Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis



Abstract
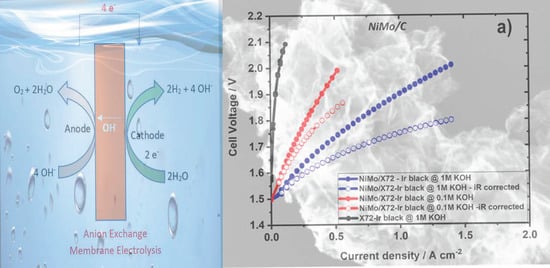
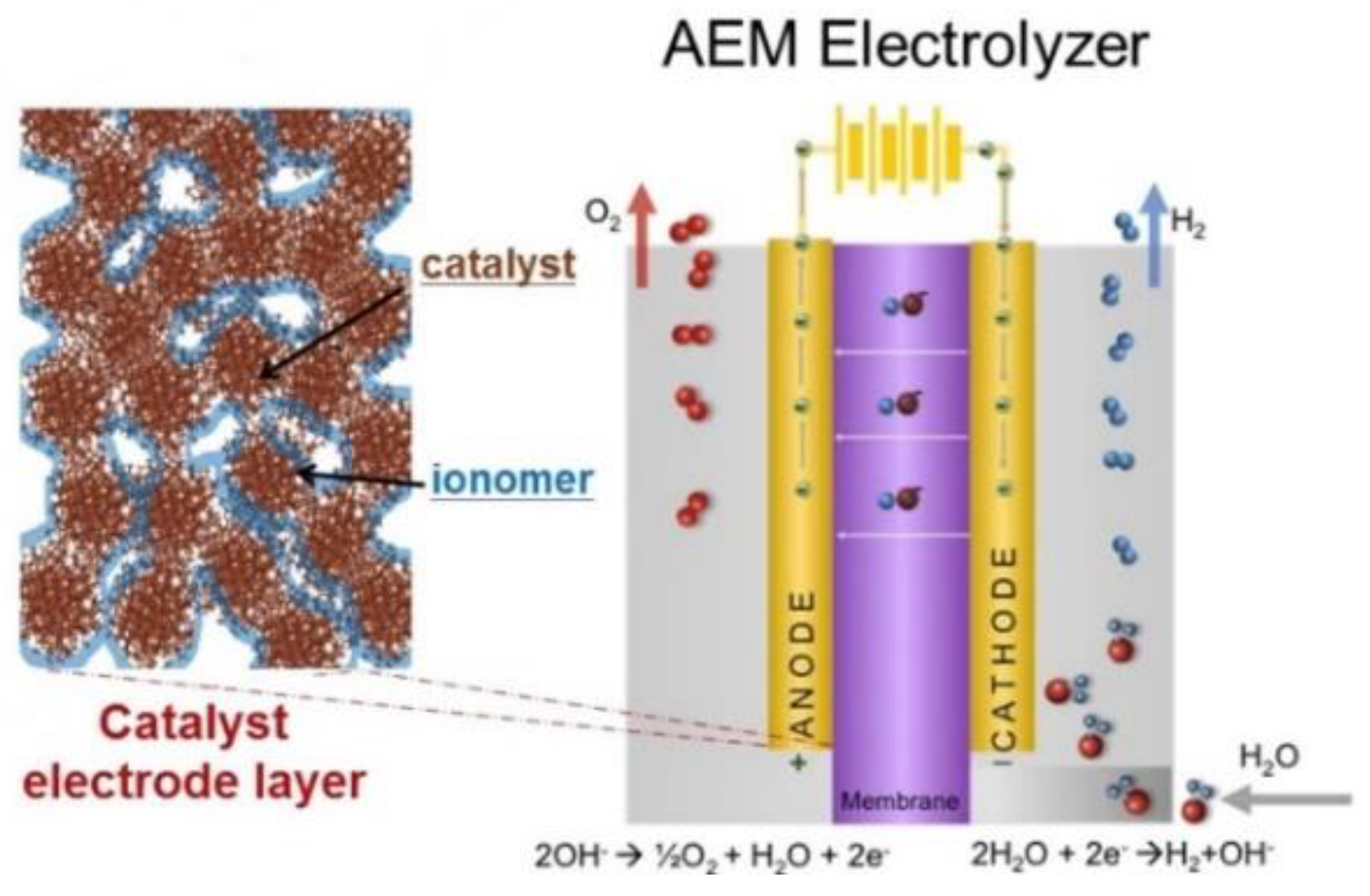
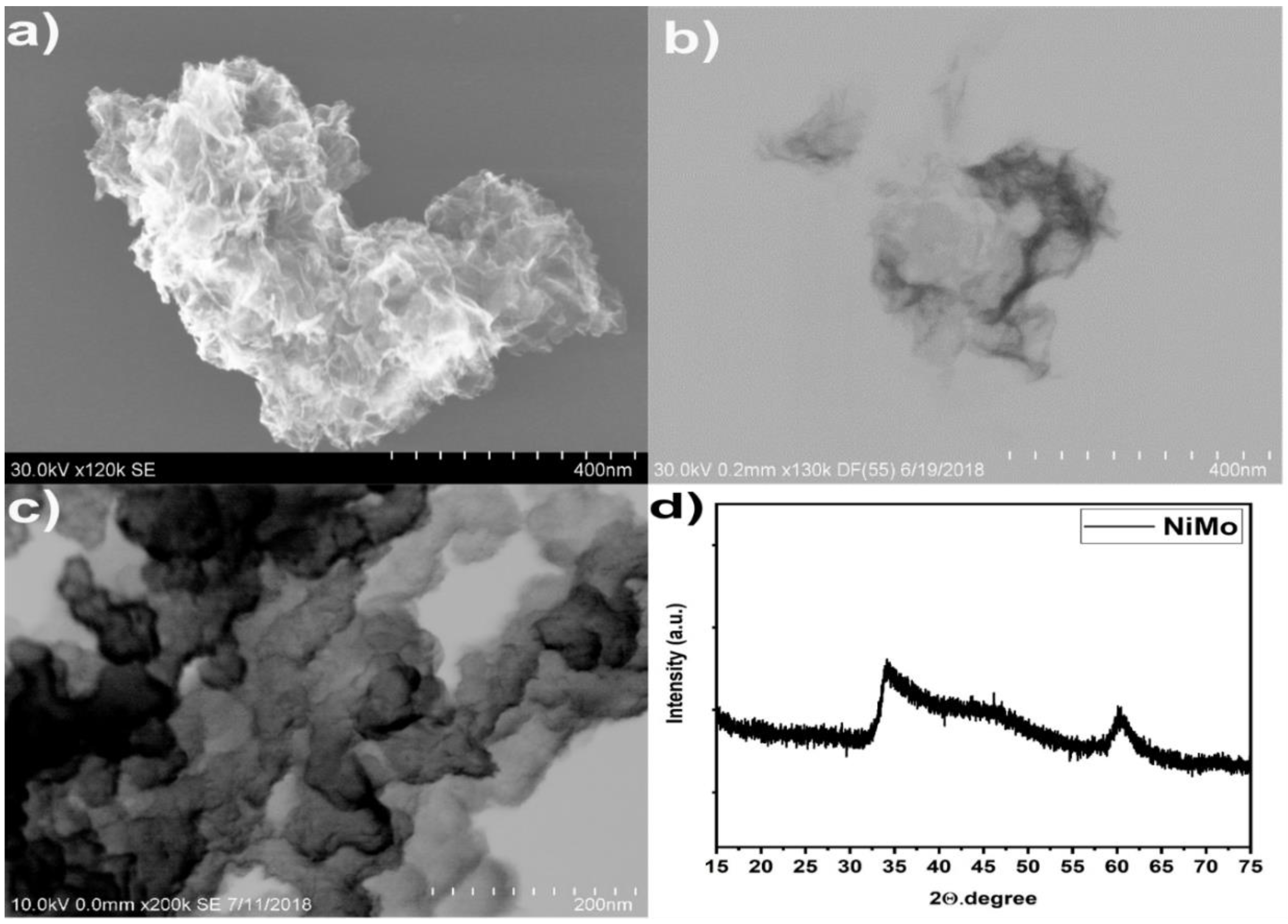
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
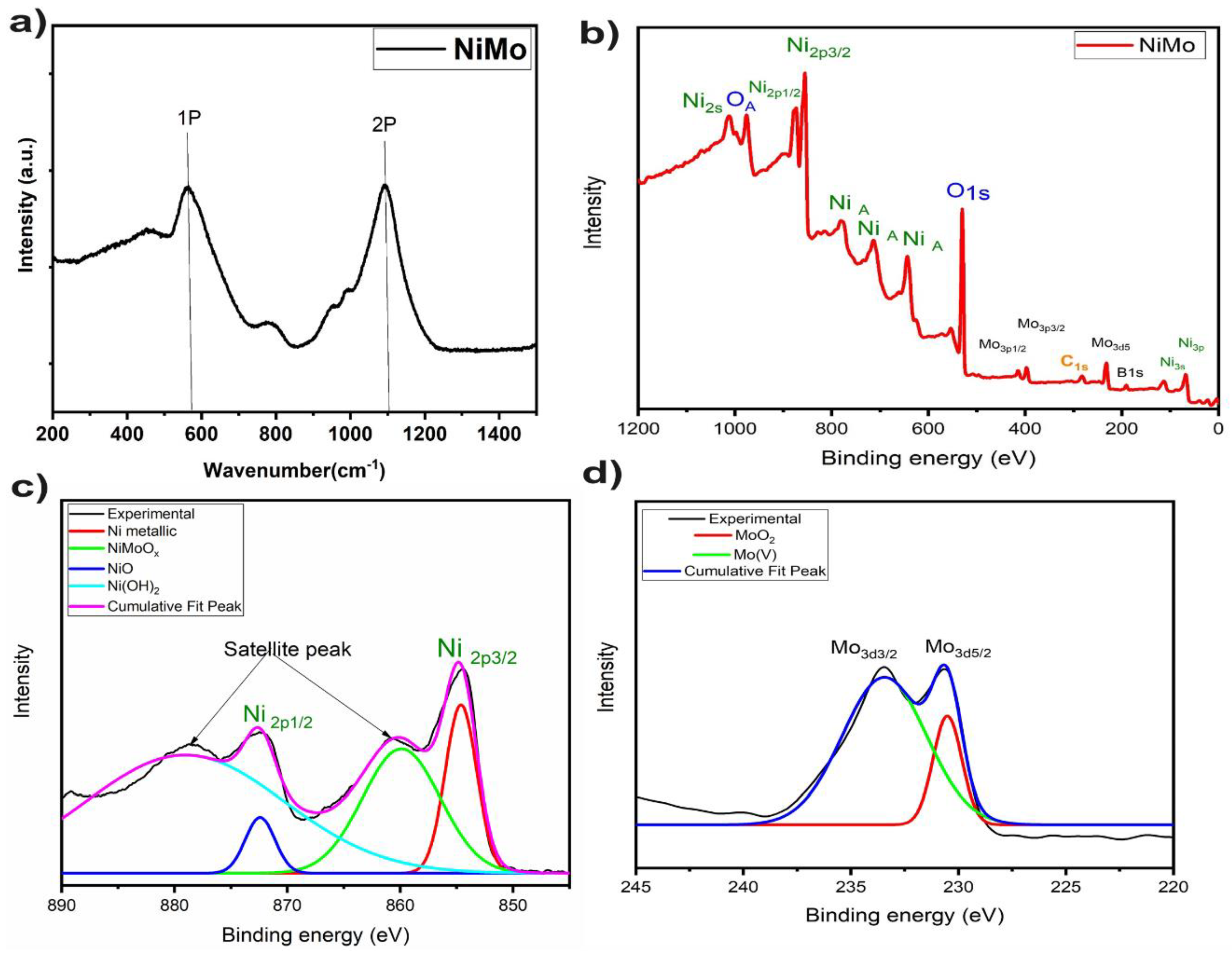
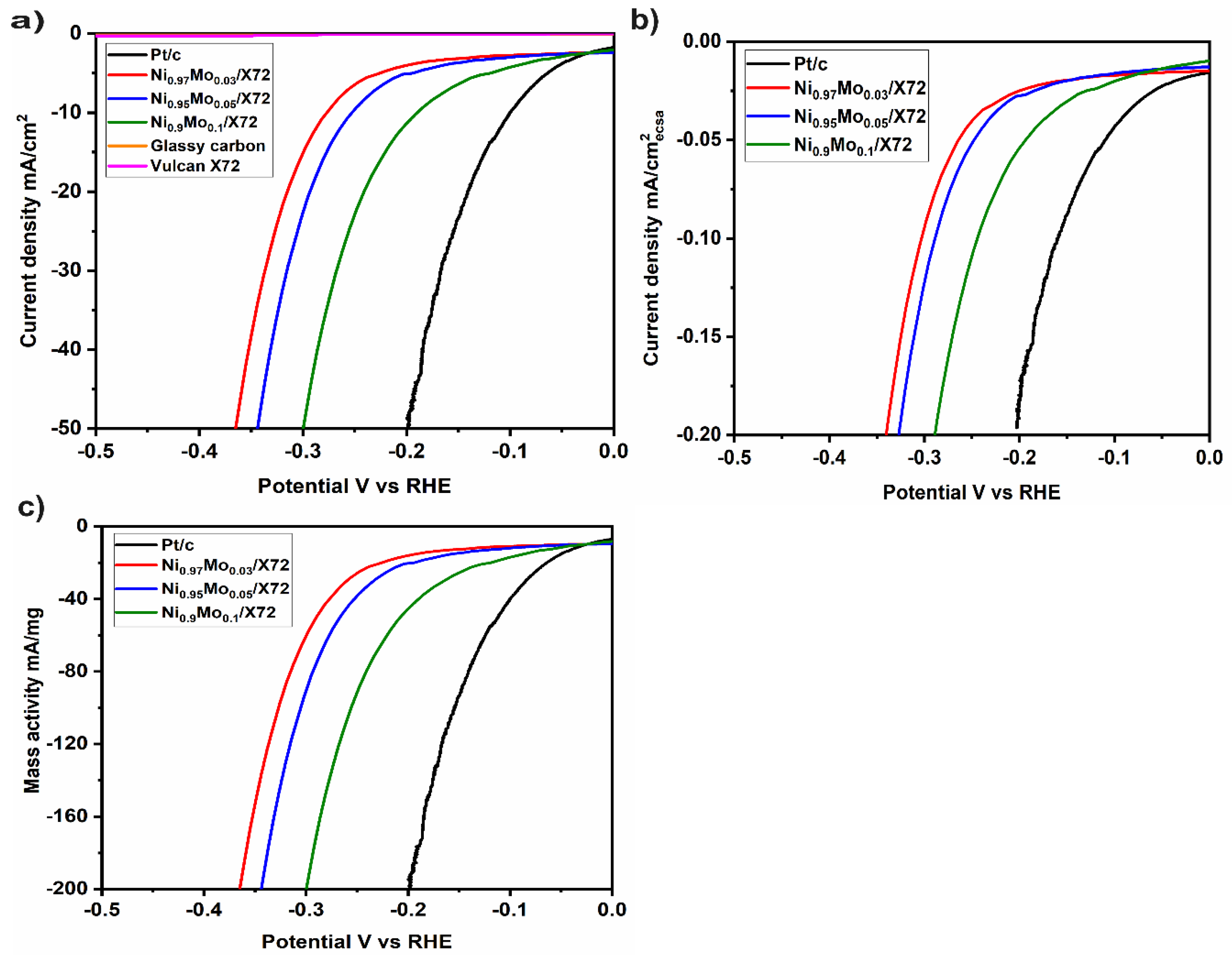
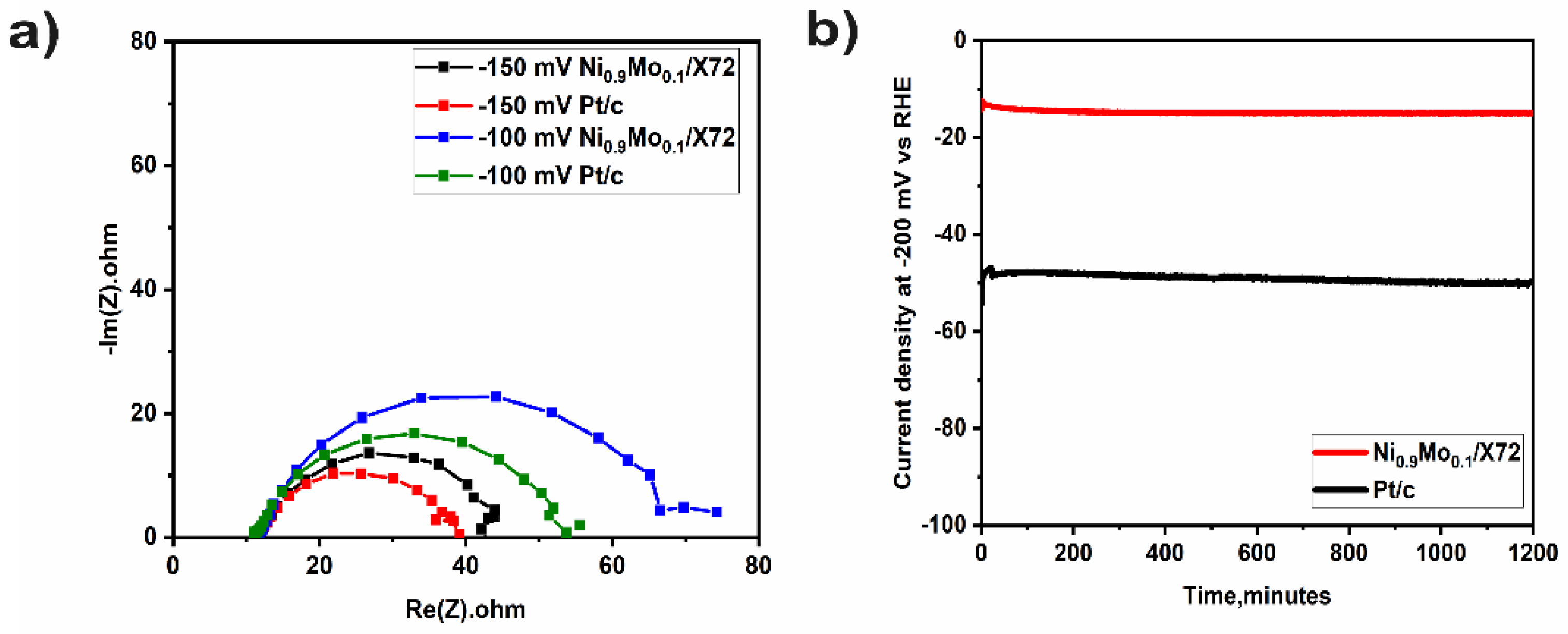
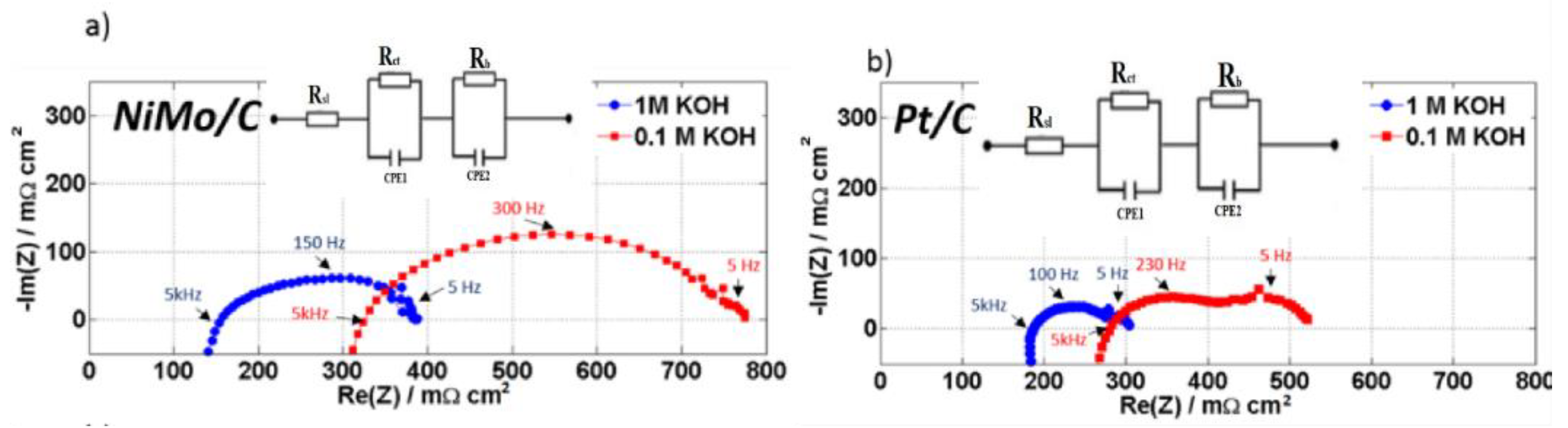
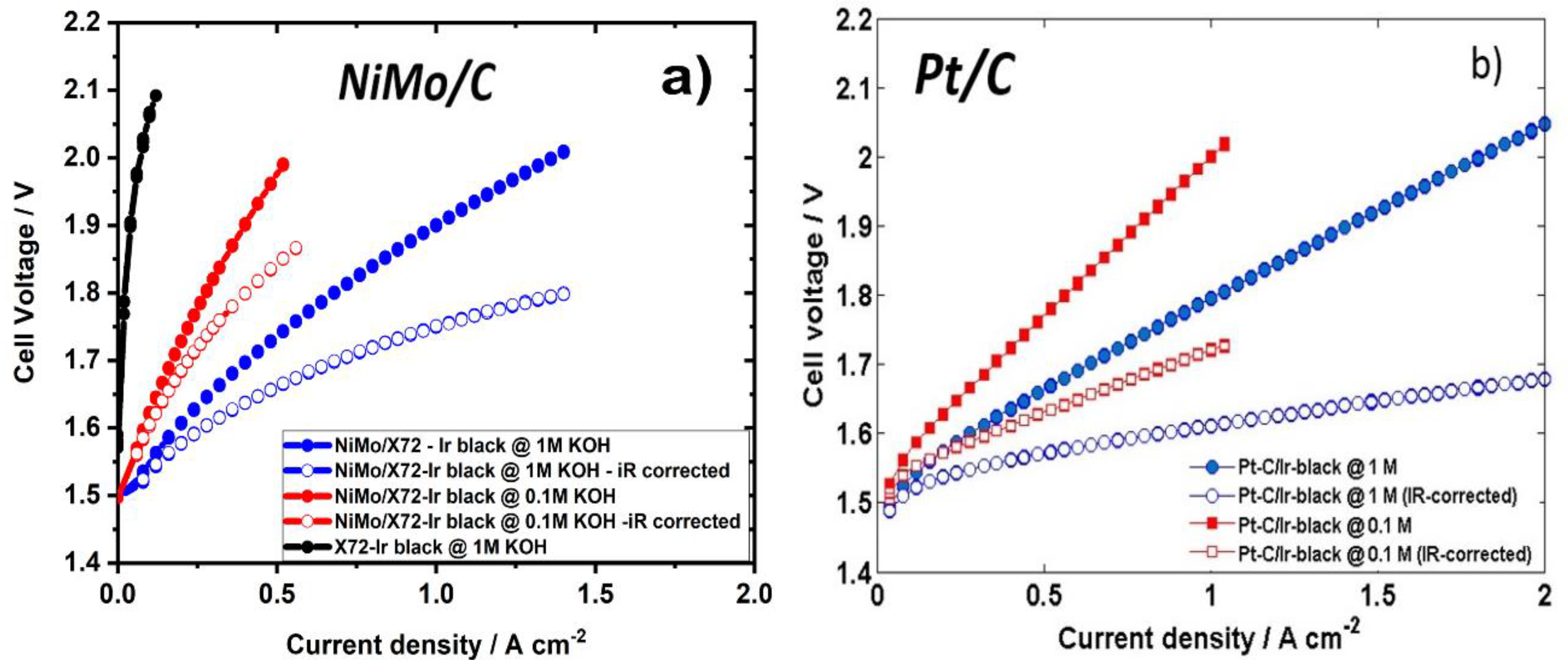
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Catalyst Characterization
3.2.1. Structural Characterization
3.2.2. Preparation of the Electrode for RDE Measurement
3.2.3. Electrochemical Characterization
3.3. MEA Preparation
3.4. Cell Testing
3.5. SEM and EDX Mapping
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vincent, I.; Kruger, A.; Bessarabov, D. Development of efficient membrane electrode assembly for low cost hydrogen production by anion exchange membrane electrolysis. Int. J. Hydrogen Energy 2017, 42, 10752–10761. [Google Scholar] [CrossRef]
- Leng, Y.; Chen, G.; Mendoza, A.J.; Tighe, T.B.; Michael, A.; Hickner, M.A.; Wang, C.-Y. Solid-state water electrolysis with an alkaline membrane. J. Am. Chem. Soc. 2012, 134, 9054–9057. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Kitty Nijmeijer, K. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef]
- Sapountzi, F.M.; Gracia, J.M.; (Kees-Jan) Weststrate, C.J.; Fredriksson, H.O.A.; (Hans) Niemantsverdriet, J.W. Electrocatalysts for the generation of hydrogen, oxygen and synthesis gas. Prog. Energy Combust. Sci. 2017, 58, 1–35. [Google Scholar] [CrossRef]
- Grond, L.; Schulze, P.; Holstein, J. Systems Analyses Power to Gas. DNV Kema 2013, GCS 13.R.2, 1–70. [Google Scholar]
- Kuckshinrichs, W.; Ketelaer, T.; Koj, J.C. Economic Analysis of Improved Alkaline Water Electrolysis. Front. Energy Res. 2017, 5. [Google Scholar] [CrossRef]
- Gong, M.; Wang, D.Y.; Chen, C.C.; Hwang, B.J.; Dai, H. A mini review on nickel-based electrocatalysts for alkaline hydrogen evolution reaction. Nano Res. 2016, 9, 28–46. [Google Scholar] [CrossRef]
- Kyung, M.; Park, H.-Y.; Choe, S.; Yoo, S.J.; Kim, J.Y.; Kim, H.-J.; Henkensmeier, D.; Lee, S.Y.; Sung, Y.-E.; Park, H.S.; Jang, J.H. Factors in electrode fabrication for performance enhancement of anion exchange membrane water electrolysis. J. Power Sources 2017, 347, 283–290. [Google Scholar]
- Phillips, R.; Dunnill, C.W. Zero gap alkaline electrolysis cell design for renewable energy storage as hydrogen gas. RSC Adv. 2016, 6, 100643–100651. [Google Scholar] [CrossRef]
- Xiang, C.; Papadantonakis, K.M.; Lewis, N.S. Principles and implementations of electrolysis systems for water splitting. Mater. Horiz. 2016, 3, 169–173. [Google Scholar] [CrossRef]
- Eftekhari, A. Electrocatalysts for hydrogen evolution reaction. Int. J. Hydrogen Energy 2017, 42, 11053–11077. [Google Scholar] [CrossRef]
- Li, X.; Hao, X.; Abudula, A.; Guan, G. Nanostructured catalysts for electrochemical water splitting: Current state and prospects. J. Mater. Chem. A 2016, 4, 11973–12000. [Google Scholar] [CrossRef]
- Bladergroen, B.; Su, H.; Pasupathi, S.; Linkov, V. Overview of Membrane Electrode Assembly Preparation Methods for Solid Polymer Electrolyte Electrolyzer. In Electrolysis; IntechOpen: London, UK, 2012. [Google Scholar]
- Artyushkova, K.; Serov, A.; Doan, H.; Danilovic, N.; Capuano, C.B.; Sakamoto, T.; Kishi, H.; Yamaguchi, S.; Mukerjee, S.; Atanassov, P. Application of X-ray photoelectron spectroscopy to studies of electrodes in fuel cells and electrolyzers. J. Electron Spectros. Relat. Phenom. 2017. [Google Scholar] [CrossRef]
- Kyung, M.; Park, H.-Y.; Lee, H.J.; Kim, H.-J.; Lim, A.; Sung, D.H.; Jin, J.Y.; So, Y.K.; Lee, Y.; Park, H.S.; Jang, J.H. Alkaline anion exchange membrane water electrolysis: Effects of electrolyte feed method and electrode binder content. J. Power Sources 2018, 382, 22–29. [Google Scholar]
- Sassin, M.B.; Garsany, Y.; Gould, B.D.; Swider-Lyons, K.E. Fabrication Method for Laboratory-Scale High-Performance Membrane Electrode Assemblies for Fuel Cells. Anal. Chem. 2017, 89, 511–518. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Zhao, T.S.; Chai, Z.H.; Tan, P.; Zeng, L. Mathematical modeling of an anion-exchange membrane water electrolyzer for hydrogen production. Int. J. Hydrogen Energy 2014, 39, 19869–19876. [Google Scholar] [CrossRef]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Wu, X.; Scott, K. A non-precious metal bifunctional oxygen electrode for alkaline anion exchange membrane cells. J. Power Sources 2012, 206, 14–19. [Google Scholar] [CrossRef]
- Pavel, C.C.; Cecconi, F.; Emiliani, C.; Santiccioli, S.; Scaffidi, A.; Catanorchi, S.; Comotti, M. Highly efficient platinum group metal free based membrane-electrode assembly for anion exchange membrane water electrolysis. Angew. Chem. Int. Ed. 2014, 53, 1378–1381. [Google Scholar] [CrossRef]
- Xiao, L.; Zhang, S.; Pan, J.; Yang, C.; He, M.; Zhuang, L.; Lu, J. First implementation of alkaline polymer electrolyte water electrolysis working only with pure water. Energy Environ. Sci. 2012, 5, 7869. [Google Scholar] [CrossRef]
- Yang, Y.; Zhuang, L.; Rufford, T.E.; Wang, S.; Zhu, Z. Efficient water oxidation with amorphous transition metal boride catalysts synthesized by chemical reduction of metal nitrate salts at room temperature. RSC Adv. 2017, 7, 32923–32930. [Google Scholar] [CrossRef]
- Nie, M.; Zou, Y.C.; Huang, Y.M.; Wang, J.Q. Ni-Fe-B catalysts for NaBH4hydrolysis. Int. J. Hydrogen Energy 2012, 37, 1568–1576. [Google Scholar] [CrossRef]
- Mironova-Ulmane, N.; Kuzmin, A.; Steins, I.; Grabis, J.; Sildos, I.; Pärs, M. Raman scattering in nanosized nickel oxide NiO. J. Phys. Conf. Ser. 2007, 93. [Google Scholar] [CrossRef]
- Wagner, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, J.F.F.; Muilenberg, G.; Stickle, W.F.; Sobol, P.; EBomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy. Surf. Interface Anal. 1979, 3, 80–90. [Google Scholar]
- Sun, Y.; Hu, X.; Luo, W.; Huang, Y. Ultrafine MoO2 nanoparticles embedded in a carbon matrix as a high-capacity and long-life anode for lithium-ion batteries. J. Mater. Chem. 2012, 22, 425–431. [Google Scholar] [CrossRef]
- Kabir, S.A.; Lemire, K.; Artyushkova, K.; Roy, A.; Odgaard, M.; Schlueter, D.; Oshchepkov, A.; Bonnefont, A.; Savinova, E.; Sabarirajan, D.C.; et al. Platinum Group Metal-free NiMo Hydrogen Oxidation Catalysts: High Performance and Durability in Alkaline Exchange Membrane Fuel Cells. J. Mater. Chem. A 2017, 5, 24433–24443. [Google Scholar] [CrossRef]
- Gupta, S.; Patel, N.; Fernandes, R.; Kadrekar, R.; Dashora, A.; Yadav, A.K.; Bhattacharyya, D.; Jha, S.N.; Miotello, A.; Kothari, D.C. Co-Ni-B nanocatalyst for efficient hydrogen evolution reaction in wide pH range. Appl. Catal. B Environ. 2016, 192, 126–133. [Google Scholar] [CrossRef]
- Gupta, S.; Patel, N.; Fernandes, R.; Hanchate, S.; Miotello, A.; Kothari, D.C. Co-Mo-B Nanoparticles as a non-precious and efficient Bifunctional Electrocatalyst for Hydrogen and Oxygen Evolution. Electrochim. Acta 2017, 232, 64–71. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Voiry, D.; Chhowalla, M.; Gogotsi, Y.; Kotov, N.A.; Li, Y.; Penner, R.M.; Schaak, R.E.; Weiss, P.S. Best Practices for Reporting Electrocatalytic Performance of Nanomaterials. ACS Nano 2018, 12, 9635–9638. [Google Scholar] [CrossRef]
- Rudi, S.; Cui, C.; Gan, L.; Strasser, P. Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupdand CO-stripping voltammetry. Electrocatalysis 2014, 5, 408–418. [Google Scholar] [CrossRef]
- Deng, X.; Öztürk, S.; Weidenthaler, C.; Tüysüz, H. Iron-Induced Activation of Ordered Mesoporous Nickel Cobalt Oxide Electrocatalyst for the Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2017, 9, 21225–21233. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nickel Price. 2018. Available online: https://markets.businessinsider.com/commodities/nickel-price (accessed on 16 November 2018).
- Mukherjee, A.; Chakrabarty, S.; Su, W.-N.; Basu, S. Nanostructured nickel ferrite embedded in reduced graphene oxide for electrocatalytic hydrogen evolution reaction. Mater. Today Energy 2018, 8, 118–124. [Google Scholar] [CrossRef]
- Saha, S.; Ojha, K.; Sharma, M.; Ganguli, A.K. Ni3Co/G alloy as an earth-abundant robust and stable electrocatalyst for the hydrogen evolution reaction. New J. Chem. 2017, 41, 5916–5923. [Google Scholar] [CrossRef]
- Franceschini, E.A.; Lacconi, G.I.; Corti, H.R. Kinetics of the hydrogen evolution on nickel in alkaline solution: New insight from rotating disk electrode and impedance spectroscopy analysis. Electrochim. Acta 2015, 159, 210–218. [Google Scholar] [CrossRef]
- Dedigama, I.; Angeli, P.; Ayers, K.; Robinson, J.B.; Shearing, P.R.; Tsaoulidis, D.; Bret, D.J.L. In situ diagnostic techniques for characterisation of polymer electrolyte membrane water electrolysers—Flow visualisation and electrochemical impedance spectroscopy. Int. J. Hydrogen Energy 2014, 39, 4468–4482. [Google Scholar] [CrossRef]
- Kraglund, M.R.; Aili, D.; Jankova, K.; Christensen, E.; Li, Q.; Jensen, J.O. Zero-Gap Alkaline Water Electrolysis Using Ion-Solvating Polymer Electrolyte Membranes at Reduced KOH Concentrations. J. Electrochem. Soc. 2016, 163, F3125–F3131. [Google Scholar] [CrossRef]
- Siracusano, S.; Trocino, S.; Briguglio, N.; Baglio, V.; Aricò, A.S. Electrochemical impedance spectroscopy as a diagnostic tool in polymer electrolyte membrane electrolysis. Materials 2018, 11, 1368. [Google Scholar] [CrossRef]
- Fleig, J. The Influence of Current Constriction on the Impedance of Polarizable Electrodes. J. Electrochem. Soc. 1997, 144, L302. [Google Scholar] [CrossRef]
- Newman, J.; Thomas, K.E. Electrochemical Systems, 3rd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Sun, S.; Shao, Z.; Yu, H.; Li, G.; Yi, B. Investigations on degradation of the long-term proton exchange membrane water electrolysis stack. J. Power Sources 2014, 267, 515–520. [Google Scholar] [CrossRef]
- Jaouen, F.; Lindbergh, G.; Wiezell, K. Transient Techniques for Investigating Mass-Transport Limitations in Gas Diffusion Electrodes. J. Electrochem. Soc. 2003, 150, A1711. [Google Scholar] [CrossRef]
- Darab, M.; Barnett, A.O.; Lindbergh, G.; Thomassen, M.S.; Sunde, S. The Influence of Catalyst Layer Thickness on the Performance and Degradation of PEM Fuel Cell Cathodes with Constant Catalyst Loading. Electrochim. Acta 2017, 232, 505–516. [Google Scholar] [CrossRef]
- Vincent, I.; Bessarabov, D. Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renew. Sustain. Energy Rev. 2018, 81, 1690–1704. [Google Scholar] [CrossRef]
- Fernandes, R.; Patel, N.; Miotello, A.; Filippi, M. Studies on catalytic behavior of Co-Ni-B in hydrogen production by hydrolysis of NaBH4. J. Mol. Catal. A Chem. 2009, 298, 1–6. [Google Scholar] [CrossRef]
- Patel, N.; Fernandes, R.; Miotello, A. Promoting effect of transition metal-doped Co-B alloy catalysts for hydrogen production by hydrolysis of alkaline NaBH4 solution. J. Catal. 2010, 271, 315–324. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faid, A.Y.; Oyarce Barnett, A.; Seland, F.; Sunde, S. Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis. Catalysts 2018, 8, 614. https://doi.org/10.3390/catal8120614
Faid AY, Oyarce Barnett A, Seland F, Sunde S. Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis. Catalysts. 2018; 8(12):614. https://doi.org/10.3390/catal8120614
Chicago/Turabian StyleFaid, Alaa Y., Alejandro Oyarce Barnett, Frode Seland, and Svein Sunde. 2018. "Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis" Catalysts 8, no. 12: 614. https://doi.org/10.3390/catal8120614
APA StyleFaid, A. Y., Oyarce Barnett, A., Seland, F., & Sunde, S. (2018). Highly Active Nickel-Based Catalyst for Hydrogen Evolution in Anion Exchange Membrane Electrolysis. Catalysts, 8(12), 614. https://doi.org/10.3390/catal8120614