium exchange in catalytic pyrolysis of biomass over zsm-5: Formation of alkyl phenols and furans. ACS Sustain. Chem. Eng. 2017, 5, 2154–2162. [Google Scholar] [CrossRef]
- Campanella, A.; Harold, M.P. Fast pyrolysis of microalgae in a falling solids reactor: Effects of process variables and zeolite catalysts. Biomass Bioenergy 2012, 46, 218–232. [Google Scholar] [CrossRef]
- Till, Z.; Varga, T.; Sója, J.; Miskolczi, N.; Chován, T. Kinetic identification of plastic waste pyrolysis on zeolite-based catalysts. Energy Convers. Manag. 2018, 173, 320–330. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, M.; Shao, J.; Yang, H.; Zeng, K.; Chen, Y.; Luo, J.; Agblevor, F.A.; Chen, H. The conversion of biomass to light olefins on fe-modified zsm-5 catalyst: Effect of pyrolysis parameters. Sci. Total Environ. 2018, 628–629, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, J.; Feng, Y.; Wang, W.; Zhang, X.; Chen, Q.; Komarneni, S.; Wang, Y. Thermally stable phosphorus and nickel modified zsm-5 zeolites for catalytic co-pyrolysis of biomass and plastics. RSC Adv. 2015, 5, 30485–30494. [Google Scholar] [CrossRef]
- Vichaphund, S.; Aht-ong, D.; Sricharoenchaikul, V.; Atong, D. Production of aromatic compounds from catalytic fast pyrolysis of jatropha residues using metal/hzsm-5 prepared by ion-exchange and impregnation methods. Renew. Energy 2015, 79, 28–37. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Gupta, R.B. Catalytic pyrolysis of biomass over h+zsm-5 under hydrogen pressure. Energy Fuels 2012, 26, 5300–5306. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Production of aromatic hydrocarbons via catalytic pyrolysis of biomass over fe-modified hzsm-5 zeolites. ACS Sustain. Chem. Eng. 2015, 3, 1623–1631. [Google Scholar] [CrossRef]
- Karnjanakom, S.; Suriya-umporn, T.; Bayu, A.; Kongparakul, S.; Samart, C.; Fushimi, C.; Abudula, A.; Guan, G. High selectivity and stability of mg-doped al-mcm-41 for in-situ catalytic upgrading fast pyrolysis bio-oil. Energy Convers. Manag. 2017, 142, 272–285. [Google Scholar] [CrossRef]
- Rizkiana, J.; Guan, G.; Widayatno, W.; Yang, J.; Hao, X.; Matsuoka, K.; Abudula, A.J.R.A. Mg-modified ultra-stable y type zeolite for the rapid catalytic co-pyrolysis of low-rank coal and biomass. RSC Adv. 2016, 6, 2096–2105. [Google Scholar] [CrossRef]
- Iliopoulou, E.F.; Stefanidis, S.D.; Kalogiannis, K.G.; Delimitis, A.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic upgrading of biomass pyrolysis vapors using transition metal-modified zsm-5 zeolite. Appl. Catal. B Environ. 2012, 127, 281–290. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Liu, X.; Zhu, S.; Hu, L.; Zhang, Q. Upgrading of bio-oil from catalytic pyrolysis of pretreated rice husk over fe-modified zsm-5 zeolite catalyst. Fuel Process. Technol. 2018, 175, 17–25. [Google Scholar] [CrossRef]
- Balasundram, V.; Ibrahim, N.; Kasmani, R.; Isha, R.; Hamid, M.K.A.; Hasbullah, H.; Ali, R.R. Catalytic upgrading of sugarcane bagasse pyrolysis vapours over rare earth metal (ce) loaded hzsm-5: Effect of catalyst to biomass ratio on the organic compounds in pyrolysis oil. Appl. Energy 2018, 220, 787–799. [Google Scholar] [CrossRef]
- Vichaphund, S.; Aht-ong, D.; Sricharoenchaikul, V.; Atong, D. Effect of cv-zsm-5, ni-zsm-5 and fa-zsm-5 catalysts for selective aromatic formation from pyrolytic vapors of rubber wastes. J. Anal. Appl. Pyrolysis 2017, 124, 733–741. [Google Scholar] [CrossRef]
- Ren, X.-Y.; Cao, J.-P.; Zhao, X.-Y.; Yang, Z.; Liu, T.-L.; Fan, X.; Zhao, Y.-P.; Wei, X.-Y. Catalytic upgrading of pyrolysis vapors from lignite over mono/bimetal-loaded mesoporous hzsm-5. Fuel 2018, 218, 33–40. [Google Scholar] [CrossRef]
- Li, P.; Li, D.; Yang, H.; Wang, X.; Chen, H. Effects of fe-, zr-, and co-modified zeolites and pretreatments on catalytic upgrading of biomass fast pyrolysis vapors. Energy Fuels 2016, 30, 3004–3013. [Google Scholar] [CrossRef]
- Ren, X.-Y.; Cao, J.-P.; Zhao, X.-Y.; Shen, W.-Z.; Wei, X.-Y. Increasing light aromatic products during upgrading of lignite pyrolysis vapor over co-modified hzsm-5. J. Anal. Appl. Pyrolysis 2018, 130, 190–197. [Google Scholar] [CrossRef]
- Lu, Q.; Guo, H.-Q.; Zhou, M.-X.; Zhang, Z.-X.; Cui, M.-S.; Zhang, Y.-Y.; Yang, Y.-P.; Zhang, L.-B. Monocyclic aromatic hydrocarbons production from catalytic cracking of pine wood-derived pyrolytic vapors over ce-mo2n/hzsm-5 catalyst. Sci. Total Environ. 2018, 634, 141–149. [Google Scholar] [CrossRef]
- Adam, J.; Antonakou, E.; Lappas, A.; Stöcker, M.; Nilsen, M.H.; Bouzga, A.; Hustad, J.E.; Øye, G. In situ catalytic upgrading of biomass derived fast pyrolysis vapours in a fixed bed reactor using mesoporous materials. Microporous Mesoporous Mater. 2006, 96, 93–101. [Google Scholar] [CrossRef]
- Antonakou, E.; Lappas, A.; Nilsen, M.H.; Bouzga, A.; Stöcker, M. Evaluation of various types of al-mcm-41 materials as catalysts in biomass pyrolysis for the production of bio-fuels and chemicals. Fuel 2006, 85, 2202–2212. [Google Scholar] [CrossRef]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Price, R. Furfural—A promising platform for lignocellulosic biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Yemiş, O.; Mazza, G. Acid-catalyzed conversion of xylose, xylan and straw into furfural by microwave-assisted reaction. Bioresour. Technol. 2011, 102, 7371–7378. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Liu, S.; Abu-Omar, M.M.; Mosier, N.S. Selective conversion of biomass hemicellulose to furfural using maleic acid with microwave heating. Energy Fuels 2012, 26, 1298–1304. [Google Scholar] [CrossRef]
- Li, Y.; Lu, X.; Yuan, L.; Liu, X. Fructose decomposition kinetics in organic acids-enriched high temperature liquid water. Biomass Bioenergy 2009, 33, 1182–1187. [Google Scholar] [CrossRef]
- Chheda, J.N.; Román-Leshkov, Y.; Dumesic, J.A. Production of 5-hydroxymethylfurfural and furfural by dehydration of biomass-derived mono- and poly-saccharides. Green Chem. 2007, 9, 342–350. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Chen, Z.; Zhu, D.; Yang, H.; Liu, P.; Li, T.; Chen, H. Catalytic fast pyrolysis of cellulose to produce furan compounds with sapo type catalysts. J. Anal. Appl. Pyrolysis 2018, 129, 53–60. [Google Scholar] [CrossRef]
- Wang, W.-L.; Ren, X.-Y.; Li, L.-F.; Chang, J.-M.; Cai, L.-P.; Geng, J. Catalytic effect of metal chlorides on analytical pyrolysis of alkali lignin. Fuel Process. Technol. 2015, 134, 345–351. [Google Scholar] [CrossRef]
- Wang, W.-L.; Ren, X.-Y.; Chang, J.-M.; Cai, L.-P.; Shi, S.Q. Characterization of bio-oils and bio-chars obtained from the catalytic pyrolysis of alkali lignin with metal chlorides. Fuel Process. Technol. 2015, 138, 605–611. [Google Scholar] [CrossRef]
- Hwang, H.; Oh, S.; Choi, I.-G.; Choi, J.W. Catalytic effects of magnesium on the characteristics of fast pyrolysis products—Bio-oil, bio-char, and non-condensed pyrolytic gas fractions. J. Anal. Appl. Pyrolysis 2015, 113, 27–34. [Google Scholar] [CrossRef]
- Kalogiannis, K.G.; Stefanidis, S.D.; Karakoulia, S.A.; Triantafyllidis, K.S.; Yiannoulakis, H.; Michailof, C.; Lappas, A.A. First pilot scale study of basic vs acidic catalysts in biomass pyrolysis: Deoxygenation mechanisms and catalyst deactivation. Appl. Catal. B Environ. 2018, 238, 346–357. [Google Scholar] [CrossRef]
- Park, H.J.; Heo, H.S.; Park, Y.-K.; Yim, J.-H.; Jeon, J.-K.; Park, J.; Ryu, C.; Kim, S.-S. Clean bio-oil production from fast pyrolysis of sewage sludge: Effects of reaction conditions and metal oxide catalysts. Bioresour. Technol. 2010, 101, S83–S85. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, Z.; Zhang, Z.; Sun, J.; Wang, Q.; Pittman, C.U. Catalytic fast pyrolysis of a wood-plastic composite with metal oxides as catalysts. Waste Manag. 2018, 79, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.M.; Guil-Lopez, R.; Fierro, J.L.G.; Mota, N.; Jiménez, S.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Catalytic fast pyrolysis of biomass over mg-al mixed oxides derived from hydrotalcite-like precursors: Influence of mg/al ratio. J. Anal. Appl. Pyrolysis 2018, 134, 362–370. [Google Scholar] [CrossRef]
- Anand, V.; Gautam, R.; Vinu, R. Non-catalytic and catalytic fast pyrolysis of schizochytrium limacinum microalga. Fuel 2017, 205, 1–10. [Google Scholar] [CrossRef]
- Lu, Q.; Xiong, W.-M.; Li, W.-Z.; Guo, Q.-X.; Zhu, X.-F. Catalytic pyrolysis of cellulose with sulfated metal oxides: A promising method for obtaining high yield of light furan compounds. Bioresour. Technol. 2009, 100, 4871–4876. [Google Scholar] [CrossRef]
- Kaewpengkrow, P.; Atong, D.; Sricharoenchaikul, V. Selective catalytic fast pyrolysis of jatropha curcas residue with metal oxide impregnated activated carbon for upgrading bio-oil. Int. J. Hydrogen Energy 2017, 42, 18397–18409. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Catalytic pyrolysis-gc/ms of lignin from several sources. Fuel Process. Technol. 2010, 91, 1446–1458. [Google Scholar] [CrossRef]
- Lu, Q.; Zhou, M.-X.; Li, W.-T.; Wang, X.; Cui, M.-S.; Yang, Y.-P. Catalytic fast pyrolysis of biomass with noble metal-like catalysts to produce high-grade bio-oil: Analytical py-gc/ms study. Catal. Today 2018, 302, 169–179. [Google Scholar] [CrossRef]
- Koike, N.; Hosokai, S.; Takagaki, A.; Nishimura, S.; Kikuchi, R.; Ebitani, K.; Suzuki, Y.; Oyama, S.T. Upgrading of pyrolysis bio-oil using nickel phosphide catalysts. J. Catal. 2016, 333, 115–126. [Google Scholar] [CrossRef]
- Sushil, S.; Batra, V.S. Catalytic applications of red mud, an aluminium industry waste: A review. Appl. Catal. B Environ. 2008, 81, 64–77. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of pinyon-juniper catalytic pyrolysis oil using red mud-supported nickel catalysts. Appl. Catal. B Environ. 2018, 236, 1–12. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Jahromi, H. Aqueous-phase synthesis of hydrocarbons from furfural reactions with low-molecular-weight biomass oxygenates. Energy Fuels 2018, 32, 8552–8562. [Google Scholar] [CrossRef]
- Yathavan, B.K.; Agblevor, F.A. Catalytic pyrolysis of pinyon–juniper using red mud and hzsm-5. Energy Fuels 2013, 27, 6858–6865. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Yang, J.; Liang, S.; Liu, K.; Xiao, K.; Deng, H.; Hu, J.; Xiao, B. Improving bromine fixation in co-pyrolysis of non-metallic fractions of waste printed circuit boards with bayer red mud. Sci. Total Environ. 2018, 639, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.A.; Akay, G. Effect of cao on tar production and dew point depression during gasification of fuel cane bagasse in a novel downdraft gasifier. Fuel Process. Technol. 2013, 106, 654–660. [Google Scholar] [CrossRef]
- Ding, K.; Zhong, Z.; Wang, J.; Zhang, B.; Fan, L.; Liu, S.; Wang, Y.; Liu, Y.; Zhong, D.; Chen, P.; et al. Improving hydrocarbon yield from catalytic fast co-pyrolysis of hemicellulose and plastic in the dual-catalyst bed of cao and hzsm-5. Bioresour. Technol. 2018, 261, 86–92. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, Z.; Ding, K.; Zhang, B.; Deng, A.; Min, M.; Chen, P.; Ruan, R. Co-pyrolysis of bamboo residual with waste tire over dual catalytic stage of cao and co-modified hzsm-5. Energy 2017, 133, 90–98. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, B.; Zhong, Z.; Ding, K.; Deng, A.; Min, M.; Chen, P.; Ruan, R. Catalytic fast co-pyrolysis of bamboo residual and waste lubricating oil over an ex-situ dual catalytic beds of mgo and hzsm-5: Analytical py-gc/ms study. Energy Convers. Manag. 2017, 139, 222–231. [Google Scholar] [CrossRef]
- Zhang, H.; Likun, P.K.W.; Xiao, R. Improving the hydrocarbon production via co-pyrolysis of bagasse with bio-plastic and dual-catalysts layout. Sci. Total Environ. 2018, 618, 151–156. [Google Scholar] [CrossRef] [PubMed]
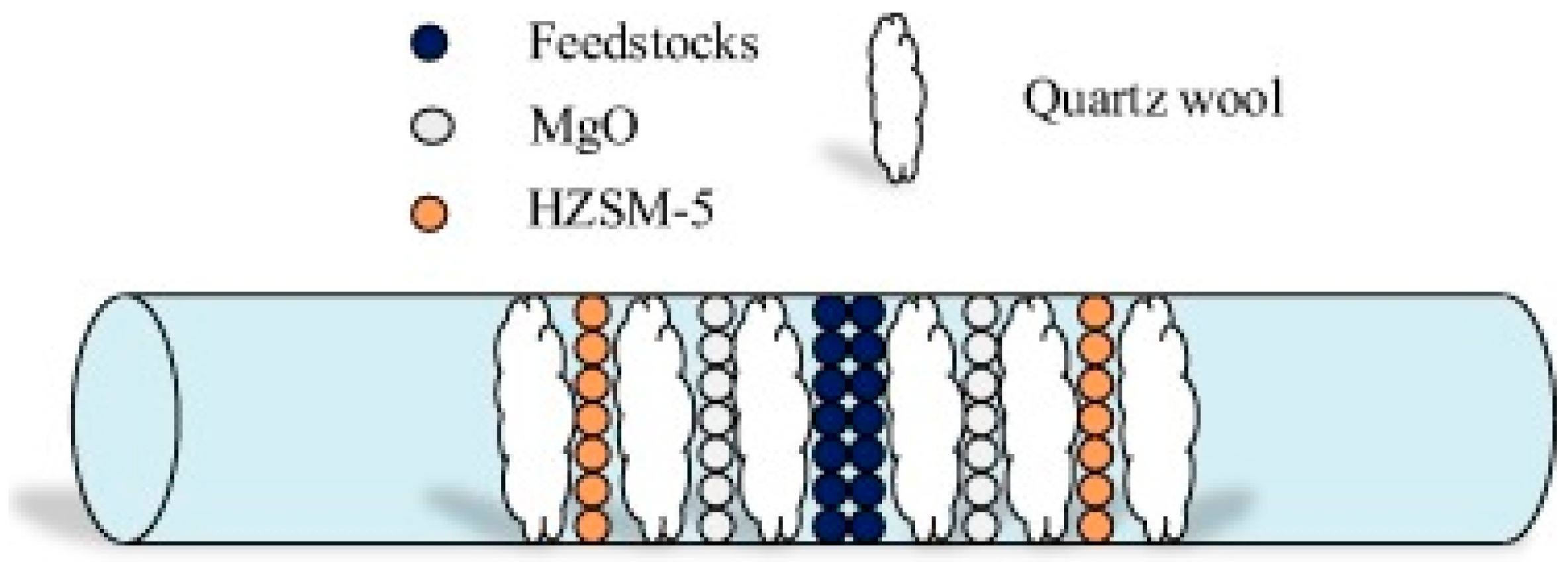
- Xue, X.; Pan, Z.; Zhang, C.; Wang, D.; Xie, Y.; Zhang, R. Segmented catalytic co-pyrolysis of biomass and high-density polyethylene for aromatics production with mgcl2 and hzsm-5. J. Anal. Appl. Pyrolysis 2018, 134, 209–217. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Ma, Z.; van Bokhoven, J.A. Deactivation and regeneration of h-usy zeolite during lignin catalytic fast pyrolysis. ChemCatChem 2012, 4, 2036–2044. [Google Scholar] [CrossRef]
- Carlson, T.R.; Vispute, T.P.; Huber, G.W. Green gasoline by catalytic fast pyrolysis of solid biomass derived compounds. ChemSusChem 2008, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Arregi, A.; Lopez, G.; Amutio, M.; Artetxe, M.; Barbarias, I.; Bilbao, J.; Olazar, M. Role of operating conditions in the catalyst deactivation in the in-line steam reforming of volatiles from biomass fast pyrolysis. Fuel 2018, 216, 233–244. [Google Scholar] [CrossRef]
- Rezaei, P.S.; Shafaghat, H.; Daud, W.M.A.W. Production of green aromatics and olefins by catalytic cracking of oxygenate compounds derived from biomass pyrolysis: A review. Appl. Catal. A 2014, 469, 490–511. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of aqueous-phase catalytic pyrolysis oil to liquid hydrocarbons using multifunctional nickel catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Palos, R.; Hita, I.; Arandes, J.M.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J.; Castaño, P. Revealing the pathways of catalyst deactivation by coke during the hydrodeoxygenation of raw bio-oil. Appl. Catal. B Environ. 2018, 239, 513–524. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Xiao, R.; Shen, D.; Zeng, J. Characterization of coke deposition in the catalytic fast pyrolysis of biomass derivates. Energy Fuels 2014, 28, 52–57. [Google Scholar] [CrossRef]
- Paasikallio, V.; Lindfors, C.; Kuoppala, E.; Solantausta, Y.; Oasmaa, A.; Lehto, J.; Lehtonen, J. Product quality and catalyst deactivation in a four day catalytic fast pyrolysis production run. Green Chem. 2014, 16, 3549–3559. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, M.; Xiao, R.; Shao, S.; Jin, B.; Xiao, G.; Zhao, M.; Liang, J. Catalytic conversion of biomass pyrolysis-derived compounds with chemical liquid deposition (cld) modified zsm-5. Bioresour. Technol. 2014, 155, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhong, Z.; Ding, K.; Xue, Z. Catalytic fast pyrolysis of mushroom waste to upgraded bio-oil products via pre-coked modified hzsm-5 catalyst. Bioresour. Technol. 2016, 212, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhong, Z.-P.; Wang, X.-B.; Ding, K.; Song, Z.-W. Catalytic upgrading of fast pyrolysis biomass vapors over fresh, spent and regenerated zsm-5 zeolites. Fuel Process. Technol. 2015, 138, 430–434. [Google Scholar] [CrossRef]
- Lerici, L.C.; Renzini, M.S.; Sedran, U.; Pierella, L.B. Tertiary recycling of low-density polyethylene by catalytic cracking over zsm-11 and beta zeolites modified with Zn2+: Stability study. Energy Fuels 2013, 27, 2202–2208. [Google Scholar] [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Hernández, F.; Navarro, R. Hzsm5 and husy deactivation during the catalytic pyrolysis of polyethylene. Appl. Catal. A 2004, 278, 37–43. [Google Scholar] [CrossRef]
- Anuar Sharuddin, S.D.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. A review on pyrolysis of plastic wastes. Energy Convers. Manag. 2016, 115, 308–326. [Google Scholar] [CrossRef]
- Barbarias, I.; Lopez, G.; Artetxe, M.; Arregi, A.; Bilbao, J.; Olazar, M. Valorisation of different waste plastics by pyrolysis and in-line catalytic steam reforming for hydrogen production. Energy Convers. Manag. 2018, 156, 575–584. [Google Scholar] [CrossRef]
- Zeaiter, J. A process study on the pyrolysis of waste polyethylene. Fuel 2014, 133, 276–282. [Google Scholar] [CrossRef]
- Serrano, D.P.; Aguado, J.; Rodríguez, J.M.; Peral, A. Catalytic cracking of polyethylene over nanocrystalline hzsm-5: Catalyst deactivation and regeneration study. J. Anal. Appl. Pyrolysis 2007, 79, 456–464. [Google Scholar] [CrossRef]
- Mullen, C.A.; Dorado, C.; Boateng, A.A. Catalytic co-pyrolysis of switchgrass and polyethylene over hzsm-5: Catalyst deactivation and coke formation. J. Anal. Appl. Pyrolysis 2018, 129, 195–203. [Google Scholar] [CrossRef]
- Vitolo, S.; Bresci, B.; Seggiani, M.; Gallo, M.G. Catalytic upgrading of pyrolytic oils over hzsm-5 zeolite: Behaviour of the catalyst when used in repeated upgrading-regenerating cycles. Fuel 2001, 80, 17–26. [Google Scholar] [CrossRef]
- Yildiz, G.; Lathouwers, T.; Toraman, H.E.; van Geem, K.M.; Marin, G.B.; Ronsse, F.; van Duren, R.; Kersten, S.R.A.; Prins, W. Catalytic fast pyrolysis of pine wood: Effect of successive catalyst regeneration. Energy Fuels 2014, 28, 4560–4572. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Zhao, X.; Julson, J. Application, deactivation, and regeneration of heterogeneous catalysts in bio-oil upgrading. Catalysts 2016, 6, 195. [Google Scholar] [CrossRef]
- Hellinger, M.; Baier, S.; Mortensen, P.M.; Kleist, W.; Jensen, A.D.; Grunwaldt, J.-D. Continuous catalytic hydrodeoxygenation of guaiacol over pt/SiO2 and pt/h-mfi-90. Catalysts 2015, 5, 1152–1166. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, H.; Xiao, R.; Li, X.; Cai, Y. Controlled regeneration of zsm-5 catalysts in the combined oxygen and steam atmosphere used for catalytic pyrolysis of biomass-derivates. Energy Convers. Manag. 2018, 155, 175–181. [Google Scholar] [CrossRef]
- Yung, M.M.; Starace, A.K.; Griffin, M.B.; Wells, J.D.; Patalano, R.E.; Smith, K.R.; Schaidle, J.A. Restoring zsm-5 performance for catalytic fast pyrolysis of biomass: Effect of regeneration temperature. Catal. Today 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zeolite | Framework Type | Pore Dimensions, Å | Ring Sizes |
|---|---|---|---|
| ZK-5 | KFI | 8-ring 3.9 × 3.9; 8-ring 3.9 × 3.9 | 8, 6, 4 |
| ZSM-23 | MTT | 10-ring 4.5 × 5.2 | 10, 6, 5 |
| Ferrierite | FER | 10-ring 4.2 × 5.4; 8-ring 3.5 × 4.8 | 10, 8, 6, 5 |
| MCM-22 | MWW | 10-ring 4.0 × 5.5; 10-ring 4.1 × 5.1 | 10, 6, 5, 4 |
| ZSM-5 | MFI | 10-ring 5.1 × 5.1; 10-ring 5.3 × 5.6 | 10, 6, 5, 4 |
| ZSM-11 | MEL | 10-ring 5.3 × 5.4 | 10, 8, 6, 5, 4 |
| Y | FAU | 12-ring 7.4 × 7.4 | 12, 6, 4 |
| Beta | BEA | 12-ring 6.6 × 6.7; 12-ring 5.6 × 5.6 | 12, 6, 5, 4 |
| Mordenite | MOR | 12-ring 6.5 × 7.0; 8-ring 3.4 × 4.8; 8-ring 2.6 × >5.7 | 12, 8, 5, 4 |
| Catalysts | Pyrolysis Conditions | Key Findings | Ref. |
|---|---|---|---|
| K/ZSM-5 | Catalysts prepared by ion-exchange method. Feedstock: lignin and switch grass. Optimum catalyst/feedstock ratio = 2.5. Pyrolysis temperature = 500 °C. | K/ZSM-5 produced more valuable oxygenated compounds, particularly alkyl phenols and 2-methylfuran than noncatalytic pyrolysis or catalytic pyrolysis with HZSM-5. Low K/ZSM-5 loading favored the formation of 2-methyl furan. | [202] |
| Fe-, Cu-, and Ni/ZSM-5 | Catalysts prepared by ion-exchange method. Feedstock: duckweed and microalgae. Weight hourly space velocity (g feed gas/g cat h) = 13 and 56 h−1. Pyrolysis temperature = 500 °C. | Pyrolytic liquid yields and qualities obtained by metal-modified ZSM-5 were lower than HZSM-5. | [203] |
| Fe/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: wood sawdust. Catalyst/feedstock ratio = 10. Pyrolysis temperature = 500–800 °C. | Fe/ZSM-5 maintained the structure of ZSM-5 and exhibited better activity in the conversion of oxygenates and formation of monocyclic aromatic hydrocarbons (MAHs) than the parent zeolite. | [196] |
| Ce-, Cu-, Fe2+-, Fe3+-Mg, Ni-, Sn- and Zn loaded on ZSM-5 and zeolite Y | Catalysts prepared by impregnation method. Feedstock: waste plastics. Catalyst/feedstock ratio = 1:9. Pyrolysis temperature = 430–500 °C. | Surface areas of zeolites decreased with introduction of metal. The catalyst efficiency increased in the order of Cu < Ce < Mg < Ni < Fe(III) < Fe(II) < Zn < Sn. | [204] |
| Fe/ZSM-5 | Catalyst prepared by impregnation method. Feedstock: hemicelluloses, cellulose, lignin and corn stalk. Catalyst/biomass = 0.5, 1, 2 and 4. Pyrolysis temperature = 600 °C. | Fe/ZSM-5 increased the production of light olefins and carbon yield of light olefins. For CFP of cellulose, 3% Fe/ZSM-5 was optimum. Total light olefins decreased in order of cellulose > corn stalk > hemicelluloses > lignin. The selectivity of C2H4 was more than 60% regardless of feedstock. | [205] |
| P/Ni/ZSM-5 | Catalysts were prepared by sequential impregnation method. Feedstock: pine wood and low-density polyethylene. Catalyst/feedstock ratio = 15. Pyrolysis temperature = 550 °C. | P/Ni-modified ZSM-5 significantly increased the yield of olefins and aromatic hydrocarbons and decreased the yields of low-value alkanes and undesired char/coke, as compared to ZSM-5. P/Ni/ZSM-5 exhibited improved hydrothermal stability and maintained comparable aromatic yields in co-feed CFP for up to 9 h. | [206] |
| Ni-, Co-, Mo-, Ga-, and Pd/HZSM-5 | Catalysts prepared by ion-exchange and impregnation methods. Feedstock: Jatropha residues. Biomass/catalyst ratios = 1:1, 1:5, and 1:10. Pyrolysis temperature = 500 °C. | Metal/HZSM-5 reduced contents of oxygenates (0.6–4.0%) and nitrogenates (1.8–4.6%). Aromatic selectivity was significantly increased up to 91–97%. The catalysts produced by metal-ion exchange showed slightly higher aromatic selectivity than the impregnated ones. | [207] |
| Ni-, Co-, Mo-, and Pt/HZSM-5 | Catalysts prepared by impregnation method. Pyrolysis hydrogen pressure ranged from 100 to 400 psi. Feedstock: pine wood. Biomass/catalyst ratios = 1:9. Pyrolysis temperature = 650 °C. | Except for Mo/HZSM-5, the aromatic hydrocarbons yield was not affected by hydrogen pressure. The presence of metal increased aromatic yield. Metal has no obvious effect on the aromatic selectivity. | [208] |
| Ni-, Zn-, and Ga/HZSM-5 | Catalyst prepared with Ga by total ion exchange. Catalysts prepared by ion exchange method. Feed stock: Eucalyptus urophylla. Biomass/catalyst ratios = 1:5 and 1:10. Pyrolysis temperature = 600 °C. | Ga-HZSM-5 increased the yield of aromatic hydrocarbons while Ni- and Zn- modified catalysts decreased the yield. Ga-HZSM-5 exhibited high selectivity toward xylenes. Ni/HZSM-5 produced more methane. Zn/HZSM-5 was the most selective for toluene production. | [199] |
| Fe/HZSM-5 | Catalysts prepared by ion exchange method. Feedstock: cellulose; cellobiose and lignin. Biomass/catalyst ratios = 1:5 and 1:10. Pyrolysis temperature = 500 °C. | Fe/HZSM-5 increased the yield of aromatic hydrocarbons for all three feedstocks examined. Fe-HZSM-5 favored the formation of naphthalenes while the selectivities toward p-xylene, ethylbenzene and trimethylbenzene decreased. | [209] |
| Mg-Al/MCM-41 | Catalysts prepared by impregnation method. Feedstock: cellulose and lignin. Biomass/catalyst ratios = 0.4:1. Pyrolysis temperature = 600 °C. | Mg/Al/MCM-41 presented high selectivity to aromatic hydrocarbons. Highest catalytic activity and stable reusability exhibited by 1 wt % Mg/Al-MCM-41. | [210] |
| K-, Na-, Mg-, Ce-, Cu-, and Fe/ultra-stable zeolite Y | Catalysts prepared by impregnation method. Feedstock: coal and cedar wood. Biomass/catalyst ratios = 1:1. Pyrolysis temperature = 600 °C. | Introduction of metal mitigated the deposition of coke on the zeolite surface. Mg/USY promoted hydrocarbon formation and showed good stability. | [211] |
| Catalysts | Pyrolysis Conditions | Key Findings | Ref. |
|---|---|---|---|
| Zn-, Ga-, Ni-, Co-, Mg- and Cu/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: Yunnan pine. Biomass/catalyst ratio = 1:2. Pyrolysis temperature = 450 °C. Catalytic upgrading temperature = 500 °C. | Presence of M-ZSM-5 decreased oil yields and increased non-condensable gas amount. Ga/ZSM-5 produced the highest oil yields and the lowest amount of coke. Zn/ZSM-5 favored the formation of single-ring aromatics especially toluene and xylenes. Ni/ZSM-5 yielded more polycyclic aromatic hydrocarbons, e.g., C10+ polycyclic aromatic hydrocarbons. Co/ ZSM-5 was the most selective for indene production. | [200] |
| Ni- and Co/ZSM-5 | Catalysts prepared by wet impregnation method. Feedstock: Lignocel HBS 150–500. Catalyst/feedstock ratio = 1.5/0.7. Pyrolysis temperature = 500 °C. | Reduced metallic Ni and Co formed during pyrolysis which favor hydrogen transfer reactions. Produced oil was enriched in aromatics and phenols. NiO/ZSM-5 was more reactive than Co3O4/ZSM-5 in decreasing the organic phase and increasing the gaseous products. | [212] |
| Fe/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: beech sawdust. Catalyst/biomass ratio = 0.5/5. Pyrolysis and catalytic upgrading temperature = 500 °C. | Bio-oil yields decreased in the presence of catalysts. Fe/ZSM-5 catalyst reveal a significant enhancement quality of the pyrolysis products in comparison with non-catalytic experiment. Catalyst increased oxygen removal from the organic phase of bio-oil and further developed the production of desirable products such as phenolics and aromatic compounds. | [197] |
| Fe/ZSM-5 | Catalysts prepared by wet impregnation method. Feedstock: rice husk. Catalyst/biomass ratio = 5:1. Pyrolysis temperature = 600 °C. Pyrolysis and catalytic upgrading temperature = 550 °C. | Increased Fe loading increased hydrocarbons content at the expense of decreasing yield of bio-oil. Optimum was 4% Fe/ZSM-5 to obtain high yield of upgraded bio-oil. | [213] |
| Ce/HZSM-5 | Catalysts prepared by impregnation method. Feedstock: sugarcane bagasse. Catalyst/biomass ratio = 1.5, 1, 1.5 and 2. Pyrolysis and catalytic upgrading temperature = 500 °C. | Introduction of Ce increased pyrolysis oil yield and decreased coke yield, in comparison to HZSM-5. The optimal ratio of 1.5/1 catalyst/biomass produced an 2.45% content of C6–C8 hydrocarbons. | [214] |
| Co, Ni, Mo, Ga, and Pd-modified HZSM-5 | Catalysts prepared by both ion-exchange and wet impregnation methods. Feedstock: Jatropha residues. Biomass/catalyst ratio = 1:1, 1:5, and 1:10. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalysts enhanced hydrocarbon production, particularly aromatics, and reduced oxygen and nitrogen containing compound contents. Mo/HZSM-5 showed the highest aromatics (97%) with low PAHs selectivity. | [207] |
| Ni-ZSM-5 | Catalysts was prepared by ion-exchange method. Feedstock: rubber waste. Rubber/catalyst ratio = 1:1, 1:5 and 1:10. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalyst enhanced the aromatic production, especially benzene, toluene and xylene compounds. | [215] |
| Co, Mo–Co and Ni–Co modified HZSM-5 | Catalysts prepared by impregnation method. Feedstock: lignite. Pyrolysis and catalytic upgrading temperature = 600 °C. | Light aromatics (e.g., benzene, toluene, ethylbenzene, xylene and naphthalene) production was significantly increased by Mo–Co or Ni–Co/HZSM-5. Yields of benzene, ethylbenzene and m, p-xylene followed the order Ni/Co-HZSM-5 >Mo/Co-HZSM-5 > Co-HZSM-5 > HZSM-5. Further NaOH treatment of bimetallic catalysts inhibited the coke formation. | [216] |
| Fe- Zr-, and Co- HZSM-5 | Catalysts were prepared by impregnation method. Feedstock: Pine sawdust. Catalyst/biomass ratio = 0.5. Pyrolysis temperature 550 °C. Catalytic upgrading temperature = 450–650 °C. | Fe- and Zr/HZSM-5 produced over 45% aromatic hydrocarbons in the resulting bio-oil. However, Co/HZSM-5 performed worse than HZSM-5 by producing the highest gas and coke yields. Zr/HZSM-5 enhanced the formation of benzene and its derivatives. Fe/HZSM-5 produced more naphthalene and its derivatives, and increased Fe loading facilitated deoxygenation and hydrocarbon formation. | [217] |
| Co/HZSM-5 | Catalysts prepared by impregnation method. Feedstock: Baiyinhua lignite. Catalyst/ lignite ratio = 1. Pyrolysis temperature = 400 to 700 °C. Catalytic upgrading temperature = 600 °C. | A significant reduction of the oxygen content and increase in total content of light aromatics was observed for 3% Co-HZSM5 at 600°C. | [218] |
| Fe-, Ce-, La-, Cu-, and Cr- Mo2N/HZSM-5 | Catalysts prepared by incipient wetness impregnation method. Feedstock: microcrystalline cellulose, xylan, pine wood and wood lignin. Catalyst/biomass ratio = 1, 3, 5, 7. Pyrolysis and catalytic upgrading temperature = 550–850 °C. | Both Mo2N/HZSM-5 and metal modified catalysts increased production of monocyclic aromatic hydrocarbons (MAHs) while decreasing the formation polycyclic aromatic hydrocarbons (PAHs), as compared to HZSM-5. Most effective in enhancing formation of MAHs and restraining the generation of PAHs was 10% Ce-Mo2N/HZSM-5. | [219] |
| Cu–Al-MCM-41 | Cu was introduced during the synthesis of Al-MCM-41. Feedstock: spruce wood and Miscanthus. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalyst produced more desirable compounds than the uncatalyzed sample, and the organic phase yield was increased close to the non-catalytic level. | [220] |
| Cu-Al, Fe-Al, and Zn-Al/MCM-41 | Catalysts prepared by ion-exchange. Feedstock: barkless spruce wood. Biomass/catalyst ratio = 1:5/0.7. Pyrolysis and catalytic upgrading temperature = 500 °C. | All catalysts, especially Fe–Al and Cu–Al-MCM-41 increased the yield of phenolic compounds. A low Si/Al ratio of MCM-41 was beneficial to improve the product yield and quality. | [221] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Bao, Z.; Xia, S.; Lu, Q.; Walters, K.B. Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts 2018, 8, 659. https://doi.org/10.3390/catal8120659
Zhang L, Bao Z, Xia S, Lu Q, Walters KB. Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts. 2018; 8(12):659. https://doi.org/10.3390/catal8120659
Chicago/Turabian StyleZhang, Laibao, Zhenghong Bao, Shunxiang Xia, Qiang Lu, and Keisha B. Walters. 2018. "Catalytic Pyrolysis of Biomass and Polymer Wastes" Catalysts 8, no. 12: 659. https://doi.org/10.3390/catal8120659
APA StyleZhang, L., Bao, Z., Xia, S., Lu, Q., & Walters, K. B. (2018). Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts, 8(12), 659. https://doi.org/10.3390/catal8120659