Acetic Acid/Propionic Acid Conversion on Metal Doped Molybdenum Carbide Catalyst Beads for Catalytic Hot Gas Filtration

 , , ,
, , ,
Abstract
:
1. Introduction
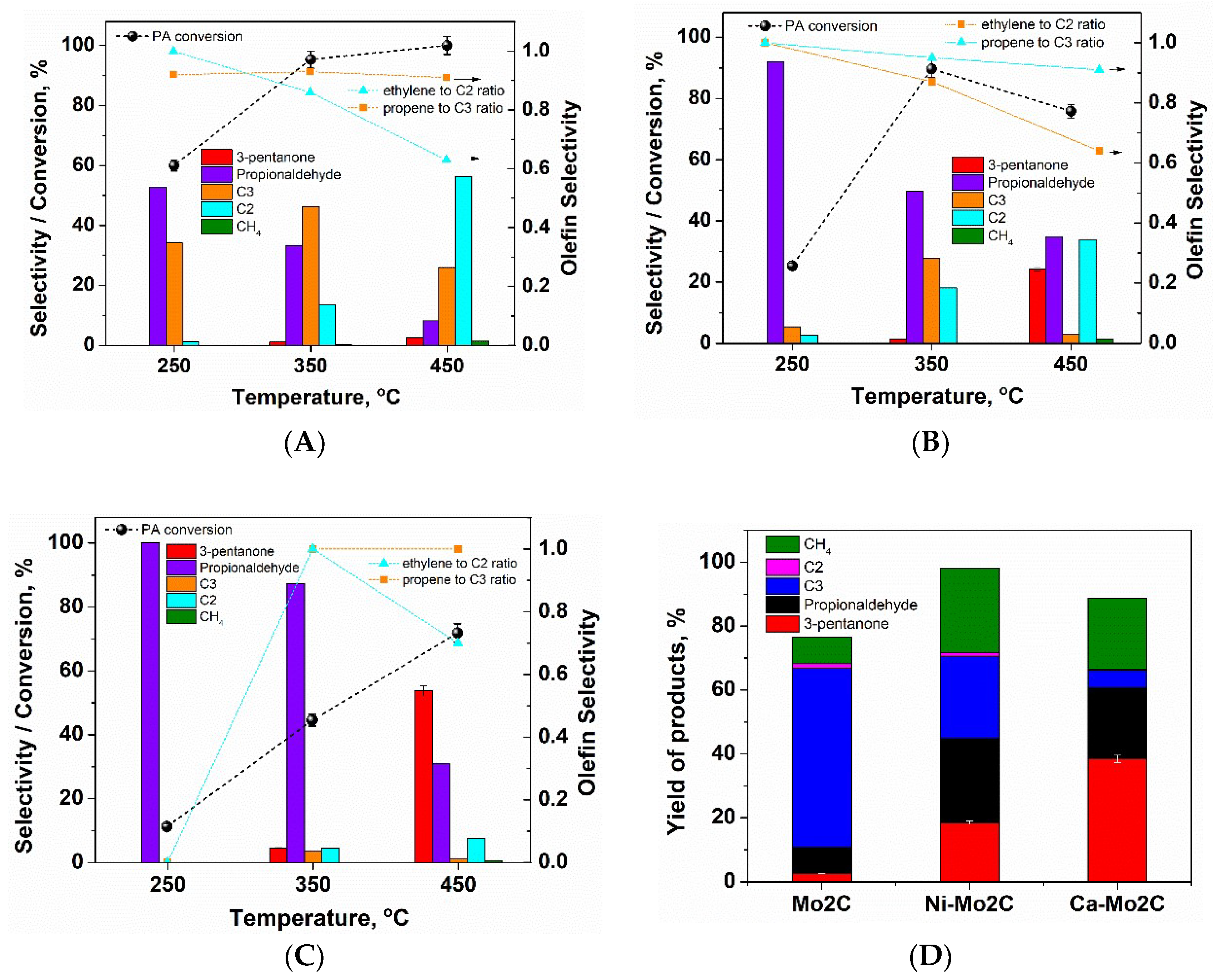
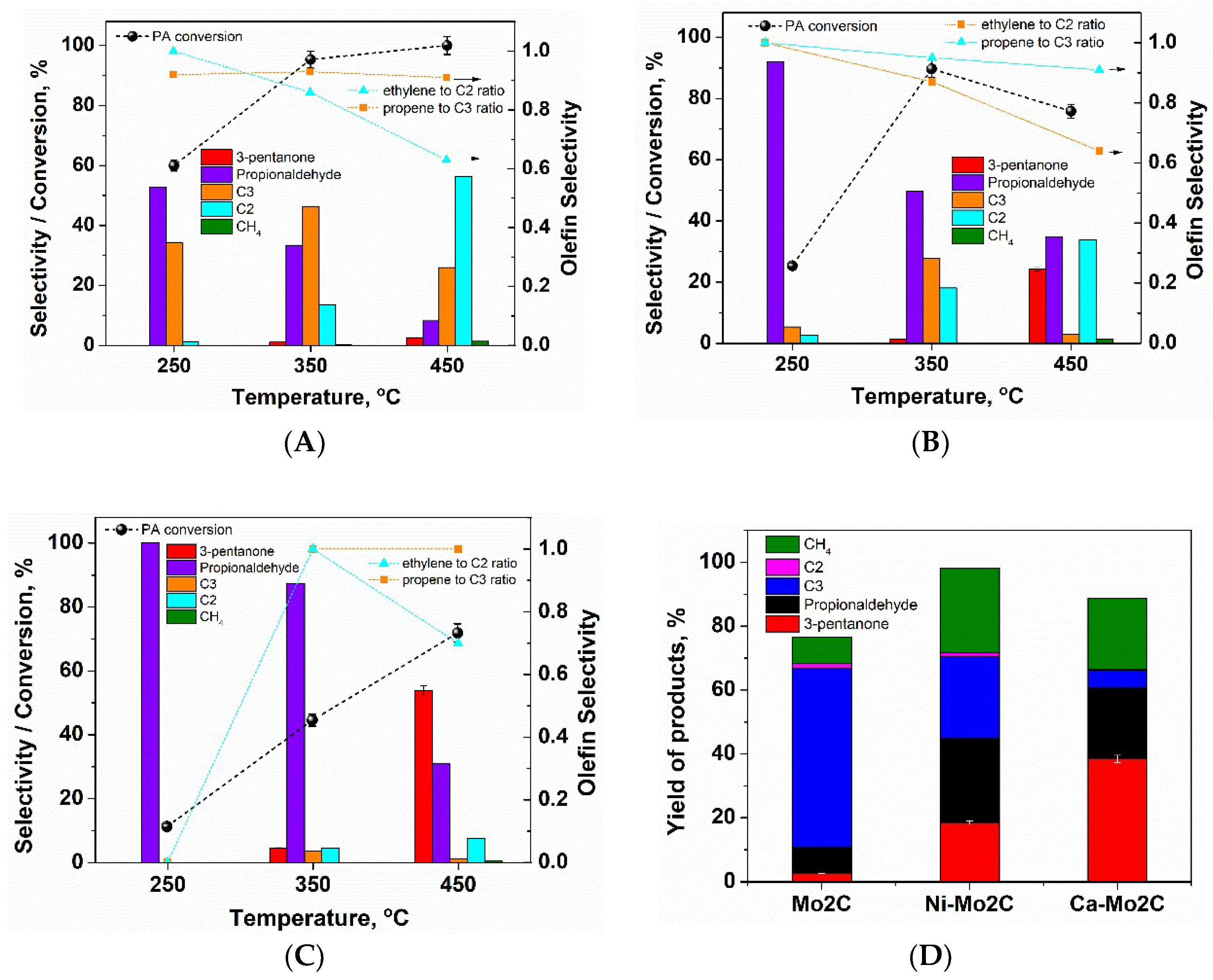
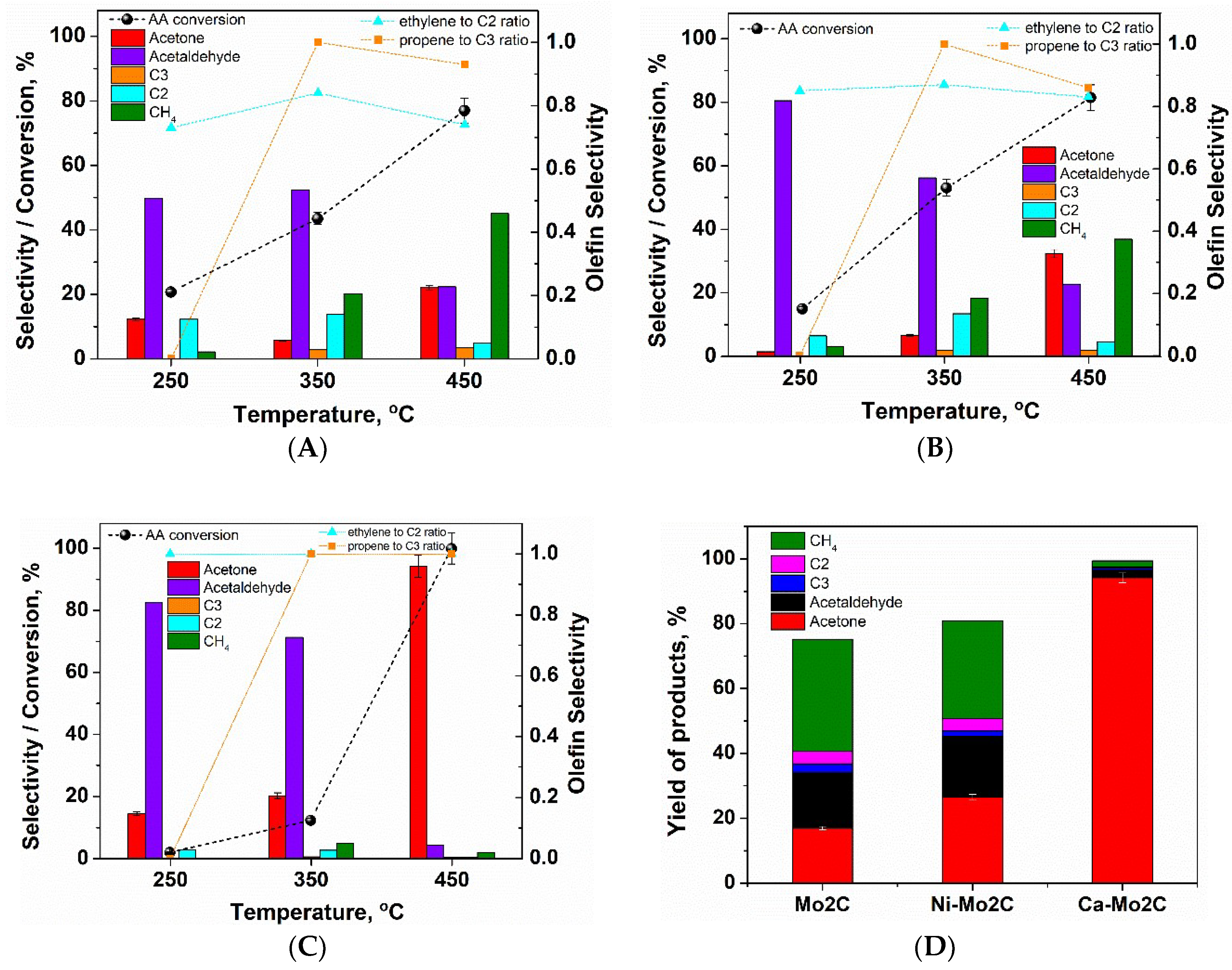
2. Results and Discussion
3. Materials and Methods
3.1. Catalyst Synthesis and Characterization
3.1.1. Carbide Catalysts Beads Synthesis
3.1.2. Specific Surface Area and Pore Size Analysis (BET)
3.1.3. Temperature Programmed Desorption of Carbon Dioxide (CO2-TPD)
3.2. Catalyst Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baldwin, R.M.; Feik, C.J. Bio-oil stabilization and upgrading by hot gas filtration. Energy and Fuels 2013, 27, 3224–3238. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Huber, G.W.; Corma, A. Synergies between bio- and oil refineries for the production of fuels from biomass. Angew. Chemie - Int. Ed. 2007, 46, 7184–7201. [Google Scholar] [CrossRef] [PubMed]
- Pallassana, V.; Neurock, M. Reaction paths in the hydrogenolysis of acetic acid to ethanol over Pd(111), Re(0001), and PdRe alloys. J. Catal. 2002, 209, 289–305. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Sengupta, D.; Raspoet, G.; Vanquickenborne, L.G. Theoretical study of the thermal decomposition of acetic acid: Decarboxylation versus dehydration. J. Phys. Chem. 1995, 99, 11883–11888. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.M.; Van Duijne, A.; Pieterse, J.A.Z.; Ponec, V. Reactions of carboxylic acids on oxides: 2. Bimolecular reaction of aliphatic acids to ketones. J. Catal. 1997, 168, 265–272. [Google Scholar] [CrossRef]
- Martinez, R.; Huff, M.C.; Barteau, M.A. Ketonization of acetic acid on titania-functionalized silica monoliths. J. Catal. 2004, 222, 404–409. [Google Scholar] [CrossRef]
- He, D.H.; Wakasa, N.; Fuchikami, T. Hydrogenation of carboxylic acids using bimetallic catalysts consisting of group 8 to 10, and group 6 or 7 metals. Tetrahedron Lett. 1995, 36, 1059–1062. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, T.; Zheng, M.; Wang, A.; Wang, H.; Wang, X.; Chen, J.G. Direct catalytic conversion of cellulose into ethylene glycol using nickel-promoted tungsten carbide catalysts. Angew. Chemie - Int. Ed. 2008, 47, 8510–8513. [Google Scholar] [CrossRef]
- Ren, H.; Yu, W.; Salciccioli, M.; Chen, Y.; Huang, Y.; Xiong, K.; Vlachos, D.G.; Chen, J.G. Selective hydrodeoxygenation of biomass-derived oxygenates to unsaturated hydrocarbons using molybdenum carbide catalysts. ChemSusChem 2013, 6, 798–801. [Google Scholar] [CrossRef]
- Xiong, K.; Yu, W.; Vlachos, D.G.; Chen, J.G. Reaction pathways of biomass-derived oxygenates over metals and carbides: From model surfaces to supported catalysts. ChemCatChem 2015, 7, 1402–1421. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon nanofiber supported transition-metal carbide catalysts for the hydrodeoxygenation of guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Li, Z.; Choi, J.-S.; Wang, H.; Lepore, A.W.; Connatser, R.M.; Lewis, S.A.; Meyer, H.M.; Santosa, D.M.; Zacher, A.H. Sulfur-Tolerant Molybdenum Carbide Catalysts Enabling Low-Temperature Stabilization of Fast Pyrolysis Bio-oil. Energy & Fuels 2017, 31, 9585–9594. [Google Scholar]
- Sullivan, M.M.; Chen, C.J.; Bhan, A. Catalytic deoxygenation on transition metal carbide catalysts. Catal. Sci. Technol. 2016, 6, 602–616. [Google Scholar] [CrossRef]
- Sullivan, M.M.; Bhan, A. Acetone Hydrodeoxygenation over Bifunctional Metallic-Acidic Molybdenum Carbide Catalysts. ACS Catal. 2016, 6, 1145–1152. [Google Scholar] [CrossRef]
- Bej, S.K.; Bennett, C.A.; Thompson, L.T. Acid and base characteristics of molybdenum carbide catalysts. Appl. Catal. A Gen. 2003, 250, 197–208. [Google Scholar] [CrossRef]
- Bej, S.K.; Thompson, L.T. Acetone condensation over molybdenum nitride and carbide catalysts. Appl. Catal. A Gen. 2004, 264, 141–150. [Google Scholar] [CrossRef]
- Hattori, H. Heterogeneous Basic Catalysis. Chem. Rev. 1995, 95, 537–558. [Google Scholar] [CrossRef]
- Hendren, T.S.; Dooley, K.M. Kinetics of catalyzed acid/acid and acid/aldehyde condensation reactions to non-symmetric ketones. Catal. Today 2003, 85, 333–351. [Google Scholar] [CrossRef]
- Mante, O.D.; Rodriguez, J. a.; Senanayake, S.D.; Babu, S.P. Catalytic conversion of biomass pyrolysis vapors into hydrocarbon fuel precursors. Green Chem. 2015, 17, 2362–2368. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Chen, P.; Lou, H.; Zheng, X.; Hong, H. Carbon-supported molybdenum carbide catalysts for the conversion of vegetable oils. ChemSusChem 2012, 5, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.T.; Kim, W.B.; Rhee, C.H.; Lee, J.S. Effects of Transition Metal Addition on the Solid-State Transformation of Molybdenum Trioxide to Molybdenum Carbides. Chem. Mater. 2004, 16, 307–314. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdneum carbide catalysts. I Synthesis of unsupported powders. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Lundberg, D. Flow Conditioners. Control Eng. 2006, 53, 2–7. [Google Scholar]
- Choi, J.S.; Zacher, A.H.; Wang, H.; Olarte, M.V.; Armstrong, B.L.; Meyer, H.M.; Soykal, I.I.; Schwartz, V. Molybdenum Carbides, Active and in Situ Regenerable Catalysts in Hydroprocessing of Fast Pyrolysis Bio-Oil. Energy and Fuels 2016, 30, 5016–5026. [Google Scholar] [CrossRef]


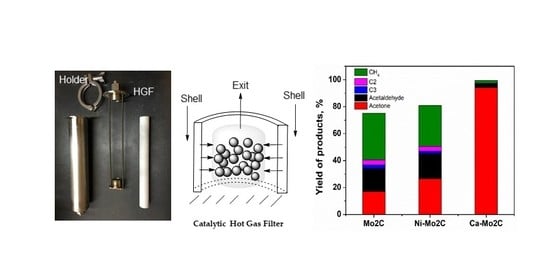
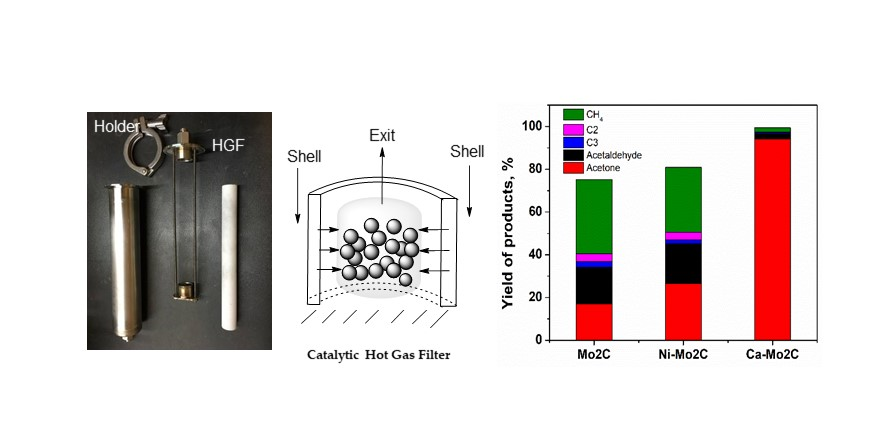{kind=link}
{kind=link}
{kind=link}
| Catalyst | aCO2 Desorbed during TPD up to 550 °C, micromoles/m2 | Surface Area, m2/g | Surface Area after Reaction *, m2/g | Total Pore Volume, cm3/g | Total Pore Volume after Reaction *, cm3/g | Average Pore Size, Å | Average Pore Size after Reaction *, Å |
|---|---|---|---|---|---|---|---|
| Mo2C | 0.86 | 23.5 | 23.7 | 0.0925 | 0.0869 | 77.2 | 73.5 |
| Ni-Mo2C | 0.79 | 18.3 | 17.2 | 0.0779 | 0.0701 | 85.2 | 81.4 |
| Ca-Mo2C | 1.77 | 9.6 | 7.1 | 0.0375 | 0.0346 | 78.5 | 97.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, M.; Lepore, A.W.; Choi, J.-S.; Li, Z.; Wu, Z.; Polo-Garzon, F.; Hu, M.Z. Acetic Acid/Propionic Acid Conversion on Metal Doped Molybdenum Carbide Catalyst Beads for Catalytic Hot Gas Filtration. Catalysts 2018, 8, 643. https://doi.org/10.3390/catal8120643
Lu M, Lepore AW, Choi J-S, Li Z, Wu Z, Polo-Garzon F, Hu MZ. Acetic Acid/Propionic Acid Conversion on Metal Doped Molybdenum Carbide Catalyst Beads for Catalytic Hot Gas Filtration. Catalysts. 2018; 8(12):643. https://doi.org/10.3390/catal8120643
Chicago/Turabian StyleLu, Mi, Andrew W. Lepore, Jae-Soon Choi, Zhenglong Li, Zili Wu, Felipe Polo-Garzon, and Michael Z. Hu. 2018. "Acetic Acid/Propionic Acid Conversion on Metal Doped Molybdenum Carbide Catalyst Beads for Catalytic Hot Gas Filtration" Catalysts 8, no. 12: 643. https://doi.org/10.3390/catal8120643
APA StyleLu, M., Lepore, A. W., Choi, J.-S., Li, Z., Wu, Z., Polo-Garzon, F., & Hu, M. Z. (2018). Acetic Acid/Propionic Acid Conversion on Metal Doped Molybdenum Carbide Catalyst Beads for Catalytic Hot Gas Filtration. Catalysts, 8(12), 643. https://doi.org/10.3390/catal8120643