Organocatalysis: A Brief Overview on Its Evolution and Applications

 , and
, and 
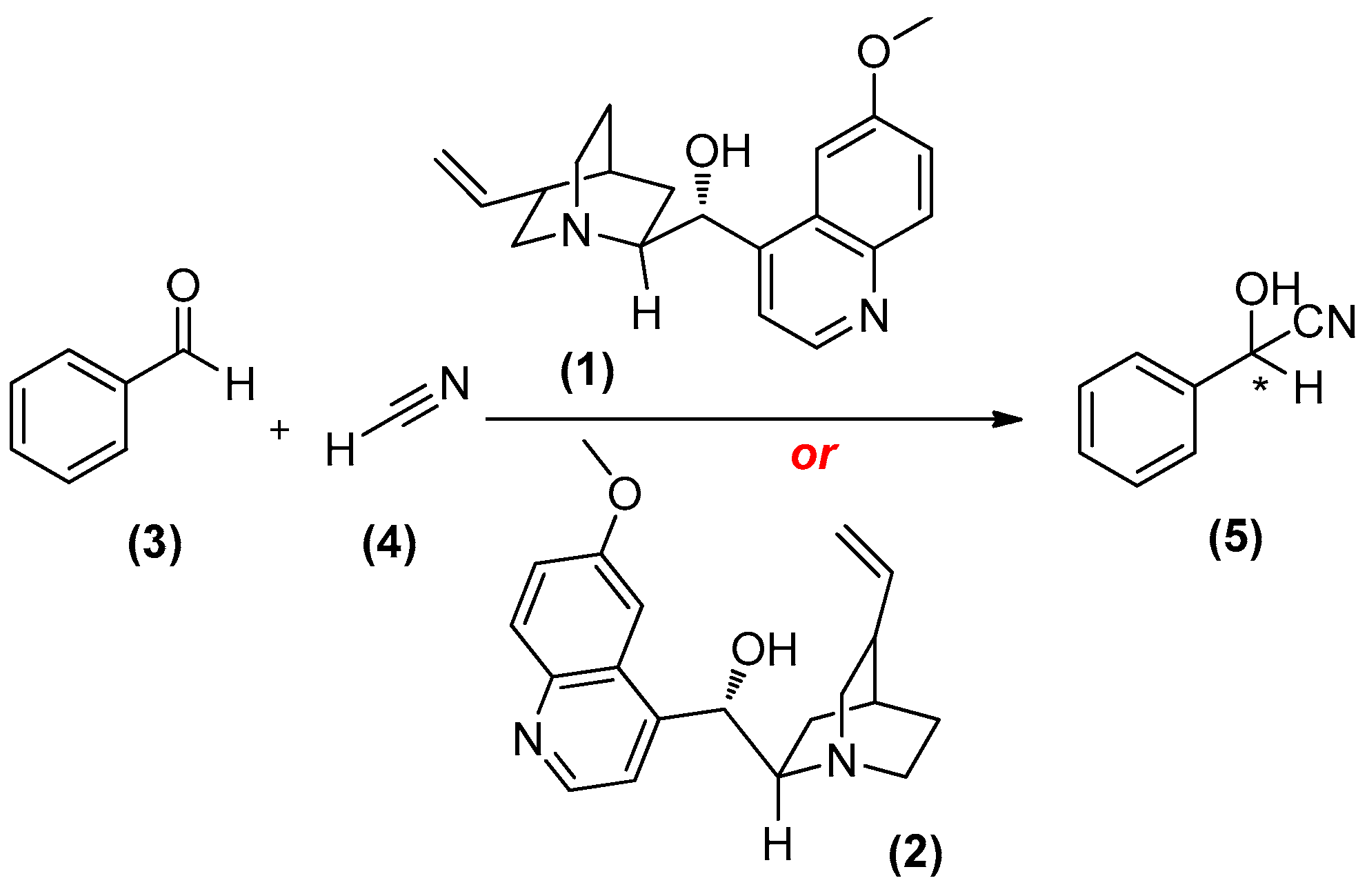{kind=link}
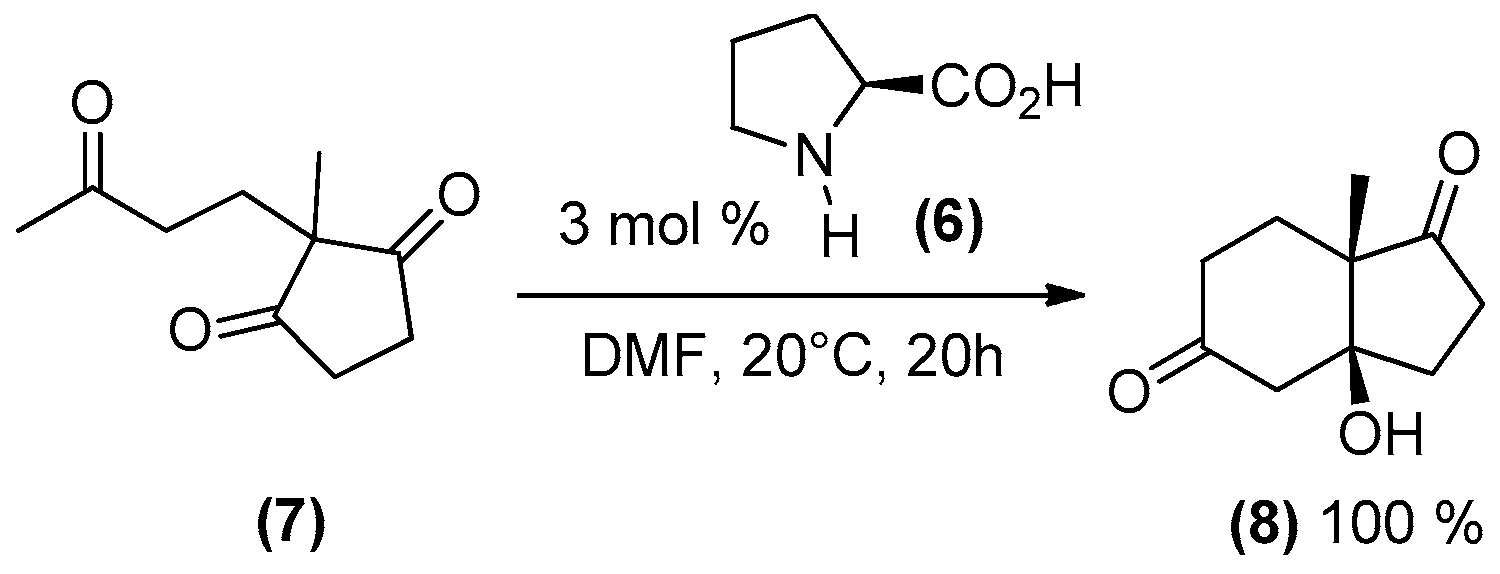{kind=link}
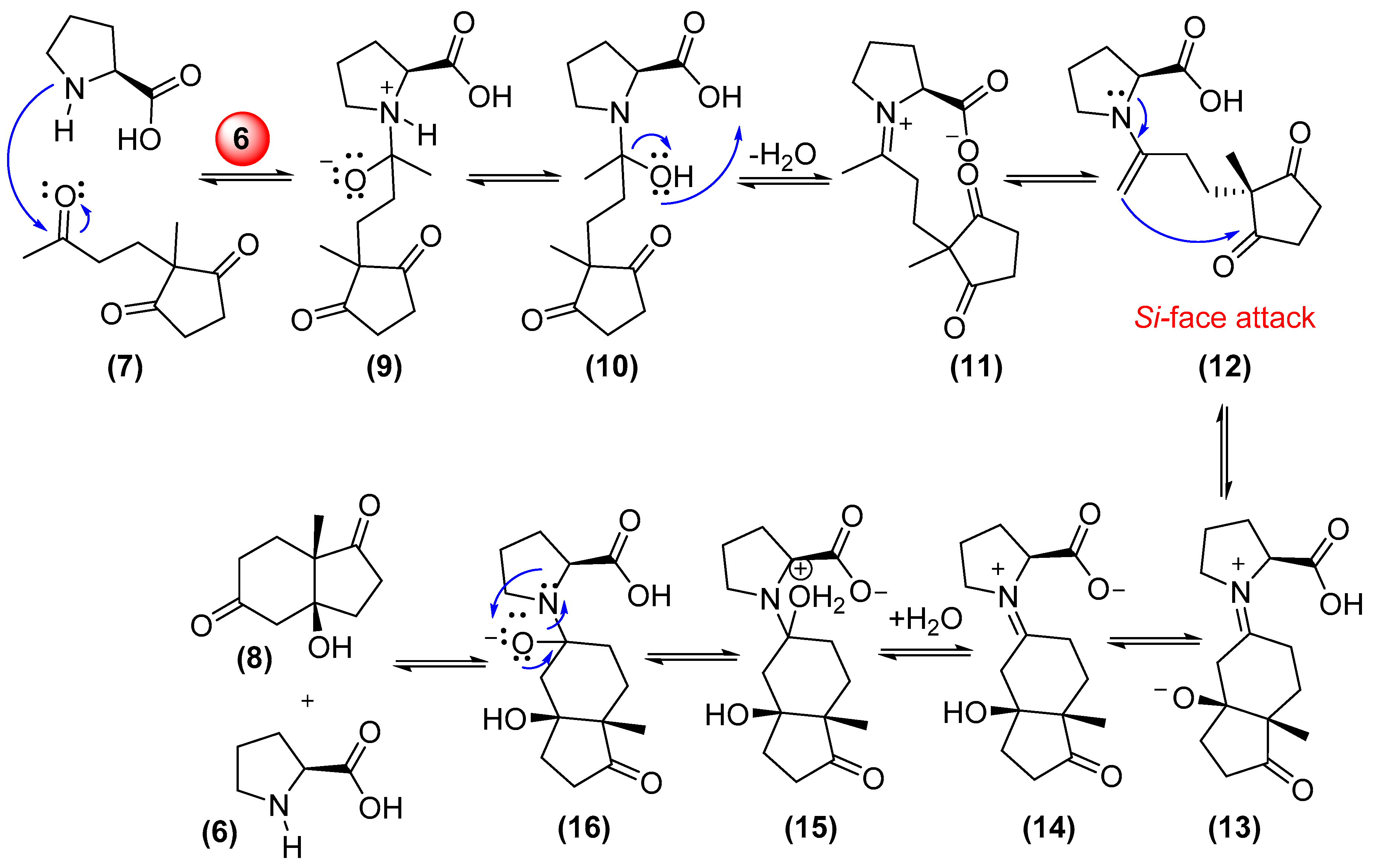{kind=link}
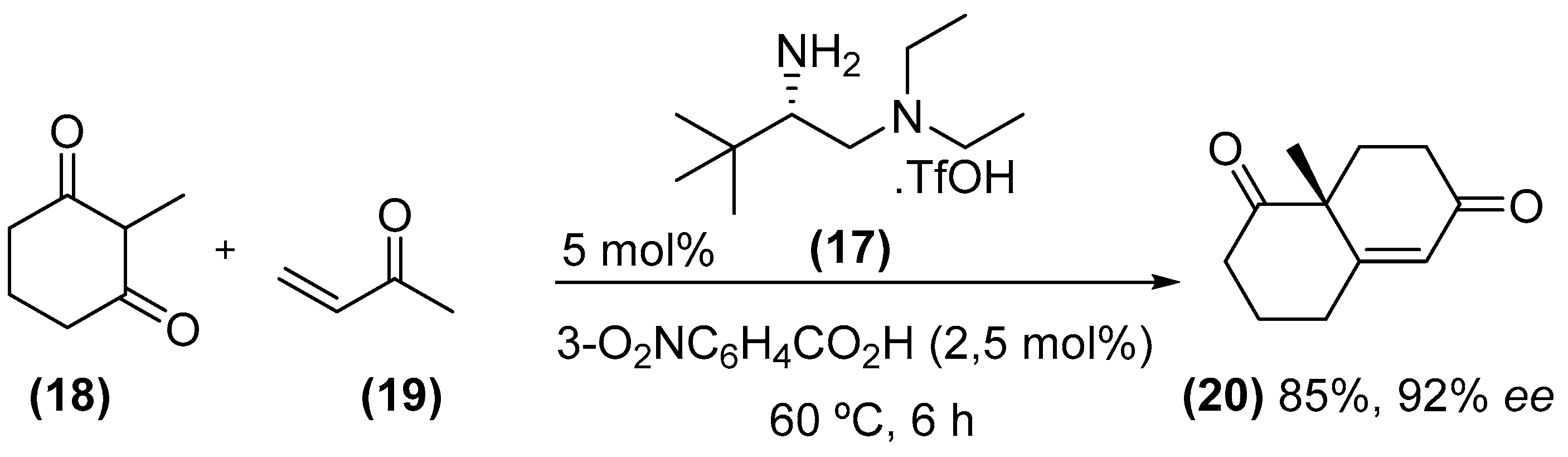{kind=link}
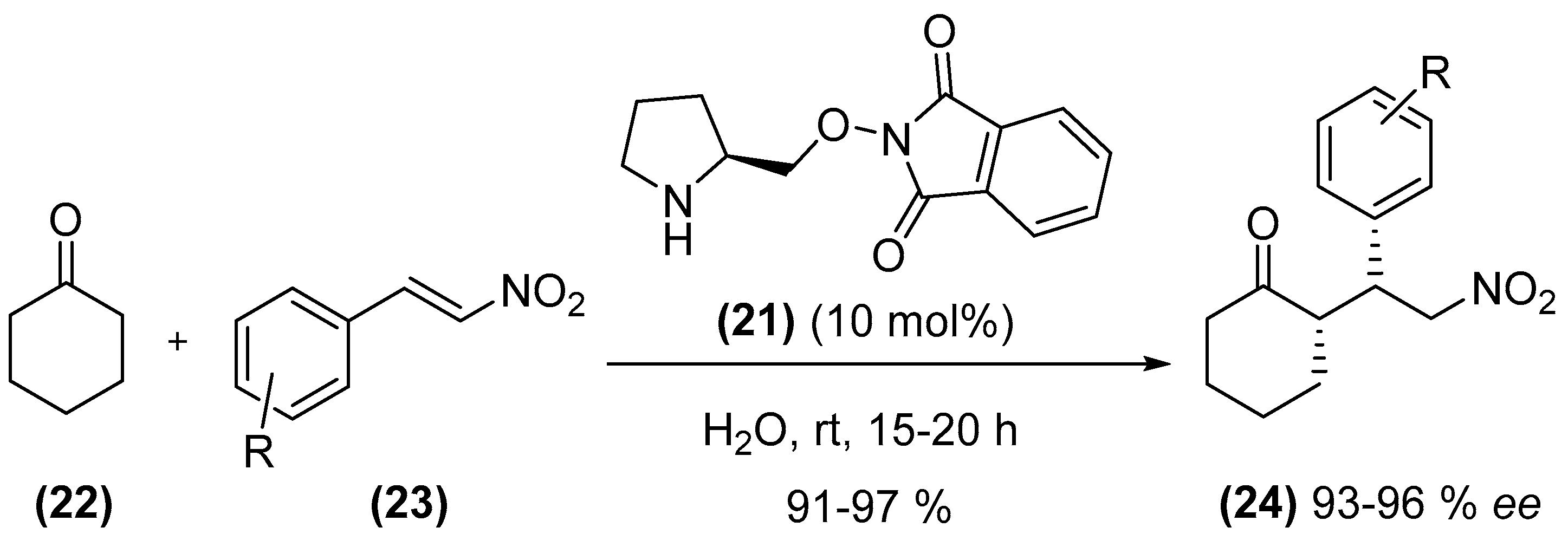{kind=link}
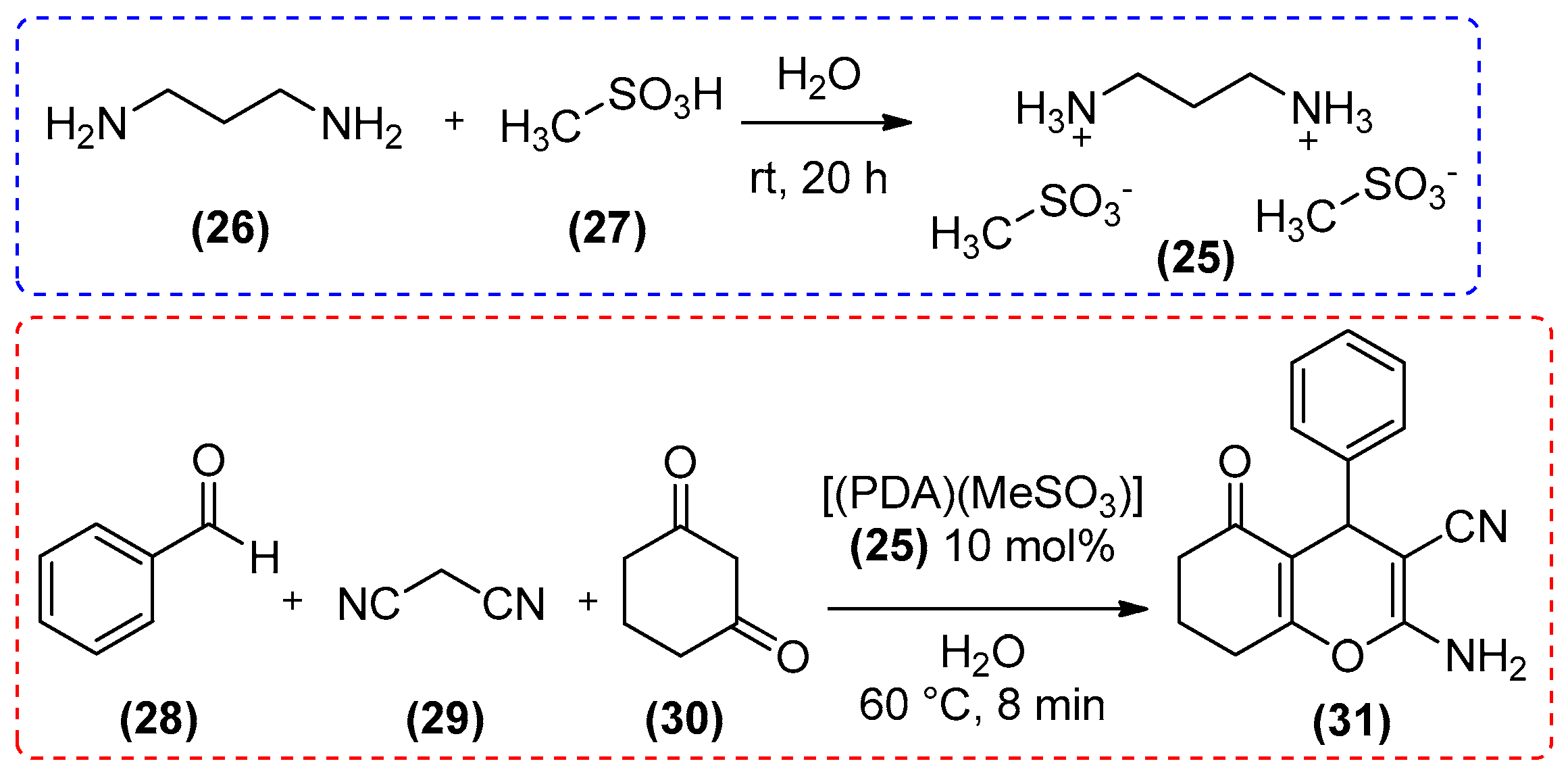{kind=link}
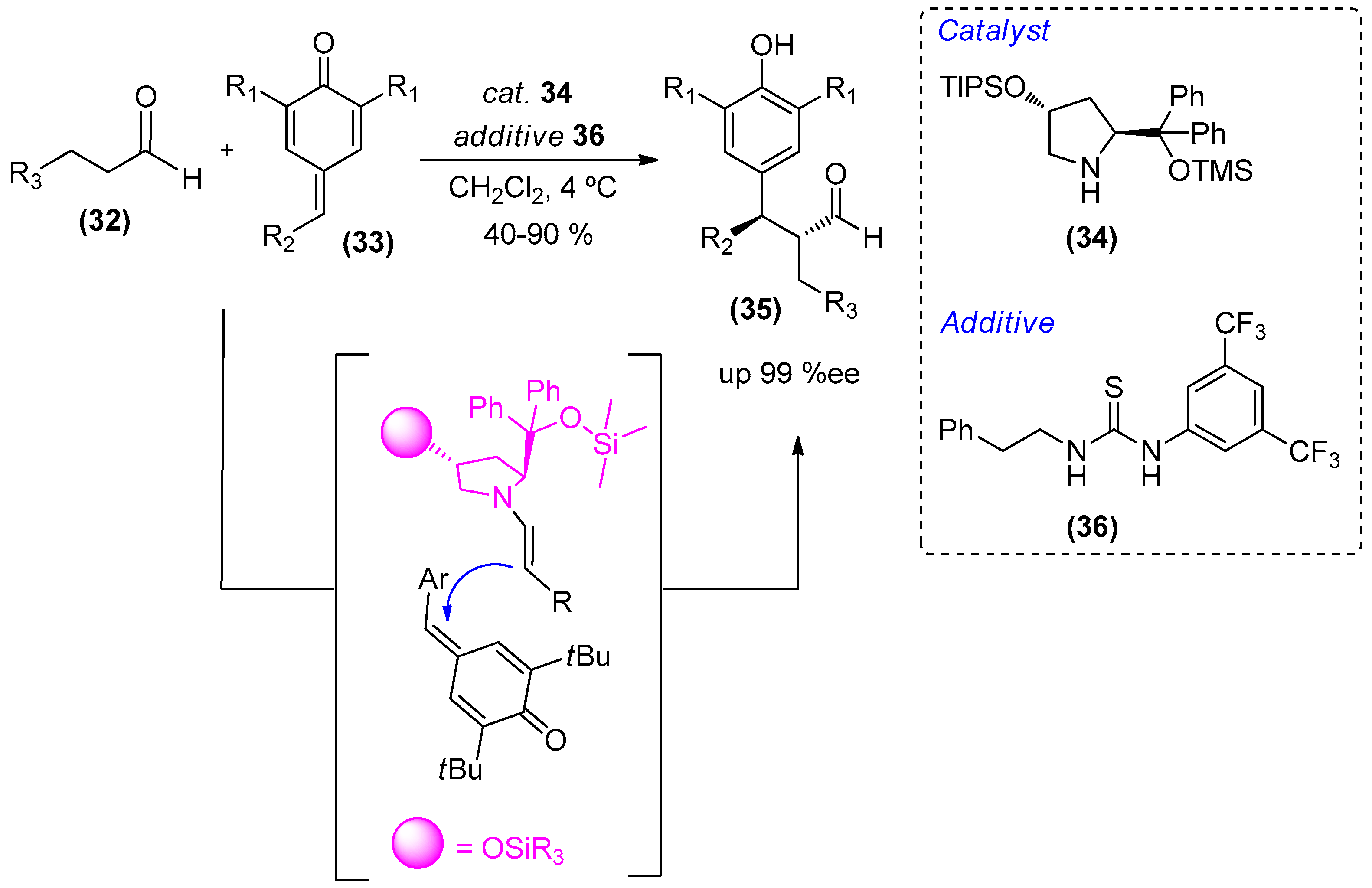{kind=link}
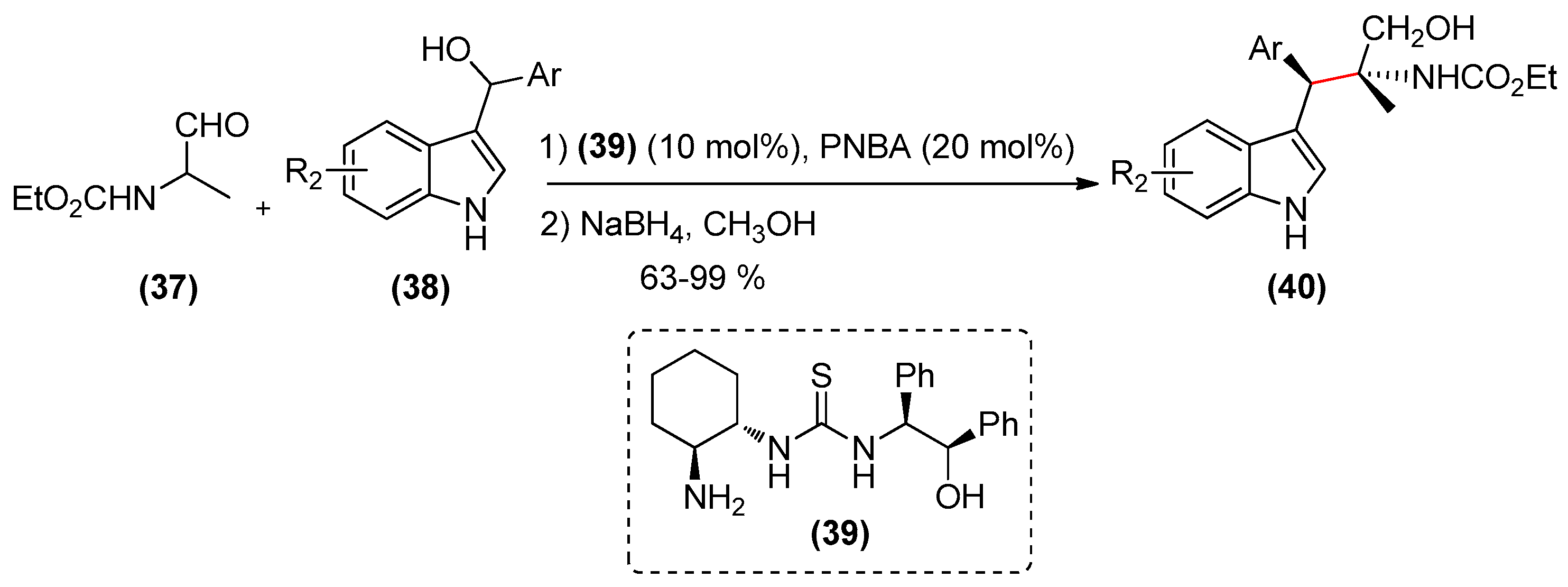{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
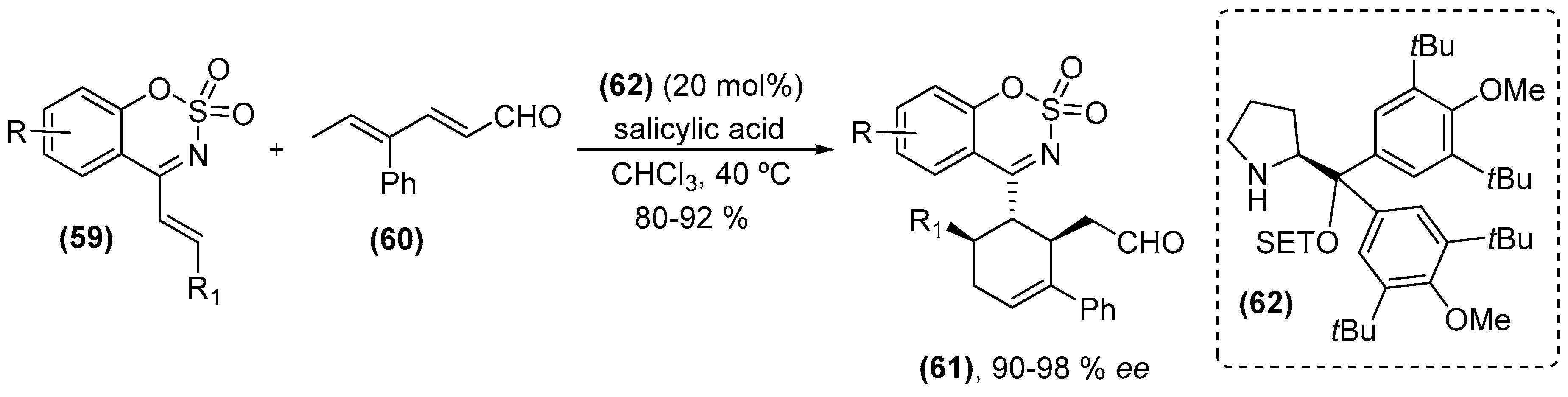{kind=link}
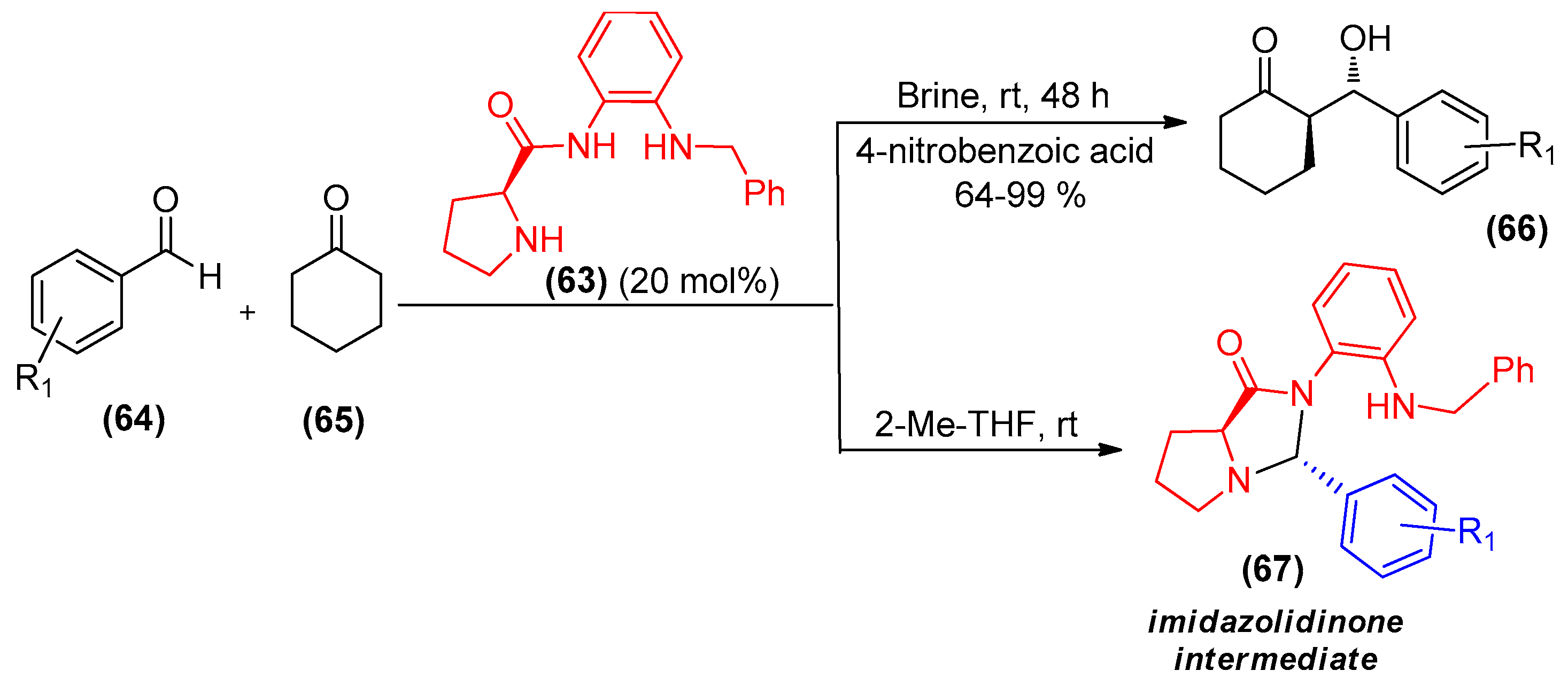{kind=link}
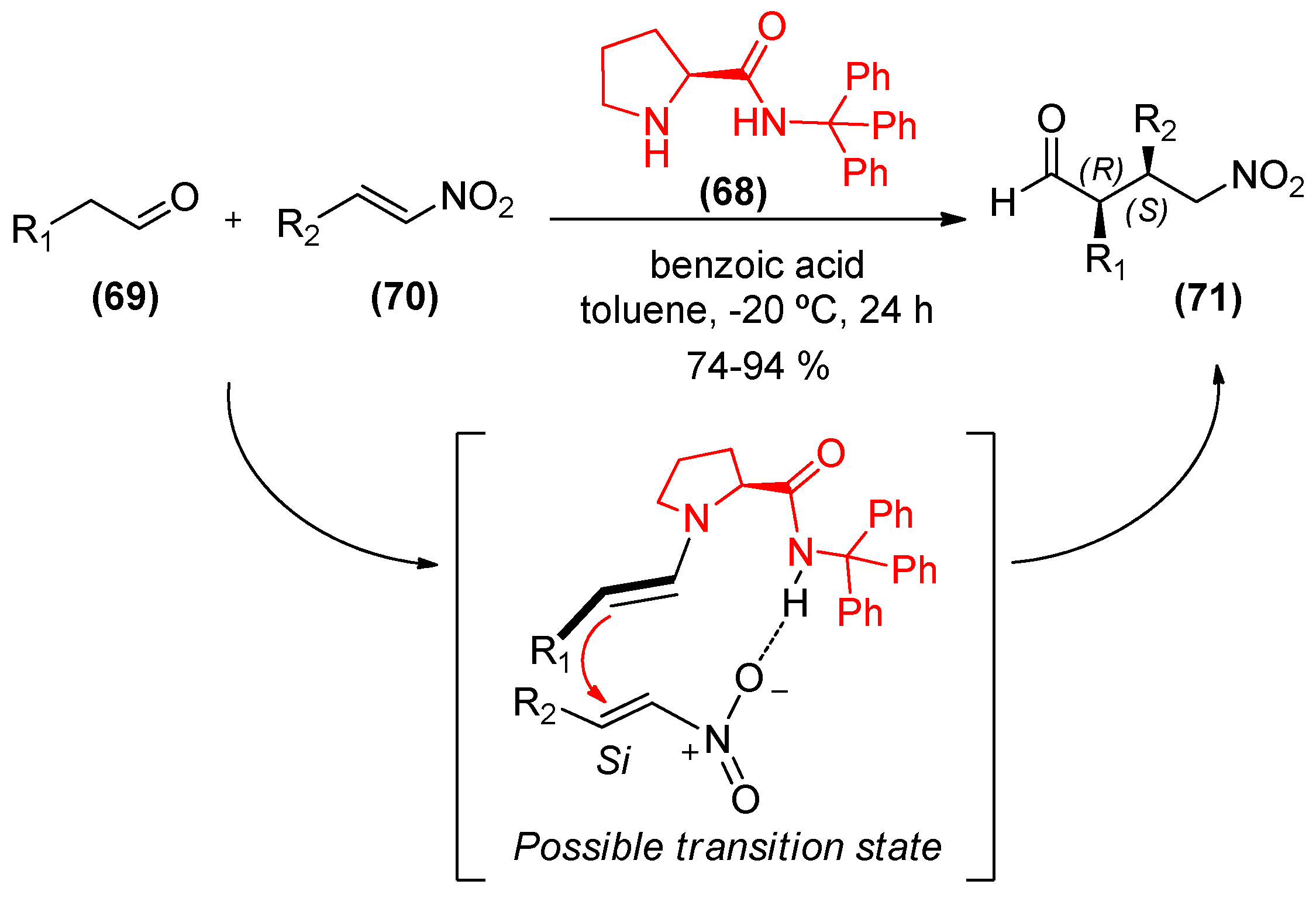{kind=link}
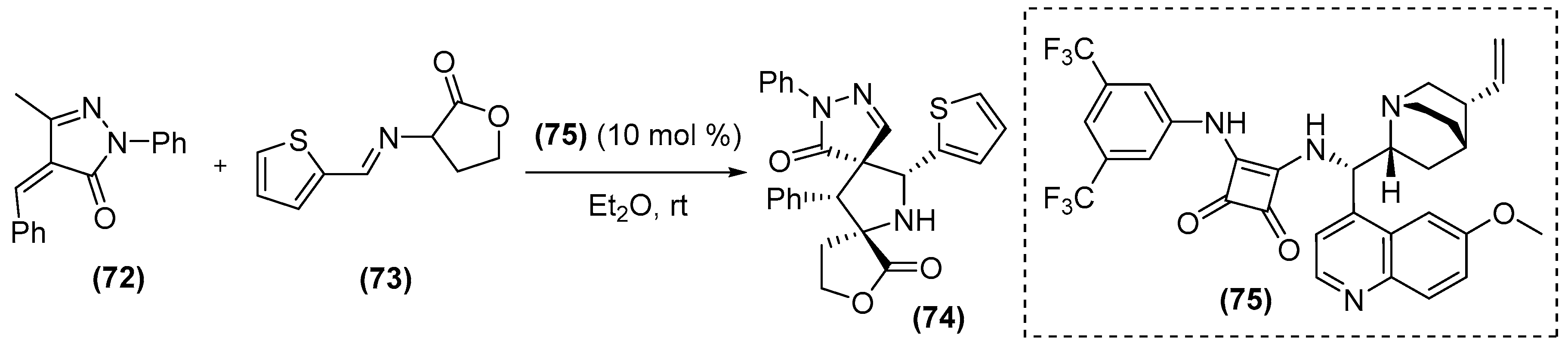{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
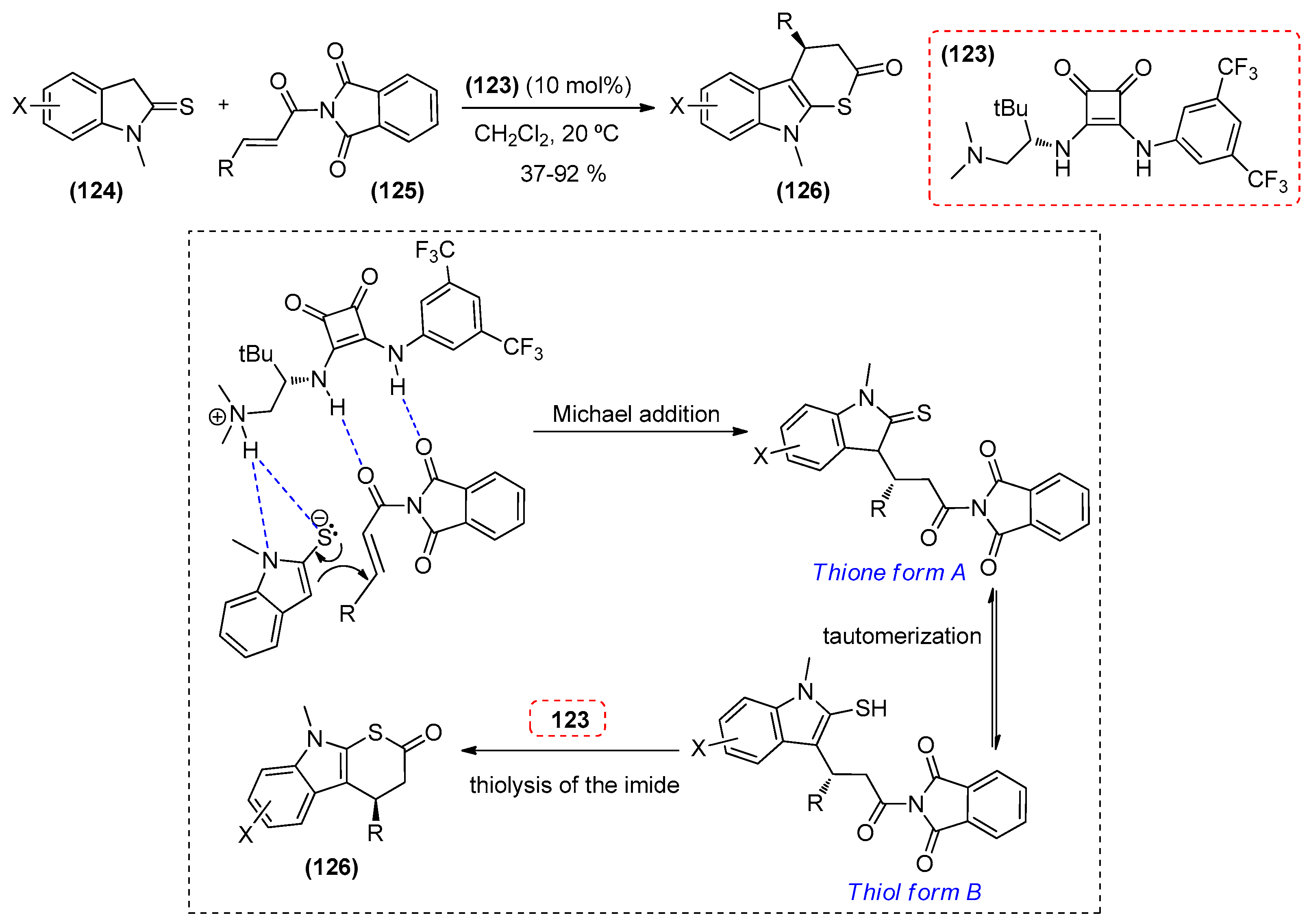{kind=link}
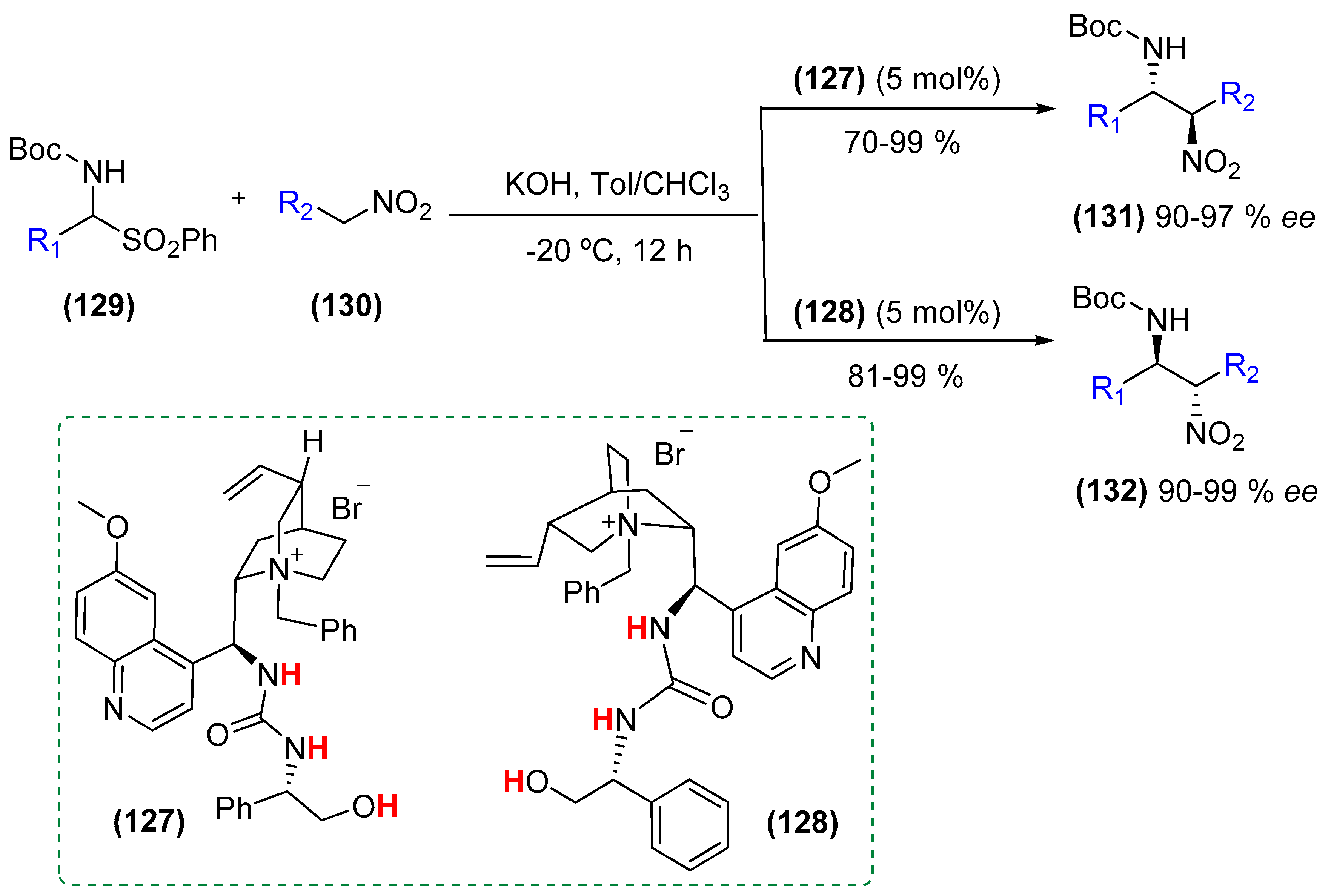{kind=link}
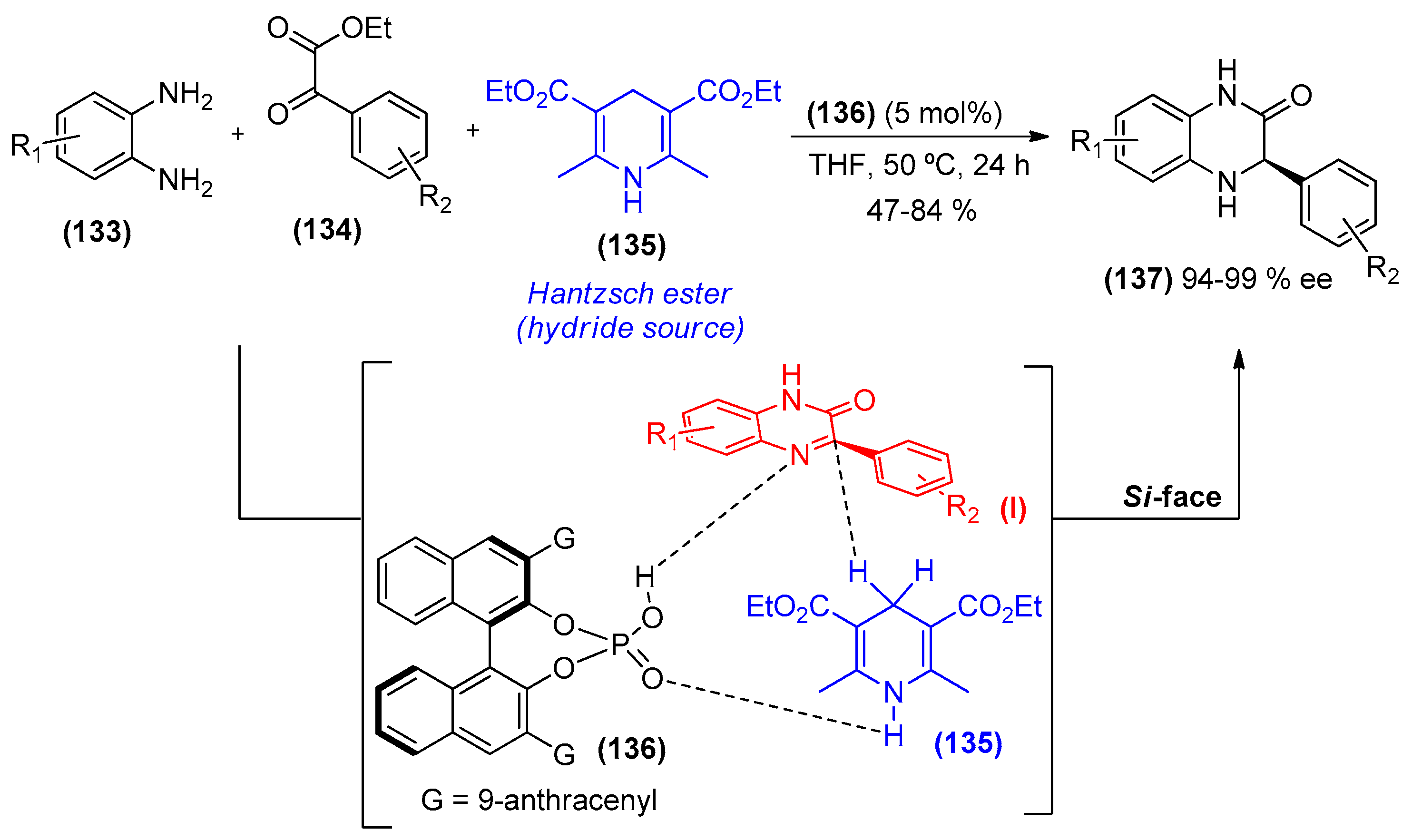{kind=link}
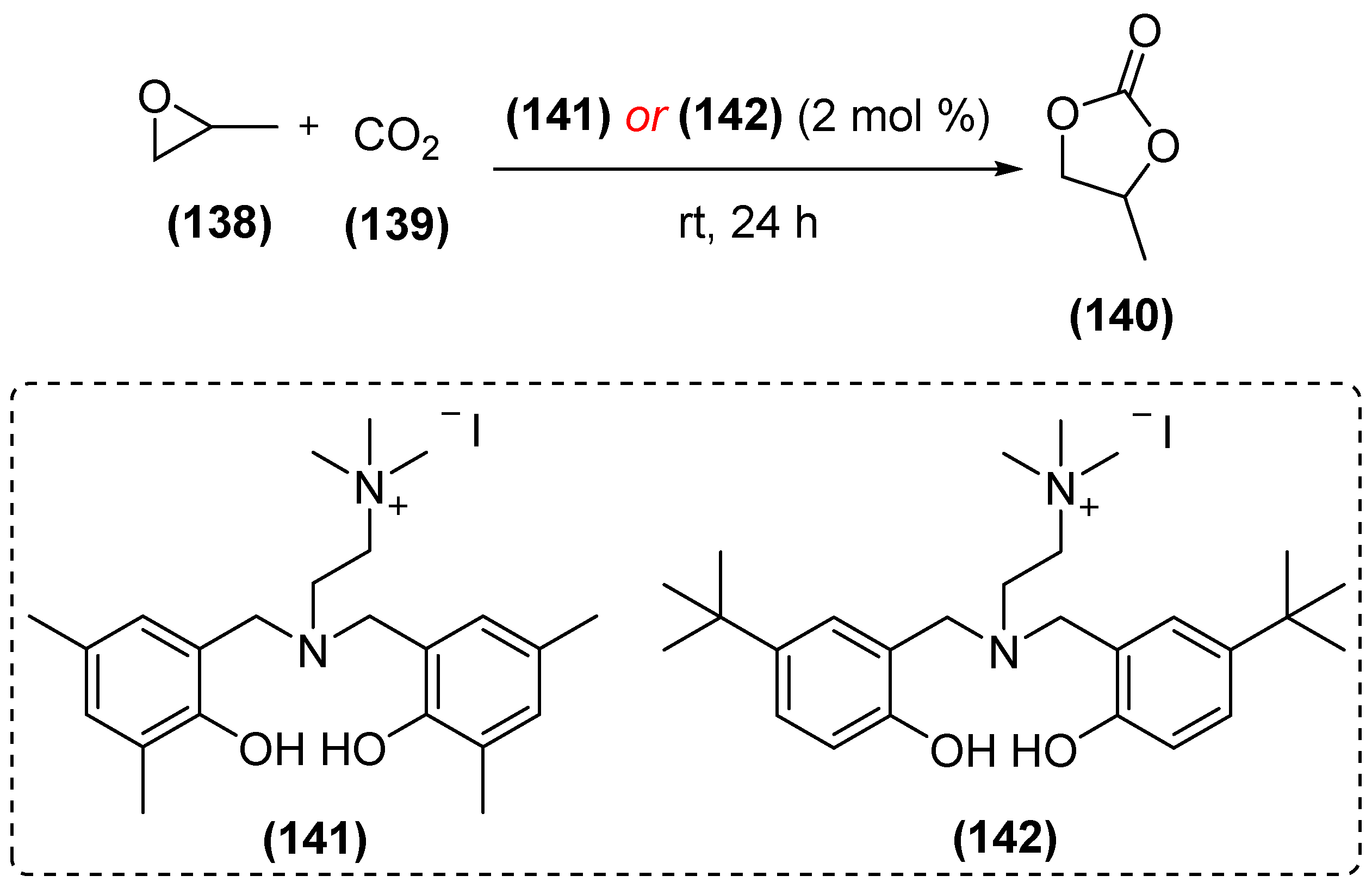{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- -
- The use of catalysts is, itself, a green chemistry principle;
- -
- It employs mild conditions, consequently saving energy;
- -
- It uses, in general, oxygen-stable reagents and does not require anhydrous conditions, reducing the cost of the synthesis [3];
- -
- It is compatible with several functional groups that could be sensitive to other processes—this reduces the need for protection groups, lowering the total number of reaction steps;
- -
- It uses less toxic and safer substances;
- -
- It prevents the formation of metallic waste and avoids traces of metals in the products, which is an essential feature for applications in medicinal chemistry.
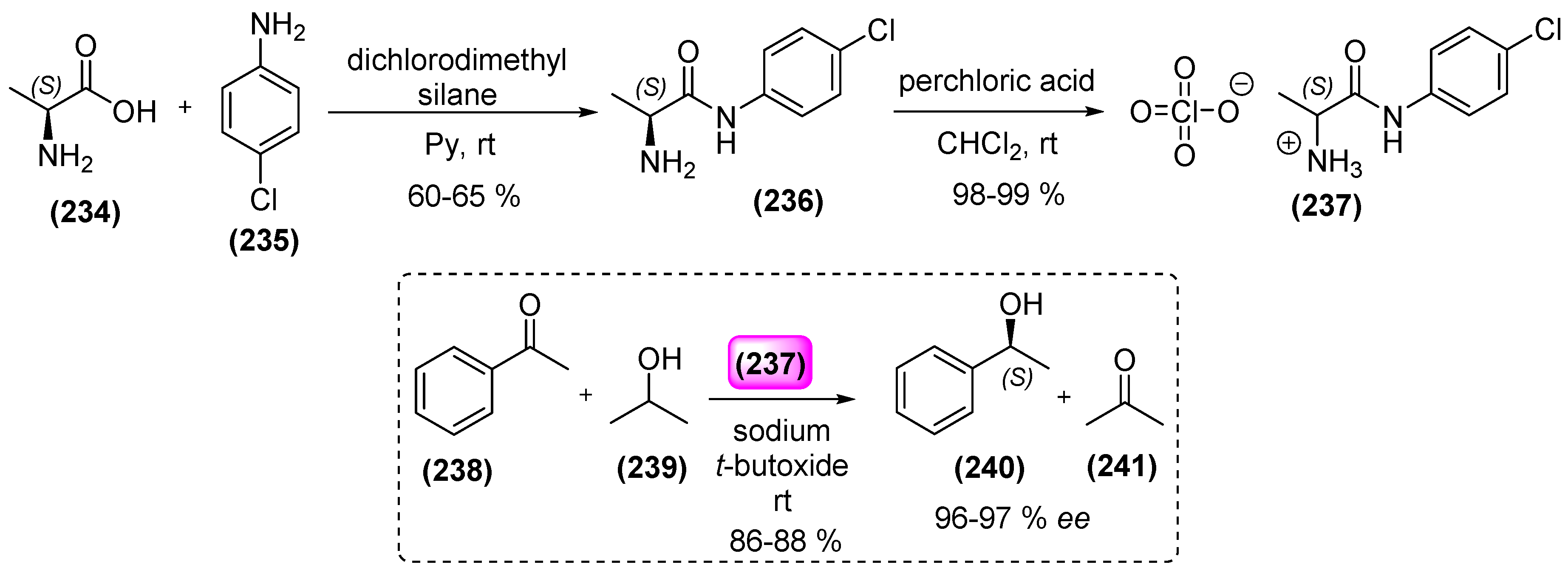
2. Amines as Organocatalysts
3. Chiral Amides as Organocatalysts
4. Iminium
5. Carbenes as Organocatalysts
6. Supramolecular Organocatalysis
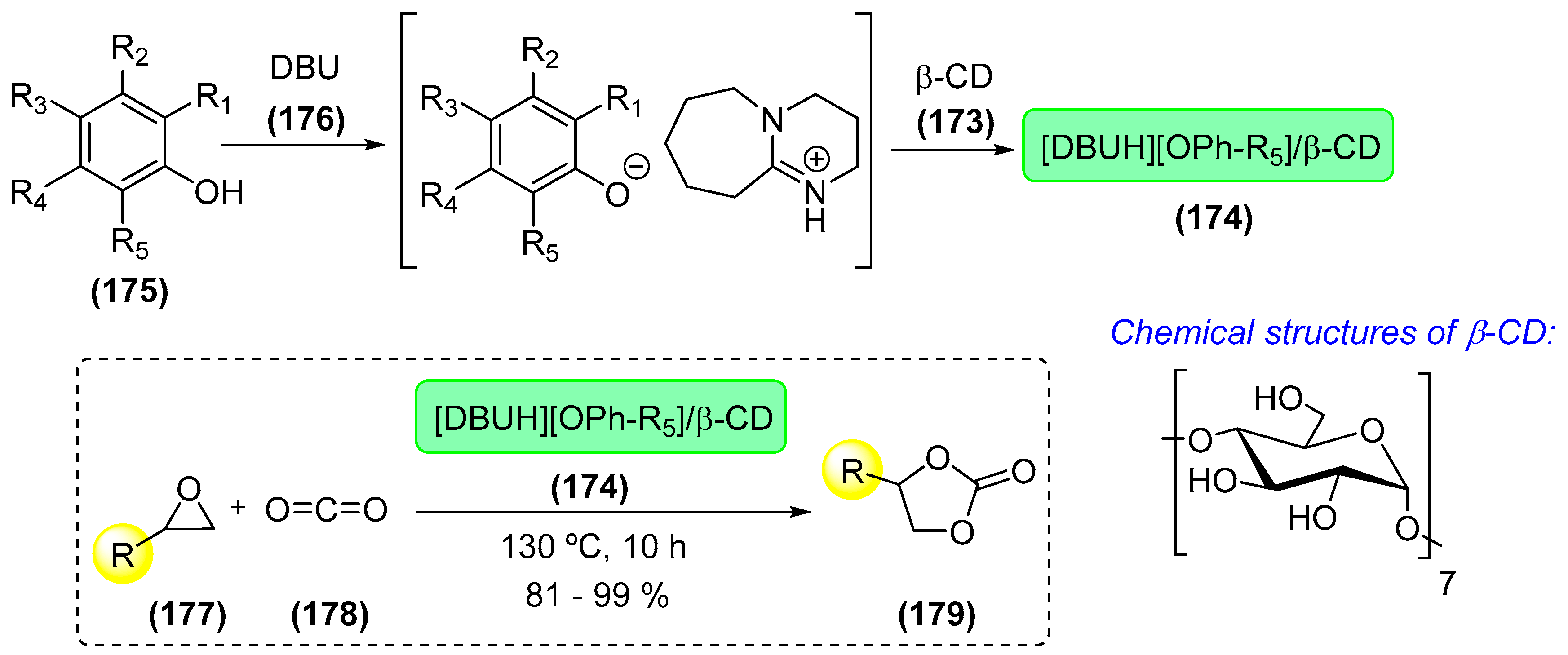
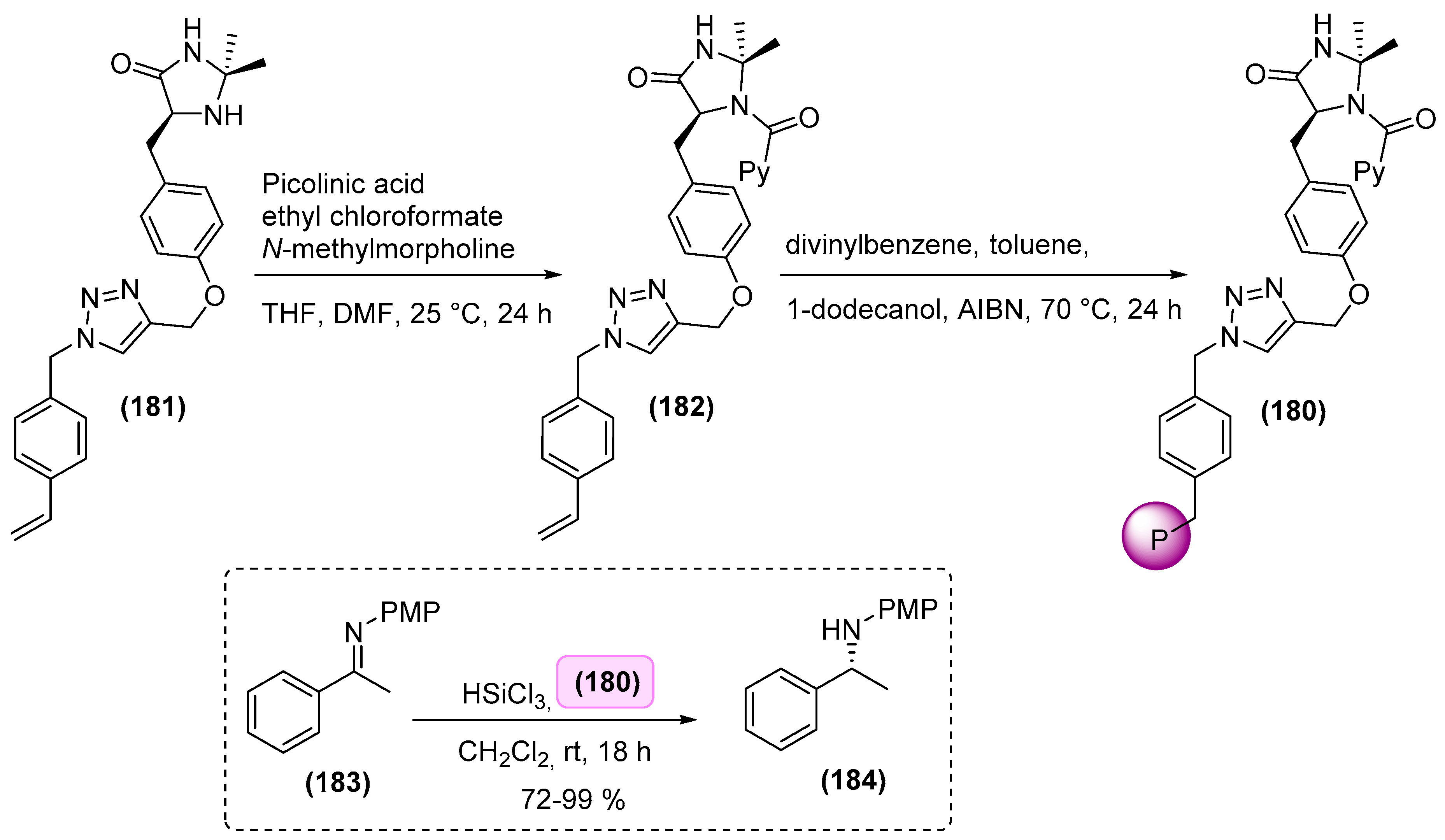
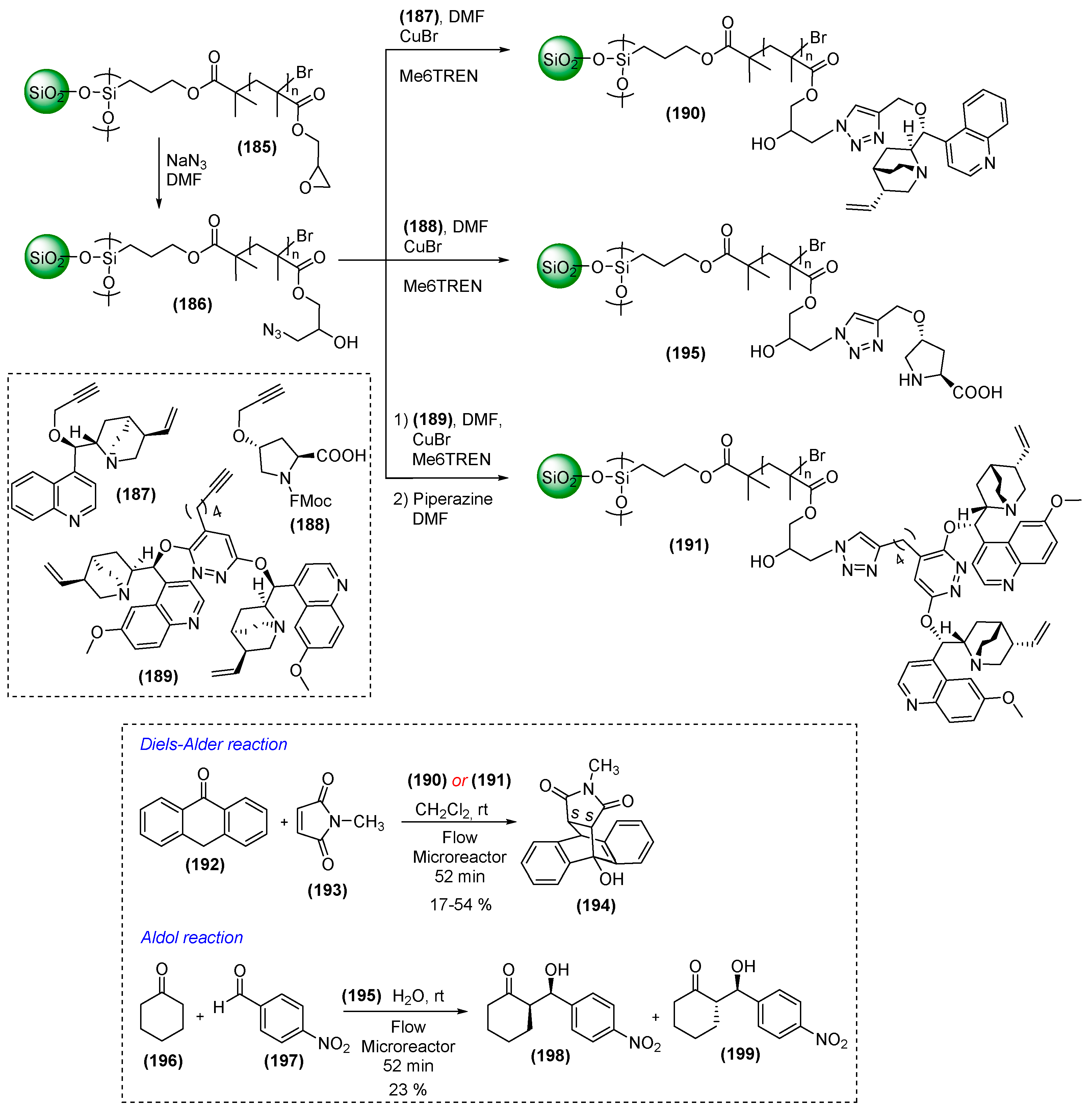
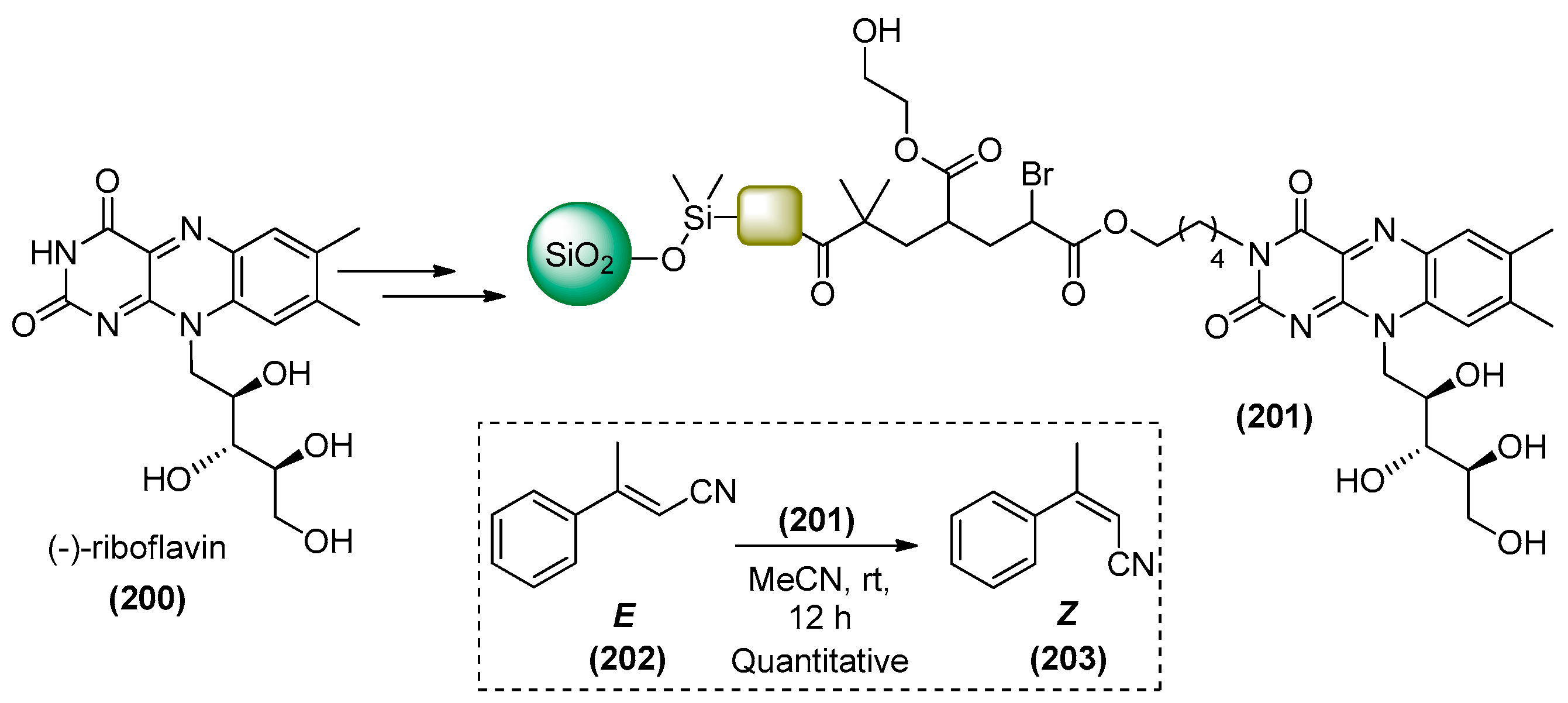
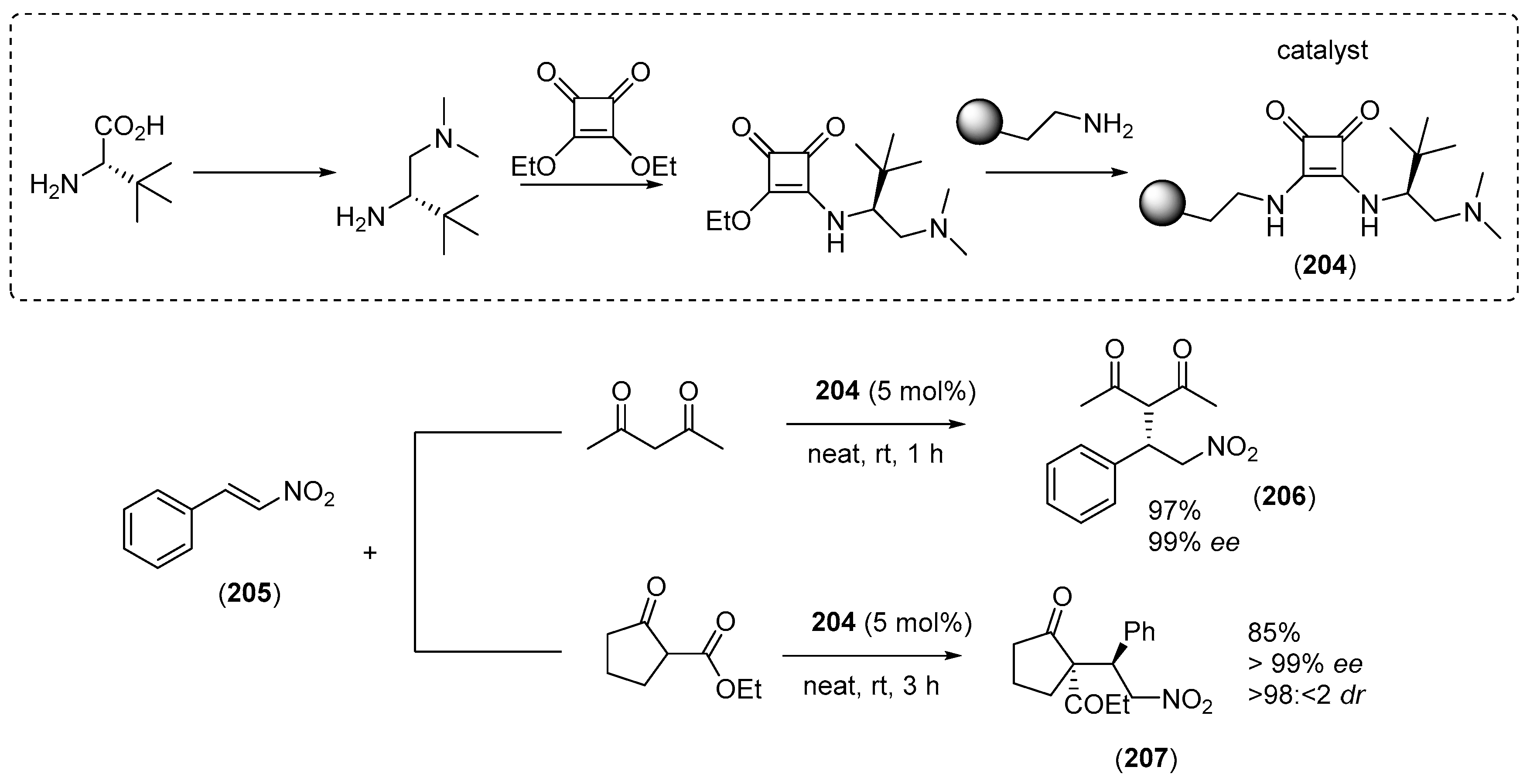
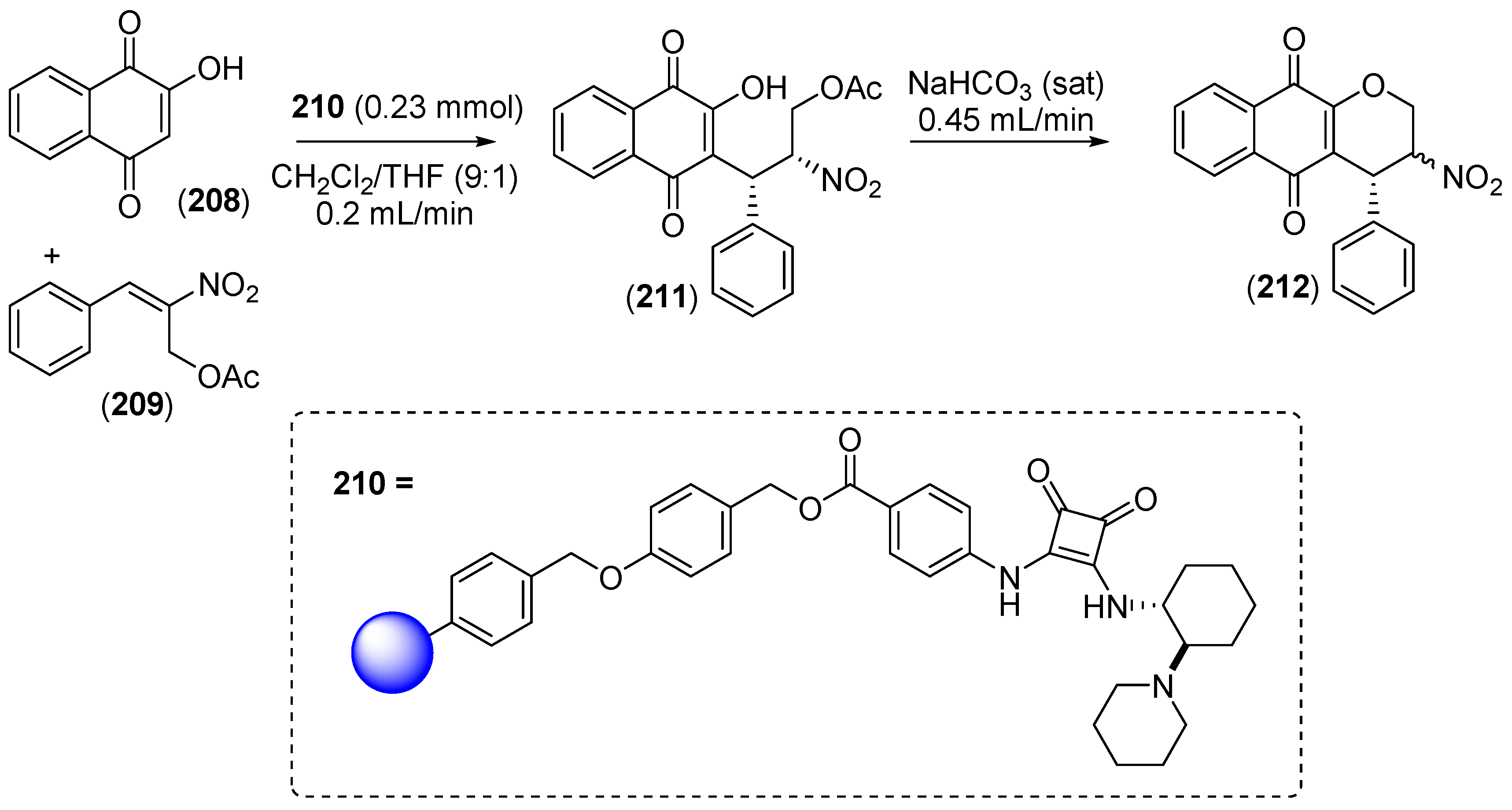
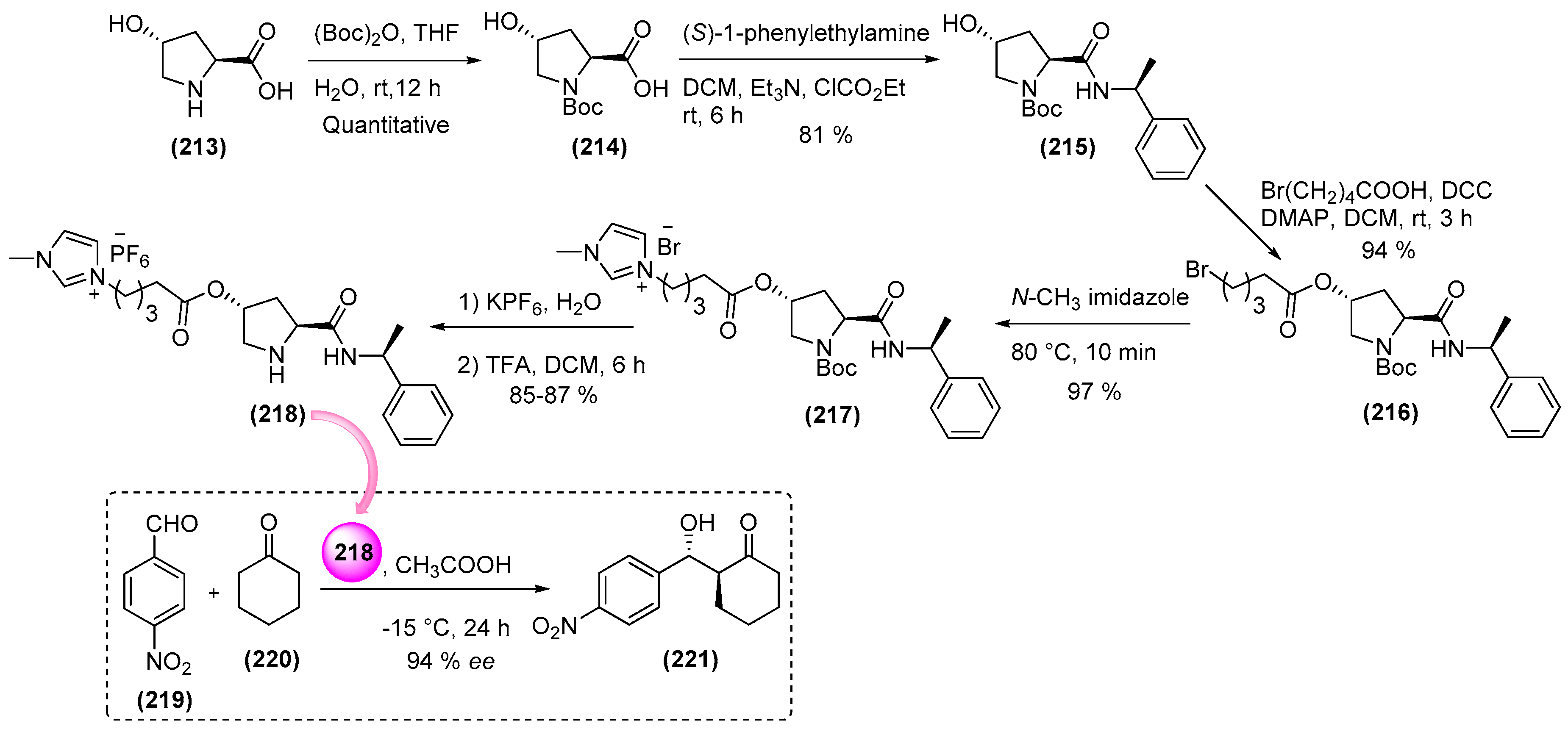
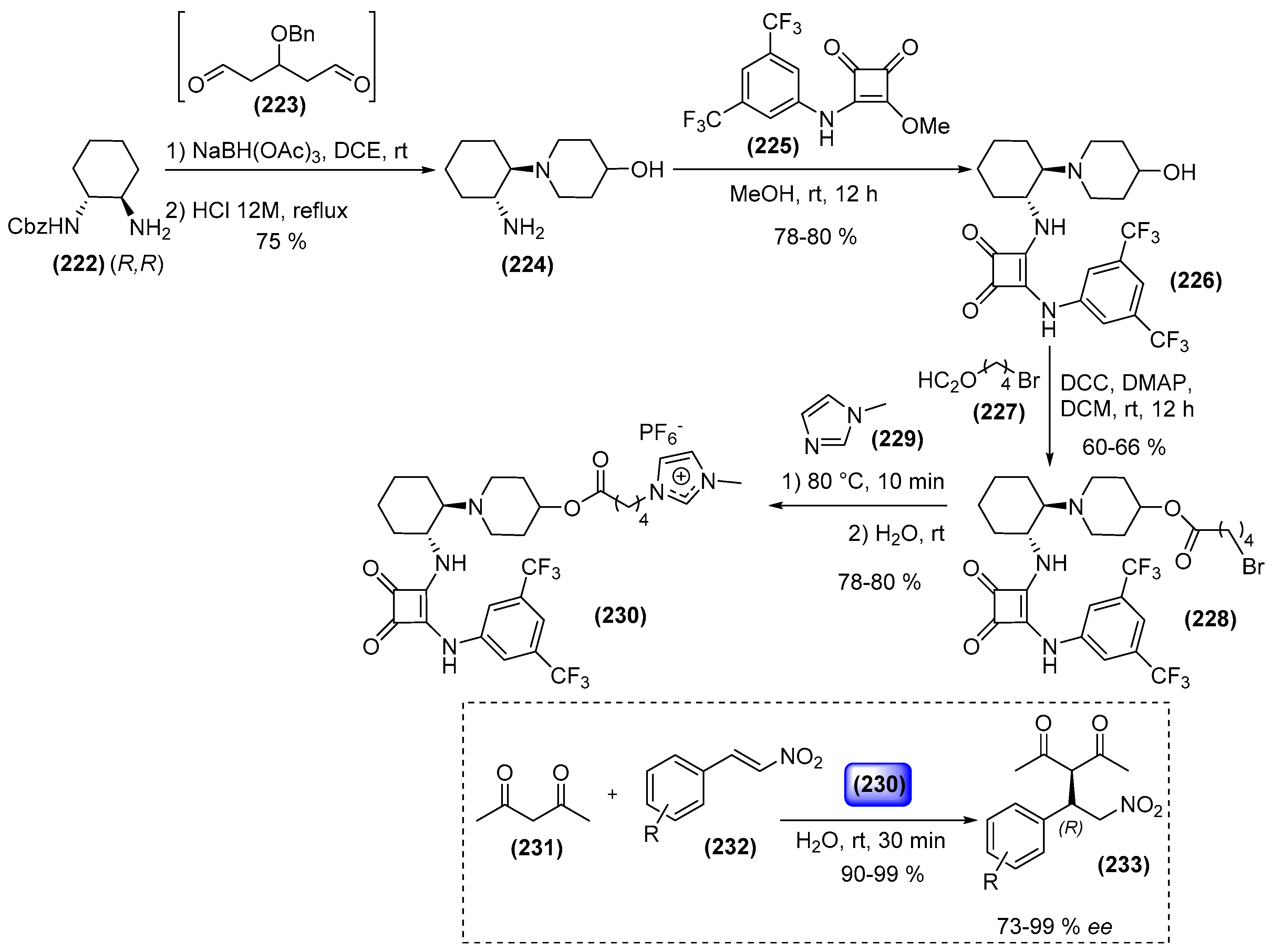
7. Immobilized Organocatalysts
8. Ionic Liquids
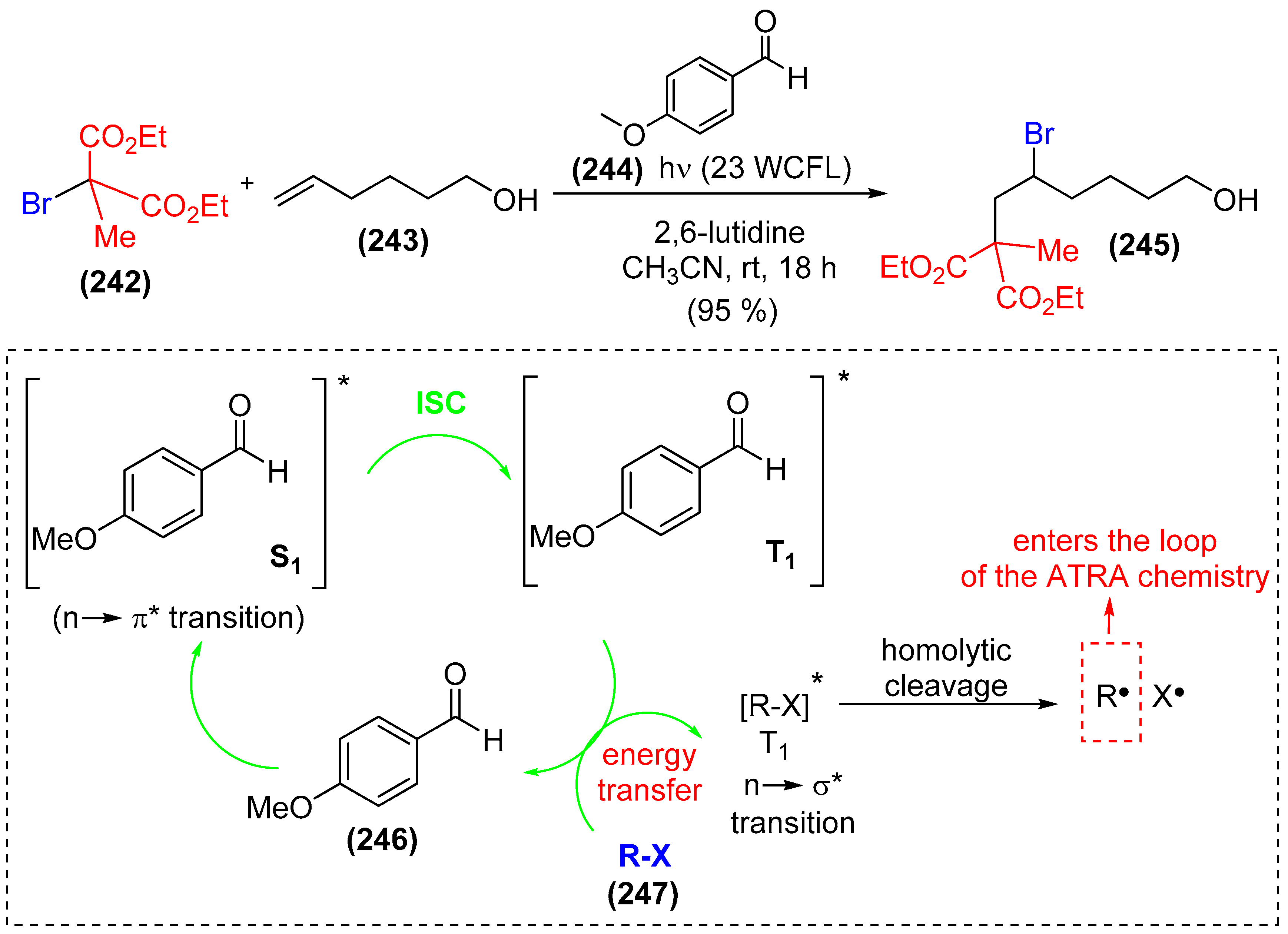
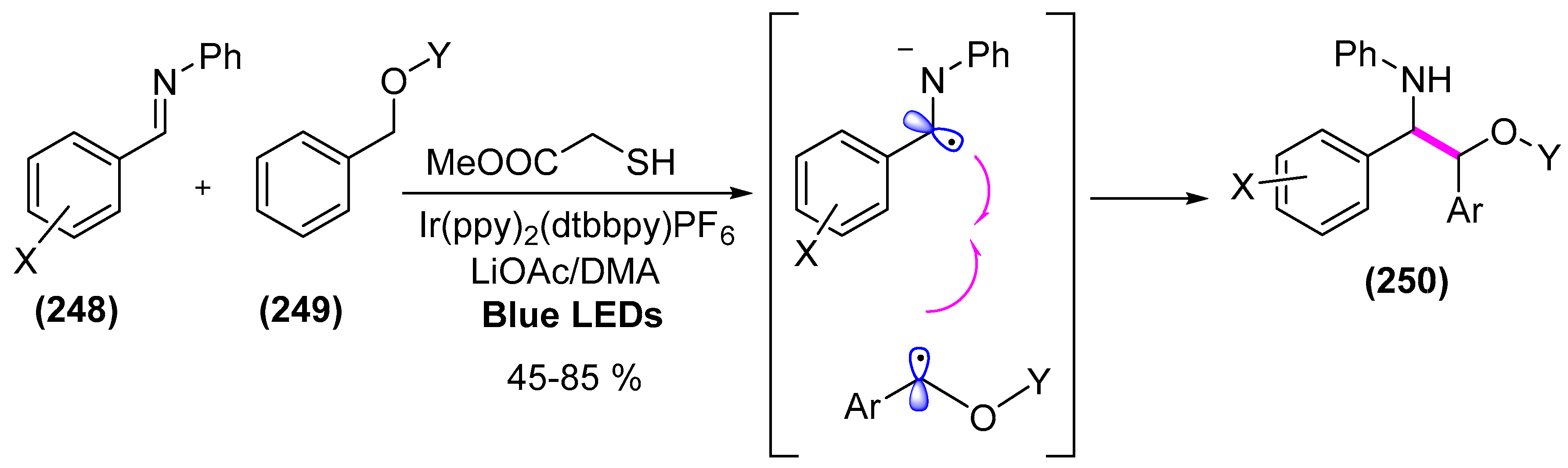
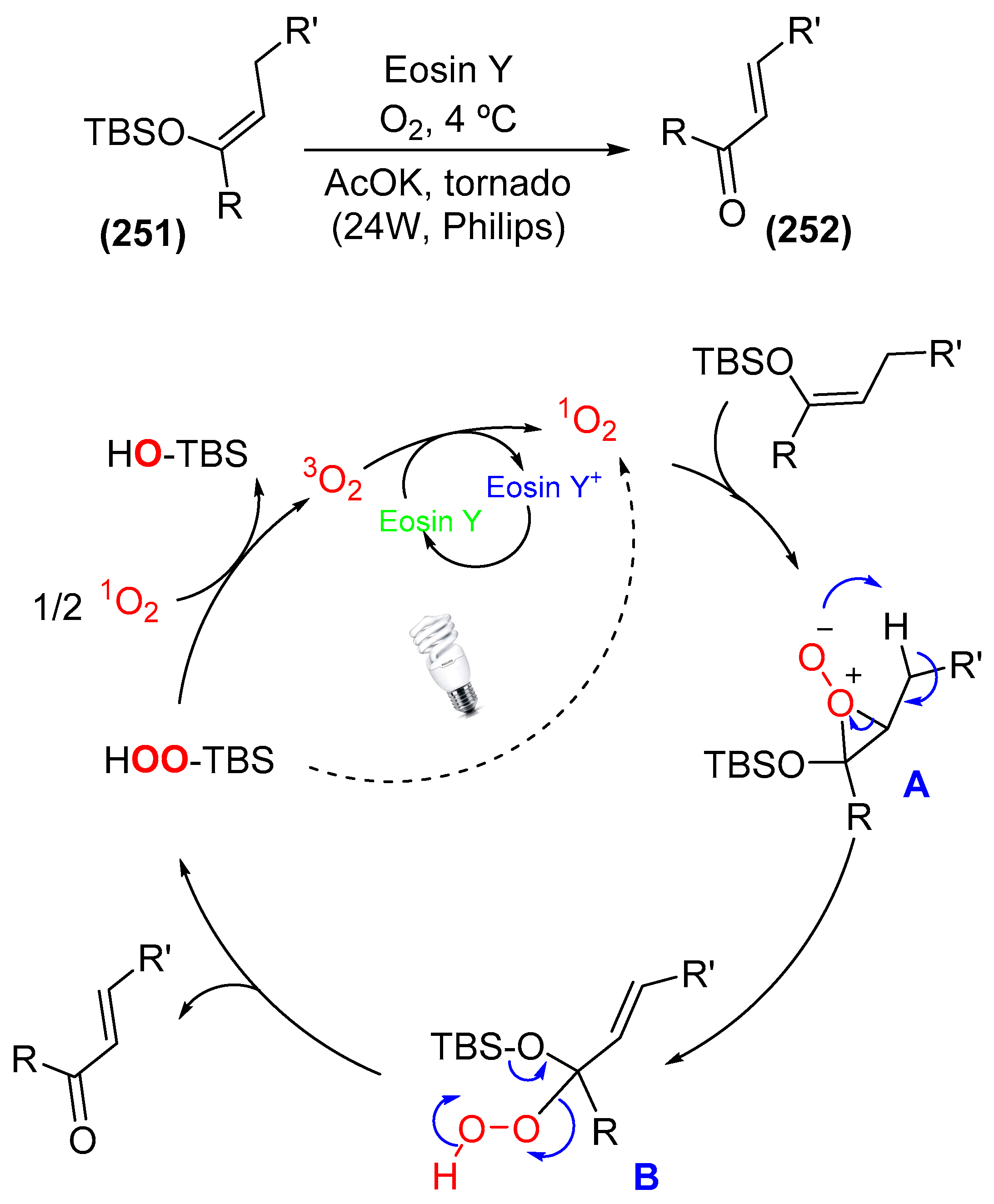
9. Photoorganocatalysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaikh, I.R. Organocatalysis: Key trends in green synthetic chemistry, challenges, scope towards Heterogenization, and importance from research and industrial point of view. J. Catal. 2014, 2014. [Google Scholar] [CrossRef]
- Govender, T.; Arvidsson, P.I.; Maguire, G.E.M.; Kruger, H.G.; Naicker, T. Enantioselective Organocatalyzed transformations of β-Ketoesters. Chem. Rev. 2016, 116, 9375–9437. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Bredig, G.; Fiske, W.S. Beiträge zur chemischen Physiologie und Pathologie. In Biochemische Zeitschrift; Springer: Berlin, Germany, 1912; Volume 46, p. 7. [Google Scholar]
- Dias, F.R.F.; Ferreira, V.F.; Cunha, A.C. Uma visão geral dos diferentes tipos de catálise em síntese orgânica. Rev. Virtual Quím. 2012, 4, 840–871. [Google Scholar]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic diels-alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Werkwijze Voor de Bereiding van 1,3-dioxycycloalkanen. German Patent DE 2,102,623, 12 August 1971. [Google Scholar]
- Hajos, Z.G.; Parrish, D.R. 2-Alkyl-2-(3-oxoalkyl)-1,3-cyclopentadiones. U.S. Patent 3,975,442, 9 December 1970. [Google Scholar]
- Eder, U.; Saeur, G.; Wiechert, R. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, L.; Zhou, P.; Luo, S.; Cheng, J. A practical protocol for asymmetric synthesis of Wieland-Miescher and Hajos-Parrish ketones catalyzed by a simple chiral primary amine. Synthesis 2013, 45, 1939–1945. [Google Scholar]
- Kumar, T.P.; Radhika, L.; Haribabu, K.; Kumar, V.N. Pyrrolidine-oxyimides: New chiral catalysts for enantioselective Michael addition of ketones to nitroolefins in water. Tetrahedron Asymmetry 2014, 25, 1555–1560. [Google Scholar] [CrossRef]
- Honarmand, M.; Naeimi, A.; Zahedifar, M. Nanoammonium salt: A novel and recyclable organocatalyst for one-pot threee-component synthesis of 2-amino-3-cyano-4H-pyran derivatives. J. Iran. Chem. Soc. 2017, 14, 1875–1888. [Google Scholar] [CrossRef]
- Caruana, L.; Kniep, F.; Johansen, T.K.; Poulsen, P.H.; Jørgensen, K.A. A new organocatalytic concept for asymmetric α-alkylation of aldehydes. J. Am. Chem. Soc. 2014, 136, 15929–15932. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-L.; Xue, J.-H.; Fu, L.-N.; Zhang, S.-E.; Guo, Q.-X. The direct asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols by enamine catalysis. Org. Lett. 2014, 16, 6472–6475. [Google Scholar] [CrossRef] [PubMed]
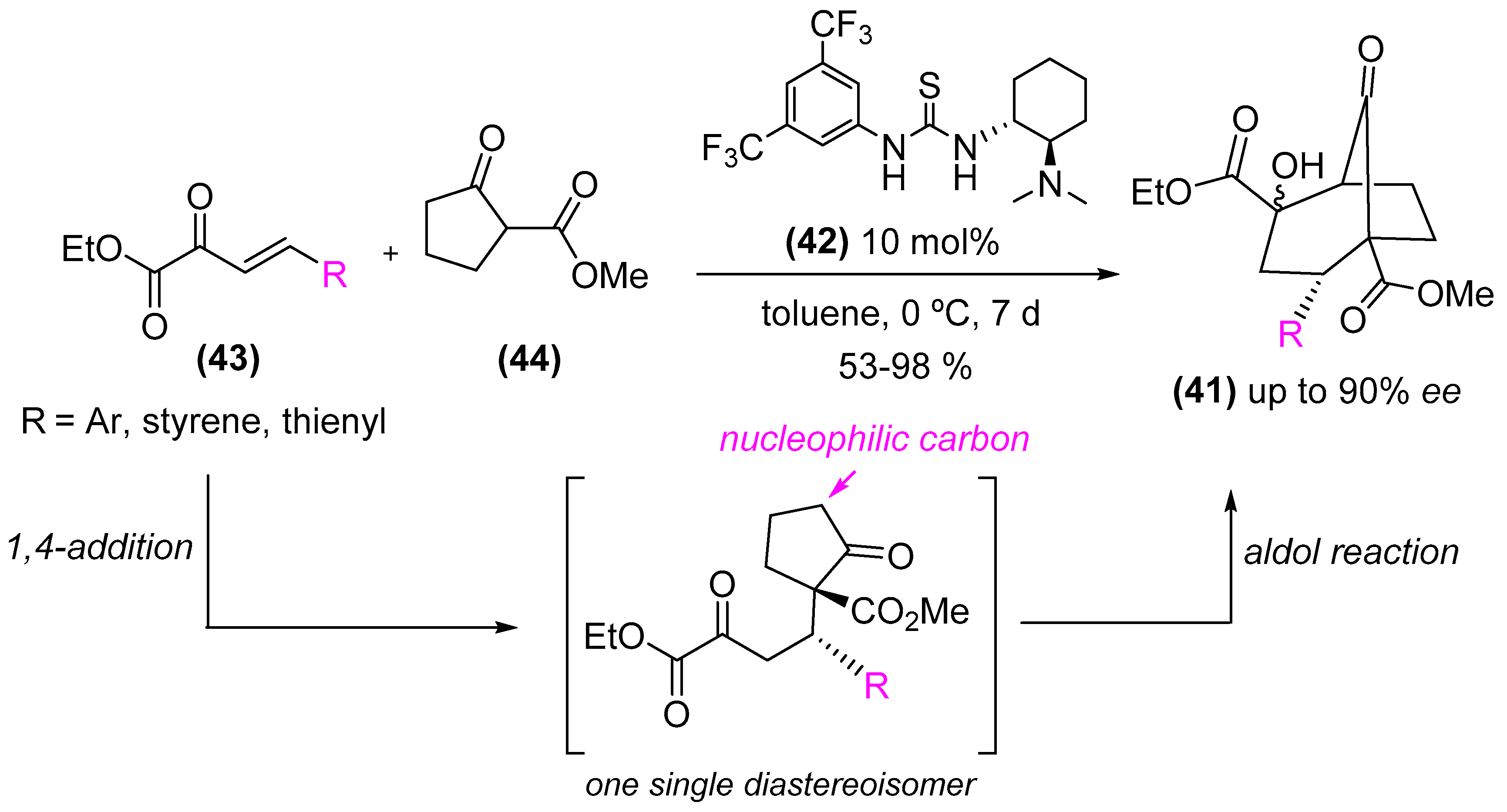
- Lefranc, A.; Gremaud, L.; Alexakis, A. Construction of bicyclo[3.2.1]octanes with four stereogenic center by organocatalytic domino Michael/Aldol reaction. Org. Lett. 2014, 16, 5242–5245. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef] [PubMed]
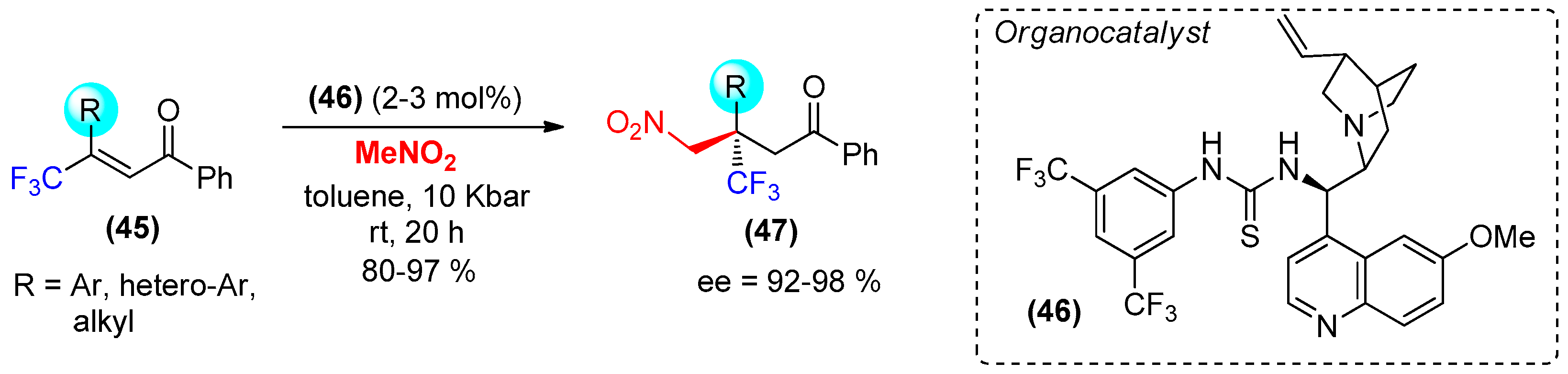
- Kwiatkowski, P.; Cholewiak, A.; Kasztelan, A. Efficient and highly enantioselective construction of trifluoromethylated quaternary stereogenic centers via high-pressure mediated organocatalytic conjugate addition of nitromethane to β,β-disubstituted enones. Org. Lett. 2014, 16, 5930–5933. [Google Scholar] [CrossRef] [PubMed]
- Portalier, F.; Bourdreux, F.; Marrot, J.; Moreau, X.; Coeffard, V.; Greck, C. Merging oxidative dearomatization and aminocatalysis: One-pot enantioselective synthesis of tricyclic architectures. Org. Lett. 2013, 15, 5642–5645. [Google Scholar] [CrossRef] [PubMed]
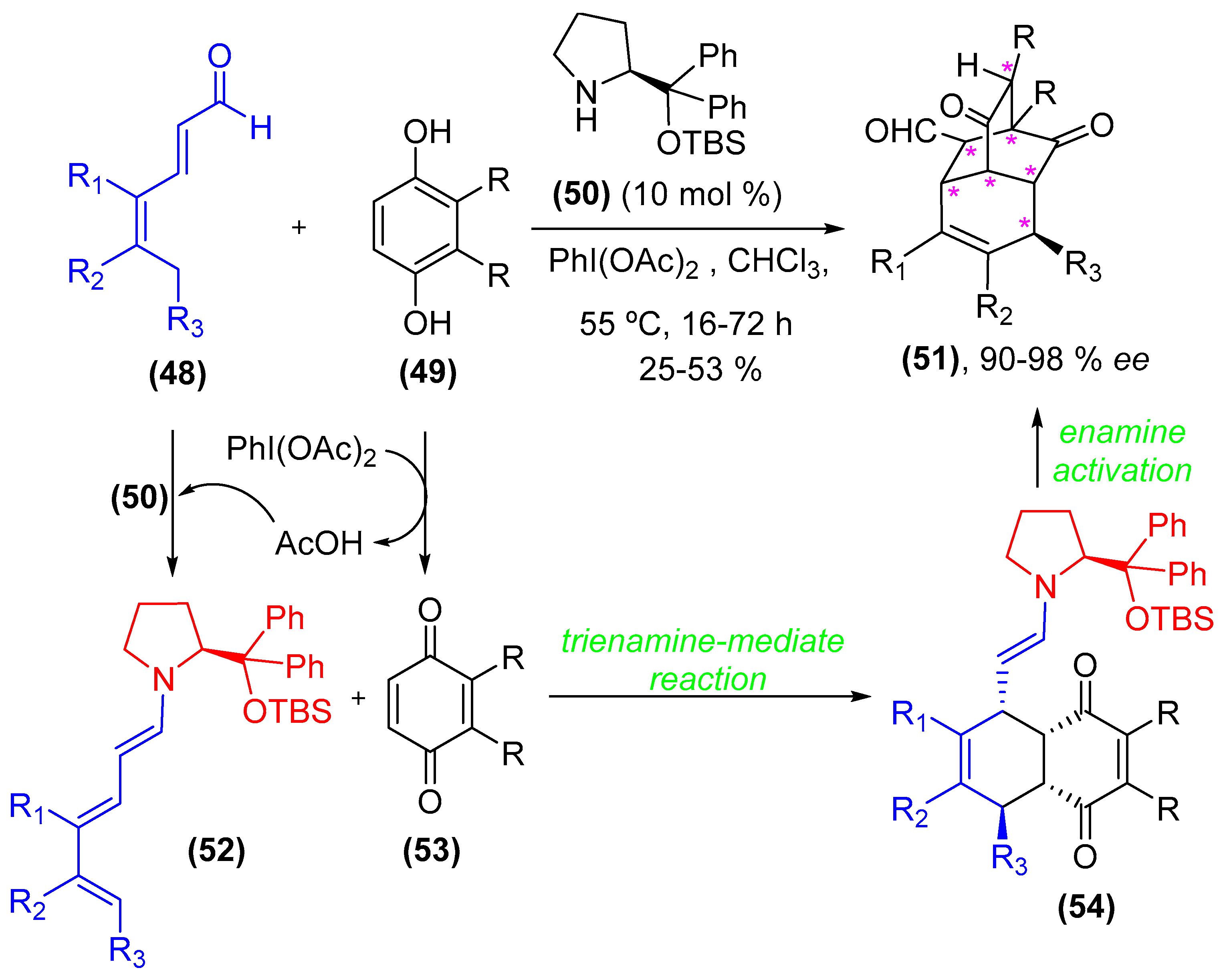
- Xiong, X.-F.; Zhou, Q.; Gu, J.; Dong, L.; Liu, T.-Y.; Chen, Y.-C. Trienamine catalysis with 2,4-dienones: Development and application in asymmetric Diels-Alder reactions. Angew. Chem. Int. Ed. 2012, 51, 4401–4404. [Google Scholar] [CrossRef] [PubMed]
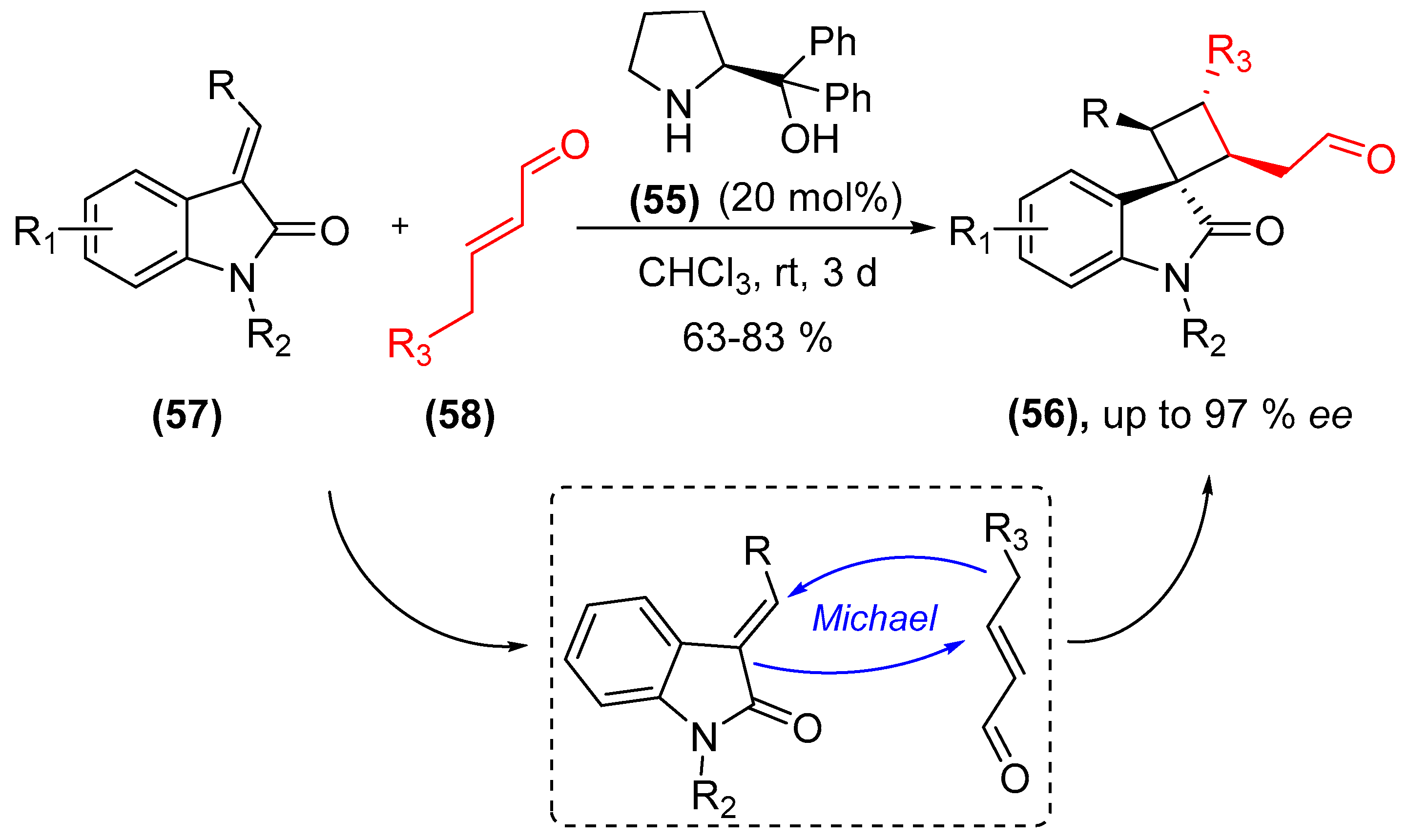
- Qi, L.-W.; Yang, Y.; Gui, Y.-Y.; Zhang, Y.; Chen, F.; Tian, F.; Peng, L.; Wang, L.-X. Asymmetric synthesis of 3,3′-spirooxindoles fused with cyclobutanes through organocatalytic formal [2+2] cycloadditions under H-bond-directing dienamine activation. Org. Lett. 2014, 16, 6436–6439. [Google Scholar] [CrossRef] [PubMed]
- Groenendaal, B.; Ruijter, E.; Orru, R.V.A. 1-Azadienes in cycloaddition and multicomponent reactions towards N-heterocycles. Chem. Commun. 2008, 5474–5489. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Gu, J.; Teng, B.; Zhou, Q.-Q.; Li, R.; Chen, Y.-C. 1-Azadienes as regio- and chemoselective dienophiles in aminocatalytic asymmetric DielsAlder reaction. Org. Lett. 2013, 15, 6206–6209. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-R.; Liu, Q.; Wang, J.; Xiao, J.-A.; Yang, H. Solvent-effects tuning the catalytic reactivity of prolinamides in asymmetric aldol reactions. Tetrahedron Asymmetry 2014, 25, 1590–1598. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, J.; Wei, K. Aromatic l-prolinamide-catalyzed asymmetric Michael addition of aldehydes to nitroalkenes. Tetrahedron Asymmetry 2014, 25, 1599–1604. [Google Scholar] [CrossRef]
- Chen, N.; Zhu, L.; Gan, L.; Liu, Z.; Wang, R.; Cai, X.; Jiang, X. Asymmetric synthesis of bispiro-[γ-butyrolactone-pyrrolidin-4,4’-pyrazolone] scaffolds containing two quaternary spirocenters via an organocatalytic 1,3-dipolar cycloaddition. Eur. J. Org. Chem. 2018, 2018, 2939–2943. [Google Scholar] [CrossRef]
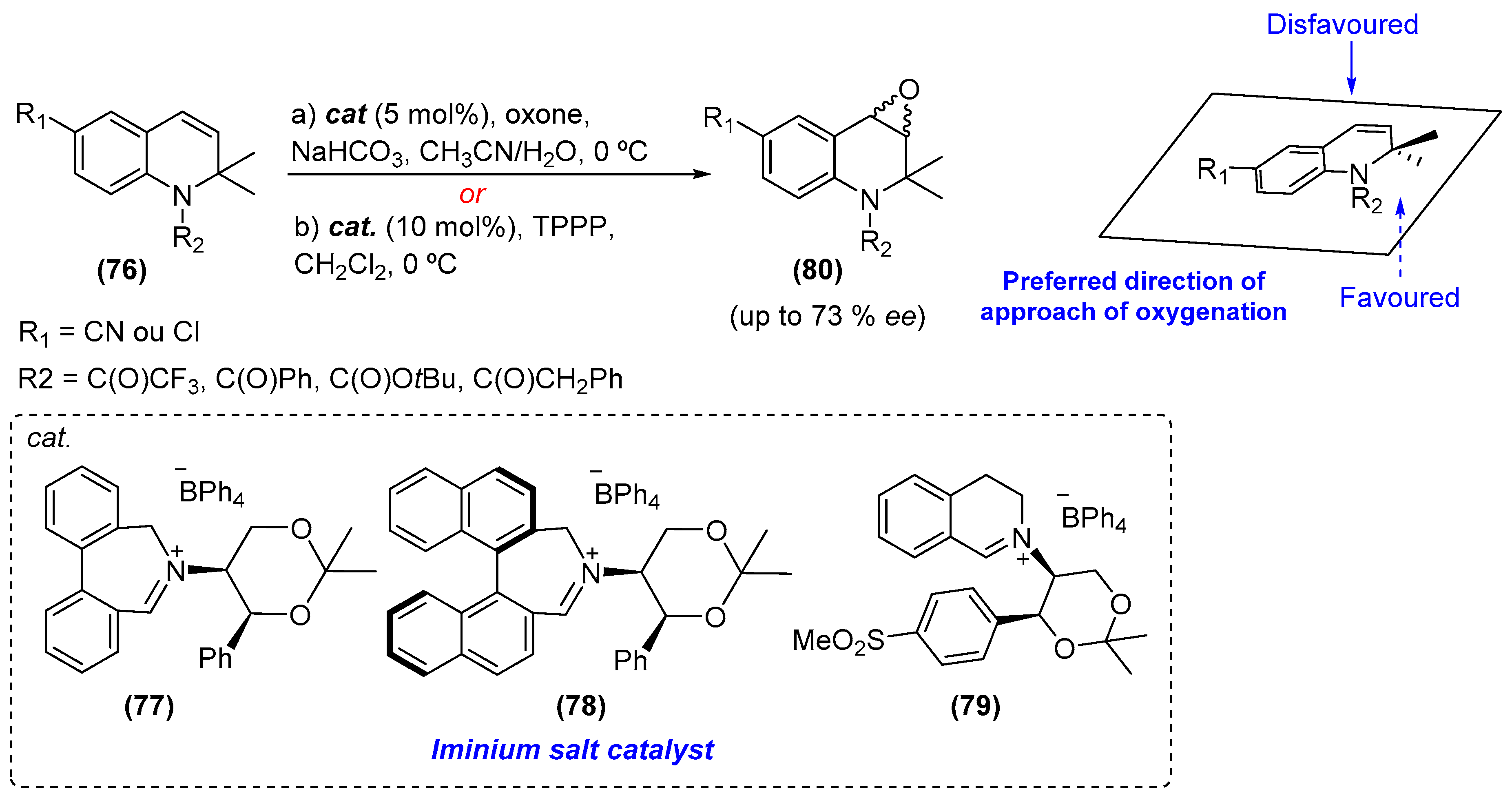
- Page, P.C.B.; Day, D.P.; Chan, Y. Enantioselective epoxidation of dihydroquinolines by using iminium salt organocatalysts. Eur. J. Org. Chem. 2014, 2014, 8029–8034. [Google Scholar] [CrossRef]
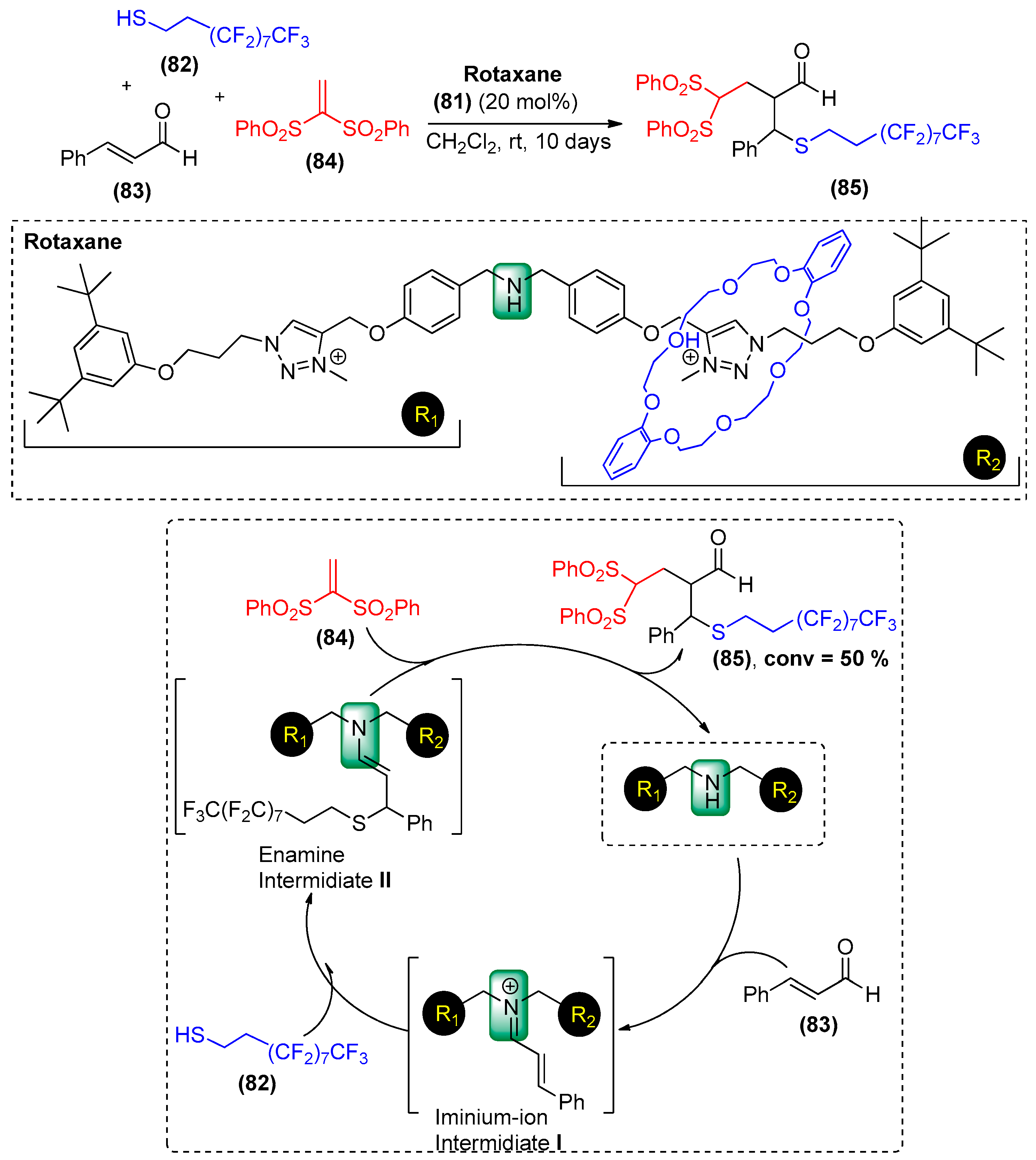
- Blanco, V.; Carlone, A.; Hänni, K.D.; Leigh, D.A.; Lewandowski, B. A rotaxane-based switchable organocatalyst. Angew. Chem. Int. Ed. 2012, 51, 5166–5169. [Google Scholar] [CrossRef] [PubMed]
- Blanco, V.; Leigh, D.A.; Lewandowska, U.; Lewandowski, B.; Marcos, V. Exploring the activation modes of a rotaxane-based switchable organocatalyst. J. Am. Chem. Soc. 2014, 136, 15775–15780. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J.; Harlow, R.L.; Hline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
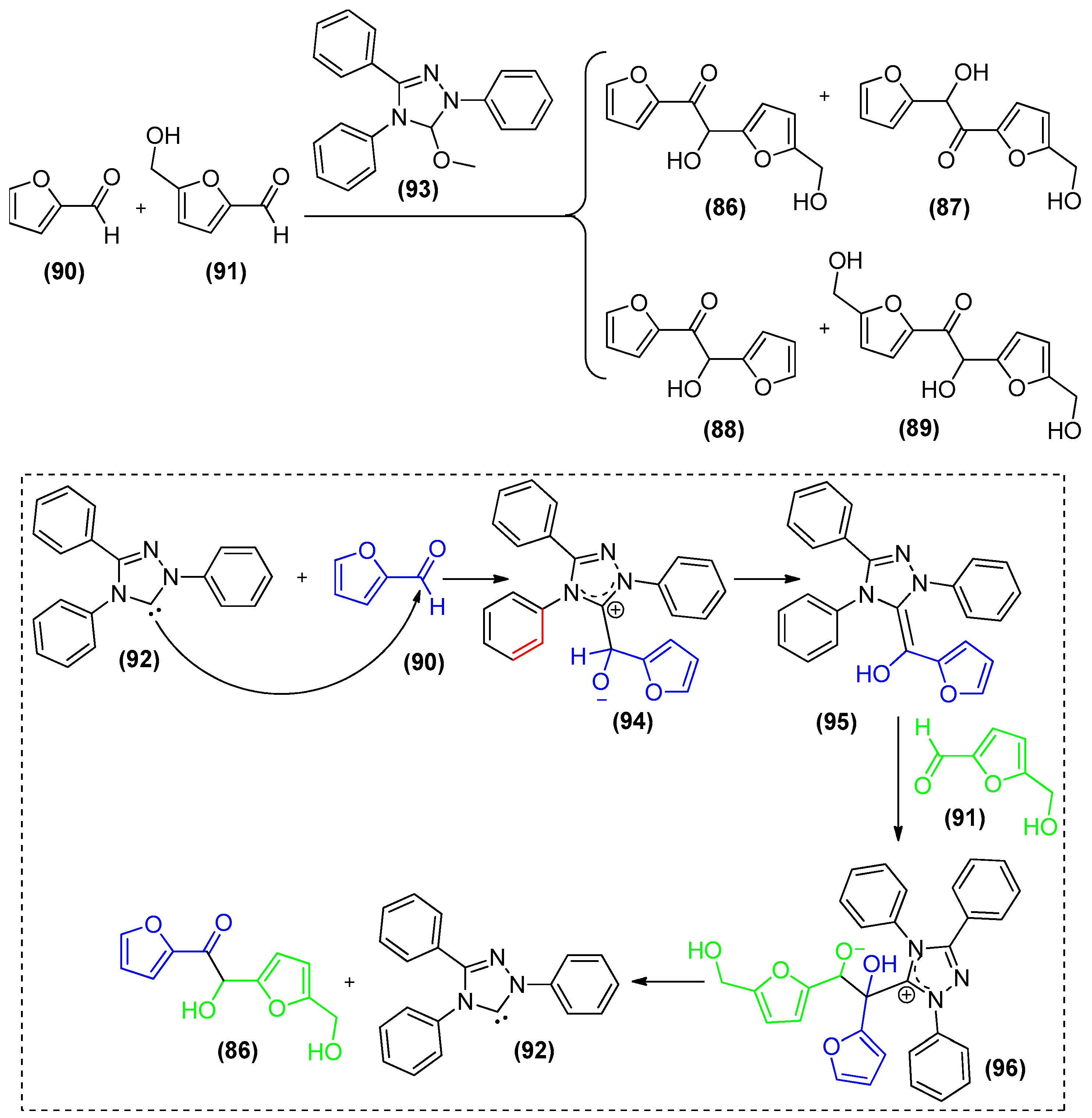
- Wilson, J.; Chen, E.Y.X. Organocatalytic cross-coupling of biofuranic to multifunctional difuranic C11 building blocks. ACS Sustain. Chem. Eng. 2016, 4, 4927–4936. [Google Scholar] [CrossRef]
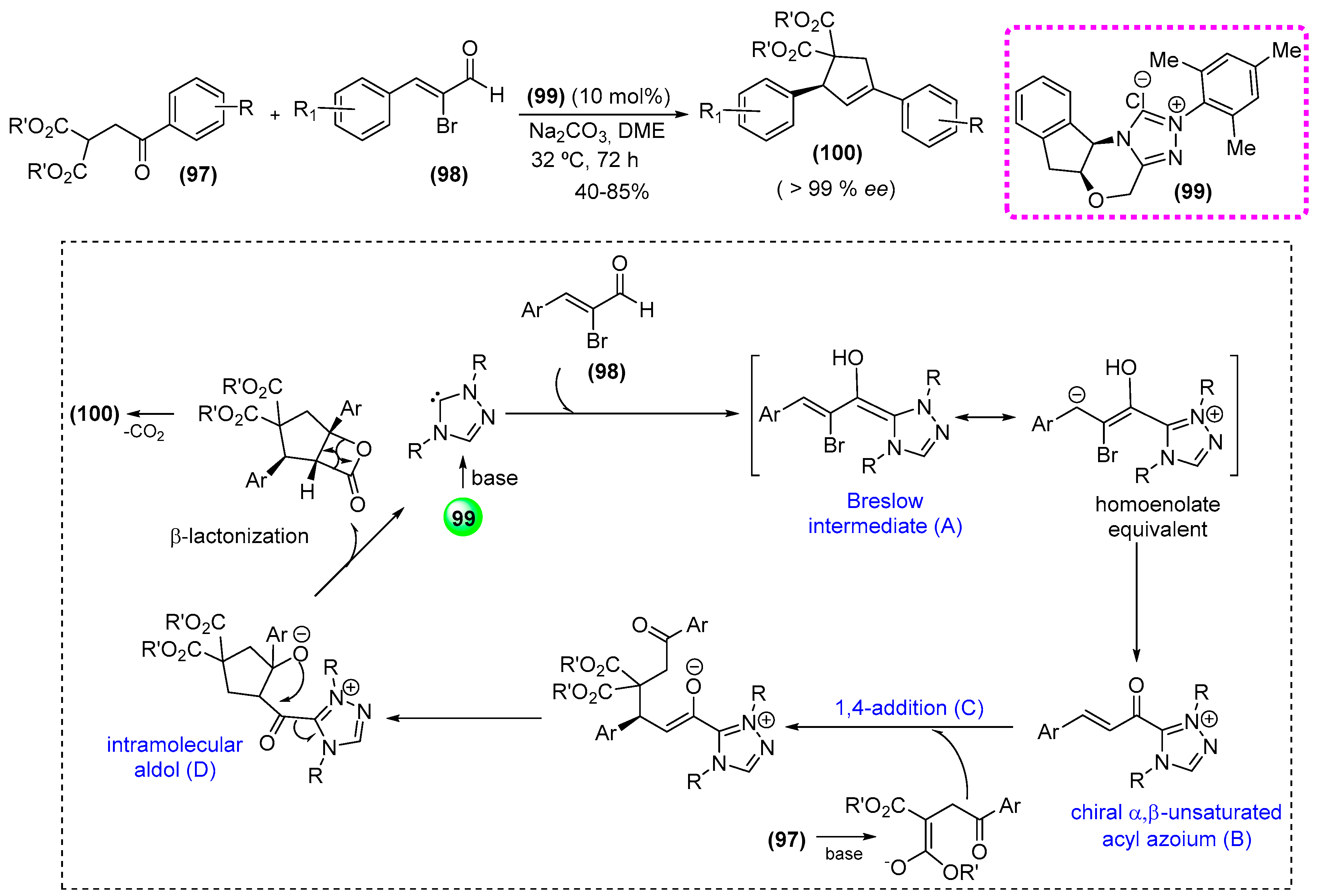
- Mondal, S.; Yetra, S.R.; Patra, A.; Kunte, S.S.; Gonnadeb, R.G.; Biju, A.T. N-Heterocyclic carbene-catalyzed enantioselective synthesis of functionalized cyclopentenes via a,b-unsaturated acyl azoliums. Chem. Commun. 2014, 50, 14539–14542. [Google Scholar] [CrossRef] [PubMed]
- Struble, J.R.; Bode, J.W. Synthesis of a N-mesityl substituted aminoindanol-derived triazolium salt [(5aS,10bR)-5a,10b-Dihydro-2-(2,4,6-trimethylphenyl)-4H,6H-indeno[2,1-b]-1,2,4-triazolo[4,3-d]-1,4-oxazinium chloride]. Org. Synth. 2010, 87, 362–376. [Google Scholar]
- Szucs, T. Cilazapril. A review. Drugs 1991, 41, 18–24. [Google Scholar] [CrossRef] [PubMed]
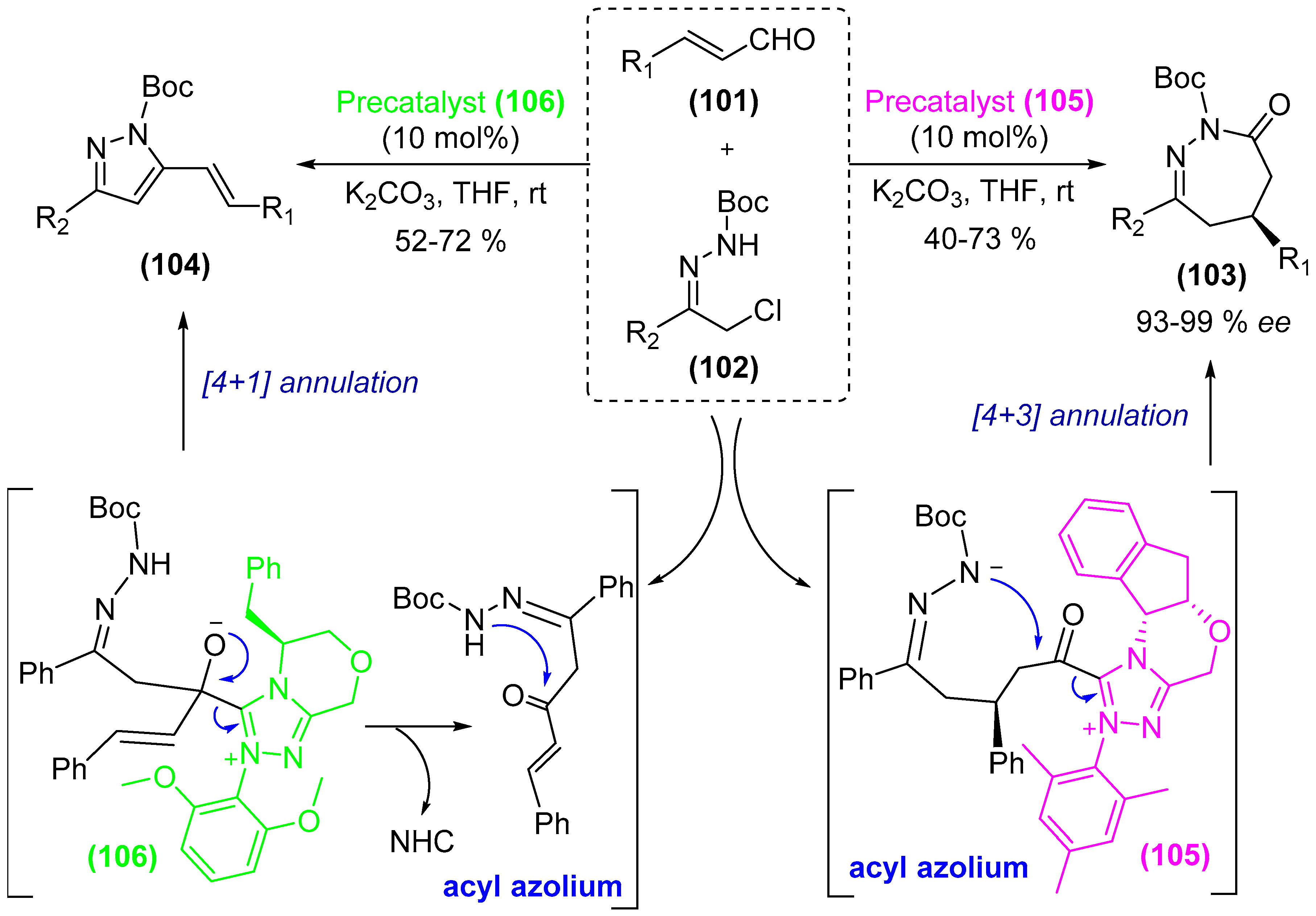
- Guo, C.; Sahoo, B.; Daniliuc, C.G.; Glorius, F. N-heterocyclic carbene catalyzed switchable reactions of enals with azoalkenes: Formal [4+3] and [4+1] annulations for the synthesis of 1,2-diazepines and pyrazoles. J. Am. Chem. Soc. 2014, 136, 17402–17405. [Google Scholar] [CrossRef] [PubMed]
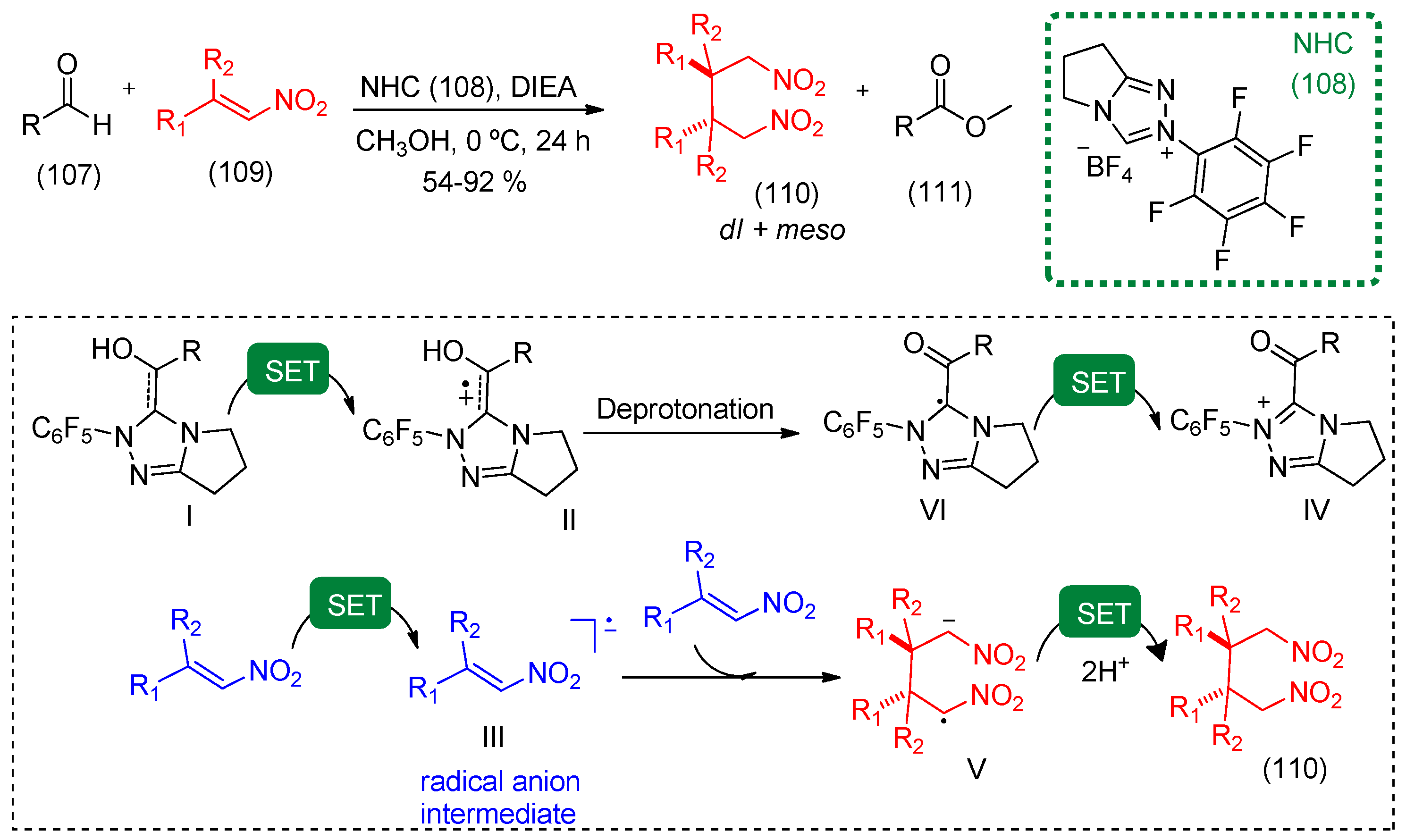
- Du, Y.; Wang, Y.; Li, X.; Shao, Y.; Li, G.; Webster, R.D.; Chi, Y.R. N-heterocyclic carbene organocatalytic reductive β,β-coupling reactions of nitroalkenes via radical intermediates. Org. Lett. 2014, 16, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef] [PubMed]
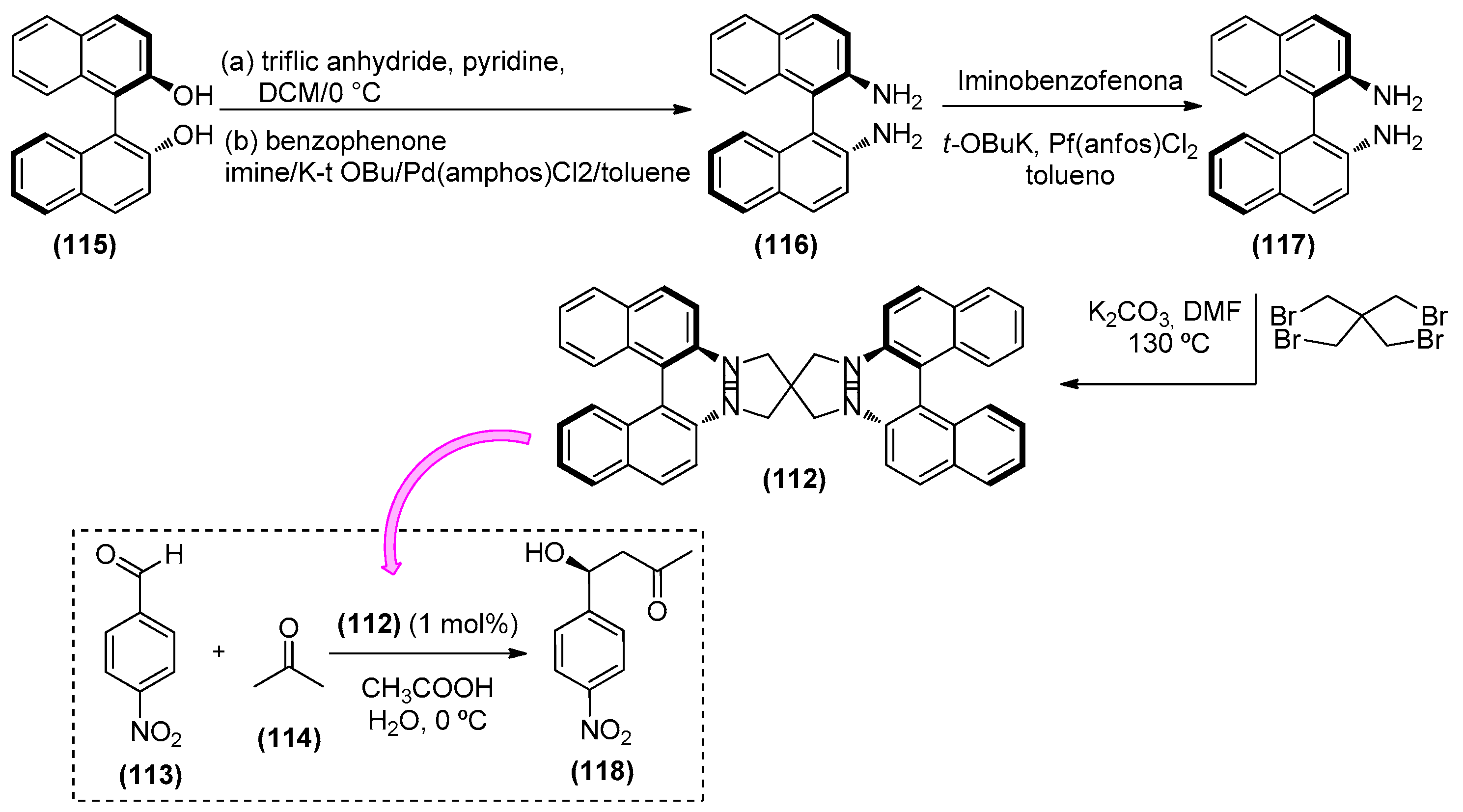
- Ashokkumar, V.; Chithiraikumar, C.; Siva, A. Binaphthyl-based chiral bifunctional organocatalysts for water mediates asymmetric List-Lerner-Barbas aldol reactions. Org. Biomol. Chem. 2016, 14, 9021–9032. [Google Scholar] [CrossRef] [PubMed]
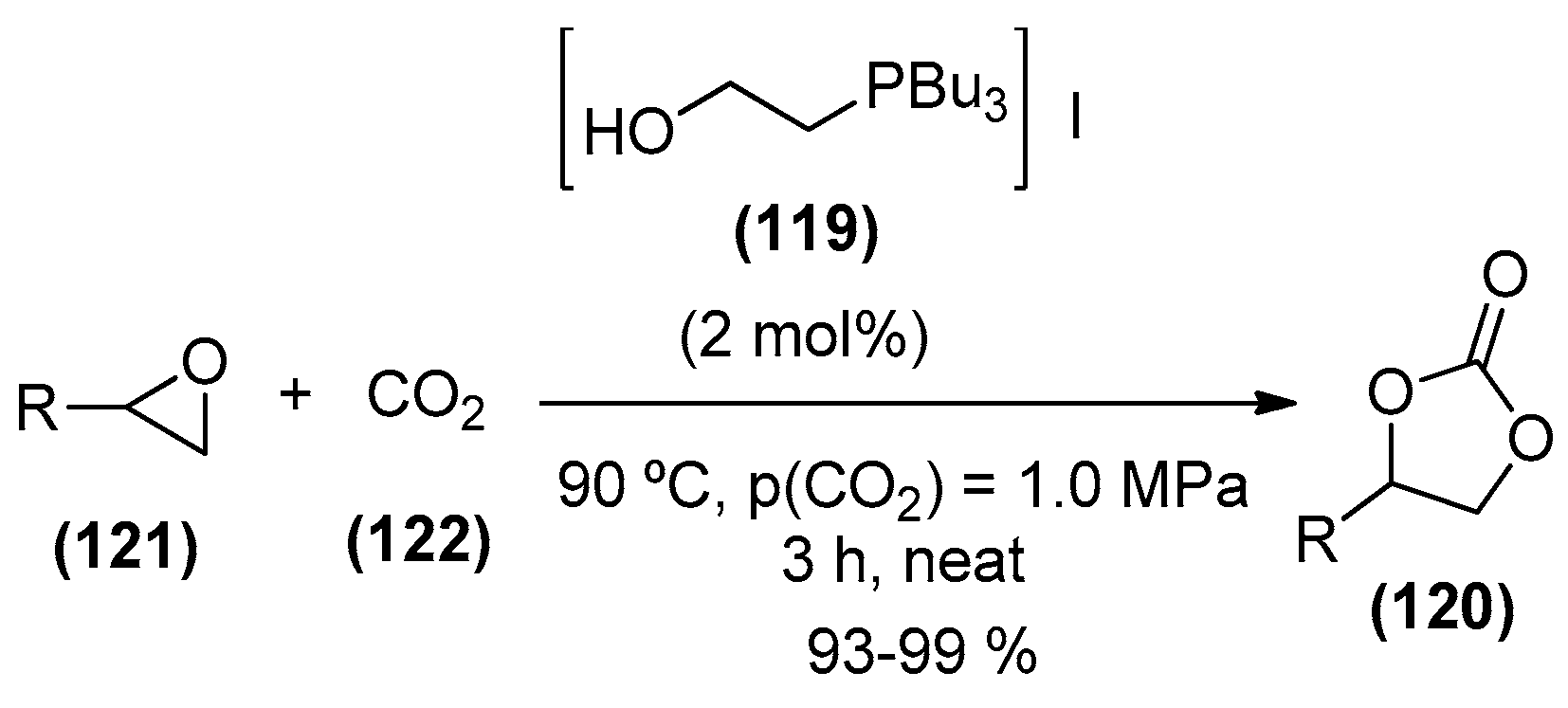
- Werner, T.; Buttner, H. Phosphorus-based bifunctional organocatalysts for the addition of carbon dioxide and epoxides. ChemSusChem 2014, 7, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Pan, J.; Wang, Y.; Zhou, Z. Stereocontrolled construction of the 3,4-dihydrothiacarbazol-2(9H)-one skeleton by using bifunctional squaramide-catalyzed cascade reactions. Eur. J. Org. Chem. 2014, 35, 7940–7947. [Google Scholar] [CrossRef]
- Sherrington, D.C.; Taskinen, K.A. Self-assembly in synthetic macromolecular systems via multiple hydrogen bonding interactions. Chem. Soc. Rev. 2001, 30, 83–93. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Sun, C.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric phase-transfer catalysts bearing multiple hydrogen-bonding donors: Highly efficient catalysts for enantio- and diastereoselective nitro-Mannich reaction of amidosulfones. Org. Lett. 2014, 16, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Tan, W.; Zhang, H.-H.; Li, M.; Ye, Q.; Ma, G.-H.; Tu, S.-J.; Lib, G. Asymmetric organocatalytic tandem cyclization/transfer hydrogenation: A synthetic strategy for enantioenriched nitrogen heterocycles. Adv. Synth. Catal. 2013, 355, 3715–3726. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer hydrogenation with hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Kim, J.; Him, H.; Cho, H.J.; Baik, M.; Kim, Y. Scorpionate catalysts for coupling CO2 and epoxides to cyclic carbonates: A rational design approach for organocatalysts. J. Org. Chem. 2018, 83, 9370–9380. [Google Scholar] [CrossRef] [PubMed]
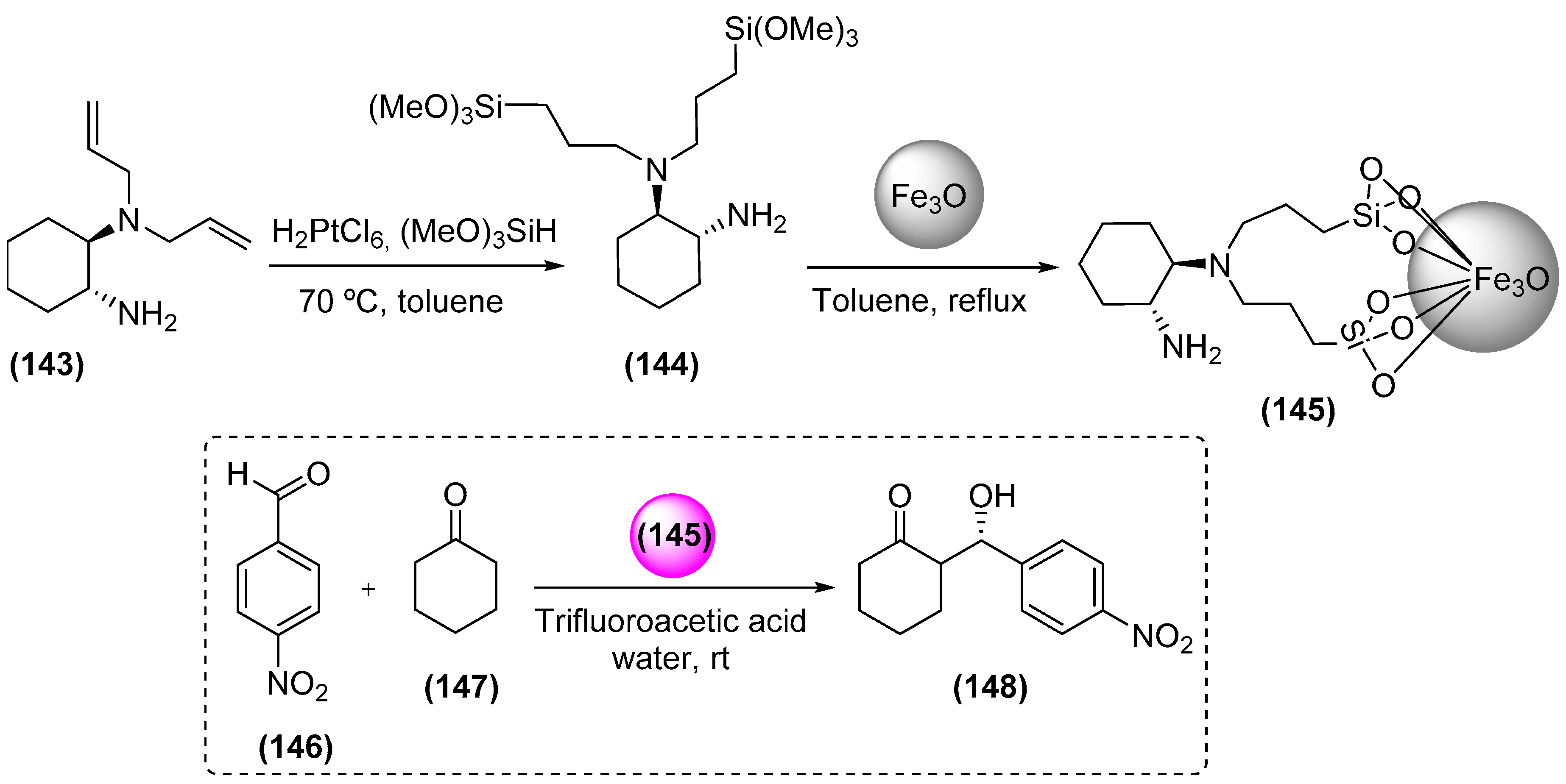
- Mrówczynski, R.; Nan, A.; Liebscher, J. Magnetic nanoparticle-supported organocatalysts- na eficiente way of recycling and use. RSC Adv. 2014, 4, 5927–5952. [Google Scholar] [CrossRef]
- Luo, S.Z.; Zheng, X.X.; Cheng, J.P. Asymmetric bifunctional primary aminocatalysis on magnetic nanoparticles. Chem. Commun. 2008, 5719–5721. [Google Scholar] [CrossRef] [PubMed]
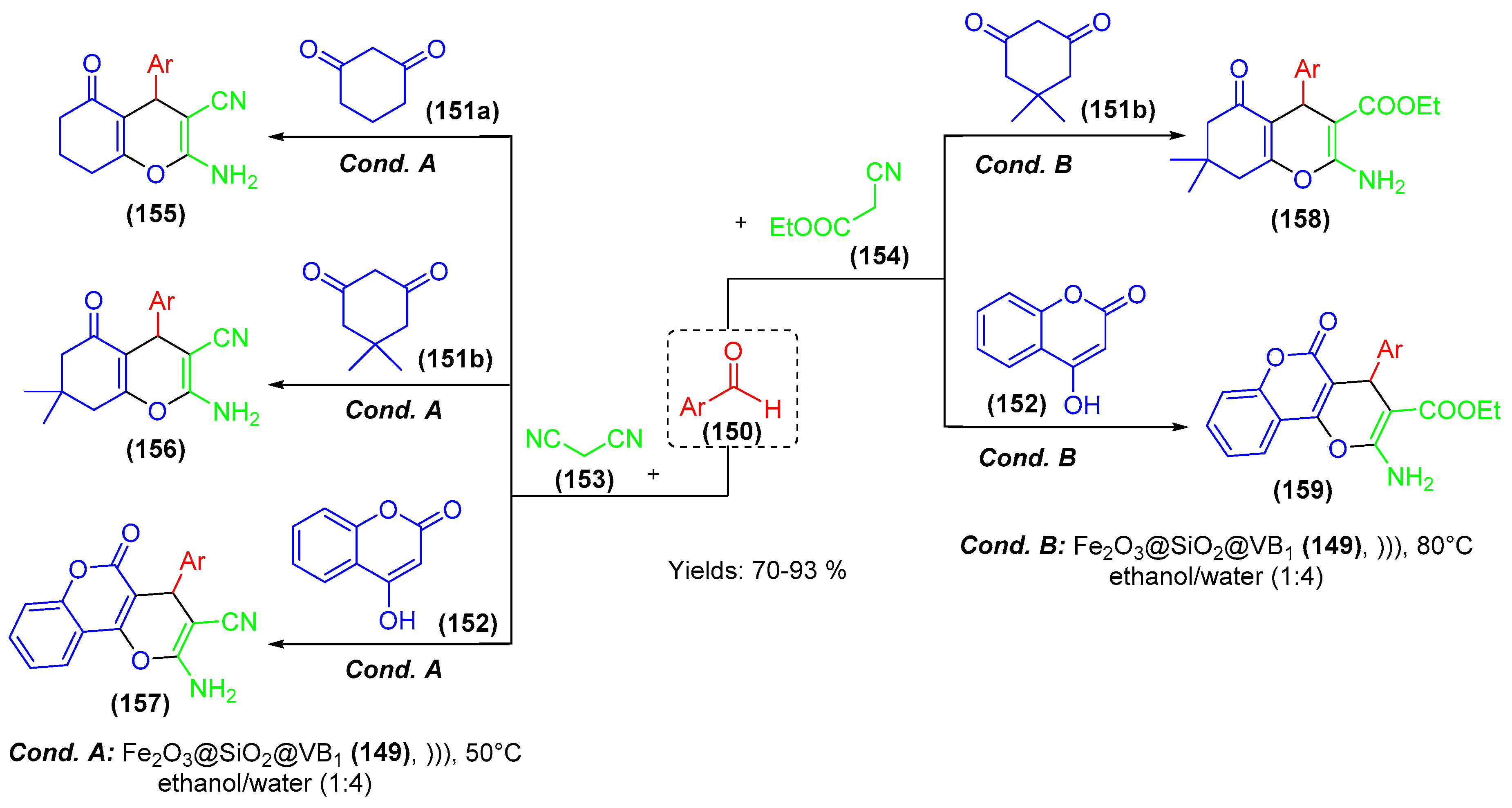
- Nongrum, R.; Nongthombam, G.S.; Kharkongor, M.; Rani, J.W.S.; Rahman, N.; Kathing, C.; Myrboh, B.; Nongkhlaw, R. A nano-organo catalyzed route towards the efficient synthesis of benzo[b]pyran derivatives under ultrasonic irradiation. RSC Adv. 2016, 6, 108384–108392. [Google Scholar] [CrossRef]
- Singh, N.G.; Lily, M.; Devi, S.P.; Rahman, N.; Ahmed, A.; Chandra, A.K.; Nongkhlaw, R. Synthetic, mechanistic and kinetic studies on the organo-nanocatalyzed synthesis of oxygen and nitrogen containing spiro compounds under ultrasonic conditions. Green Chem. 2016, 18, 4216–4227. [Google Scholar] [CrossRef]
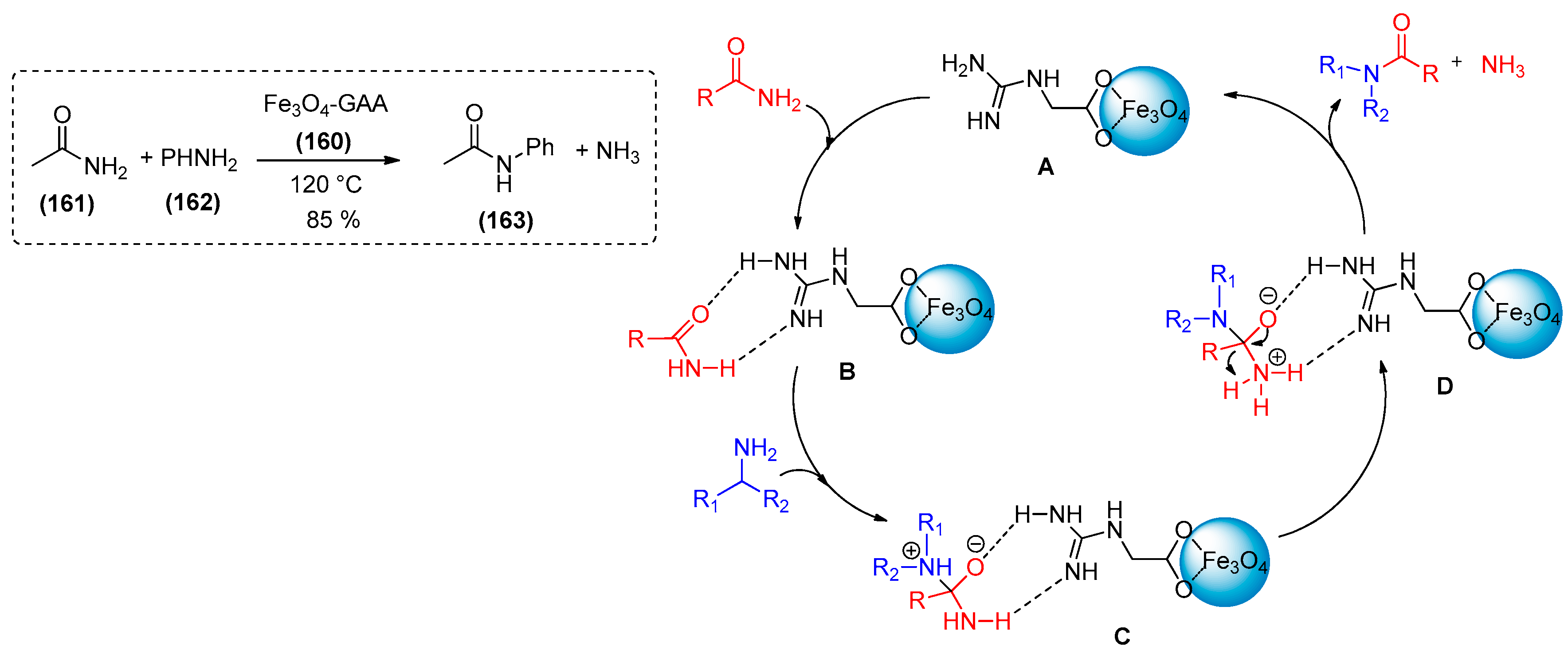
- Miraki, M.K.; Arefi, M.; Yazdani, E.; Abbasi, S.; Karimi, M.; Azizi, K.; Heydari, A. Guanidine acetic acid functionalized magnetic nanoparticles: Recoverable green catalyst for transamidation. Chem. Sel. 2016, 1, 6328–6333. [Google Scholar]
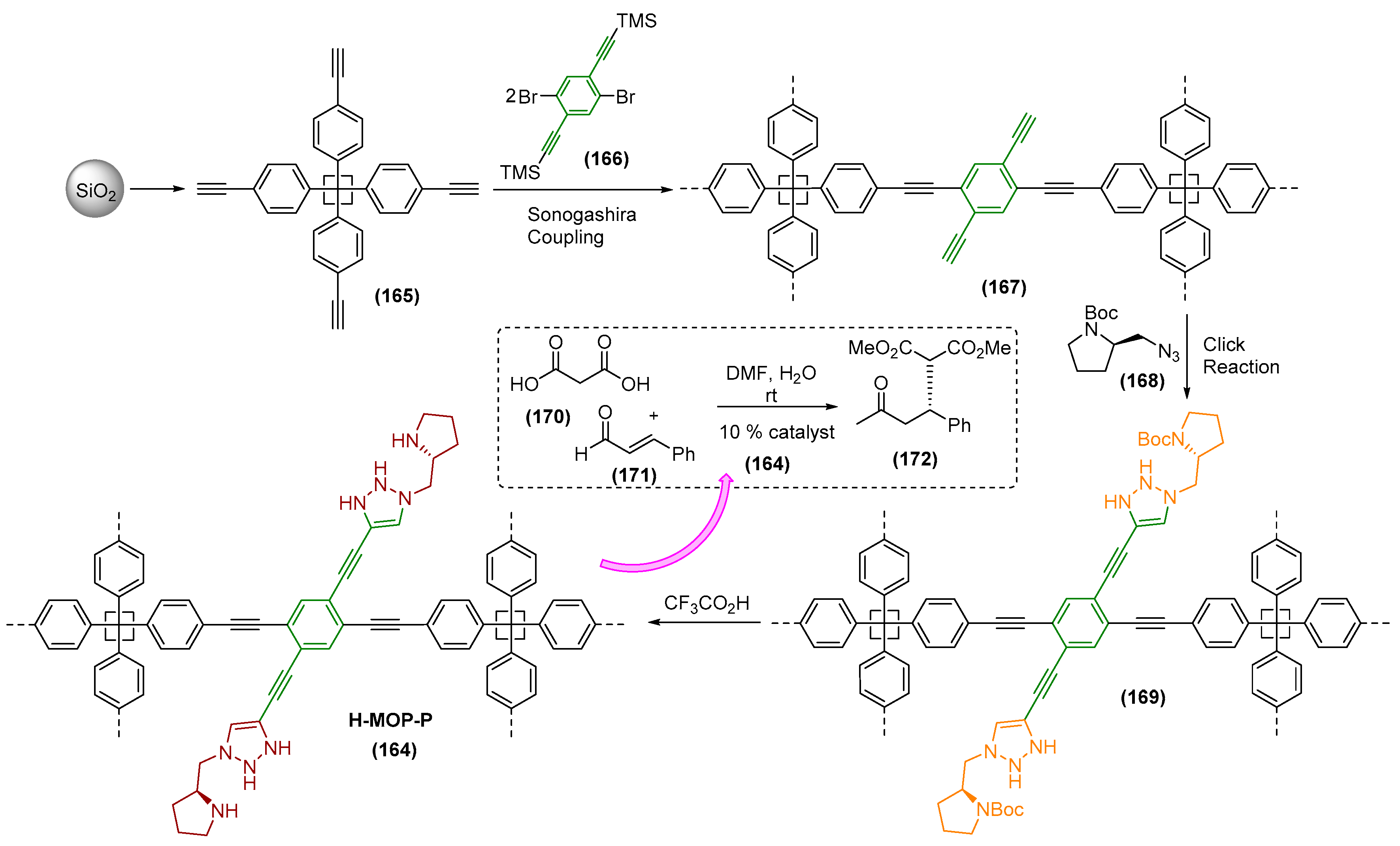
- Cho, K.; Yoo, J.; Noh, H.; Lee, S.M.; Kim, H.J.; Ko, Y.; Jang, H.; Son, S. Hollow structural effect of microporous organocatalytic polymers with pyrrolidines: Dramatic enhancement of catalytic performance. J. Mater. Chem. A 2017, 5, 8922–8926. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bonh, E. Controlled growth of monodispersed silica spheres in the micro size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Li, K.; Wu, X.; Gu, Q.; Zhao, X.; Yuan, M.; Ma, W.; Ni, W.; Hou, Z. Inclusion complexes of organic salts with β-cyclodextrin as organocatalysts for CO2 cycloaddition with epoxides. RSC Adv. 2017, 7, 14721–14732. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Annunziata, R.; Puglisi, A.; Celentano, G. Solid supported chiral n-picolylimi dazolidinones: Recyclable catalysts fot the enantioselective, metal- and hydrogen-free reduction of imines in batch and in flow mode. Adv. Synth. Catal. 2017, 359, 2375–2382. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A.; Mandoli, A.; Gualandi, A.; Cozzi, P.G. A catalytic reactor for the organocatalyzed enantioselective continuous flow alkylation of aldehydes. ChemSusChem 2014, 7, 3534–3540. [Google Scholar] [CrossRef] [PubMed]
- Munirathinam, R.; Leoncini, A.; Huskens, J.; Wormeester, H.; Verboom, W. Wall-coated polymer brushes as support for chiral organocatalysts in microreactors. J. Flow Chem. 2015, 5, 37–42. [Google Scholar] [CrossRef]
- Metternich, J.B.; Sagebiel, S.; Lückener, A.; Lamping, S.; Ravoo, B.J.; Gilmour, R. Covalent immobilization of (−)-riboflavin on polymer functionalized silica particles: Application in the photocatalytic E/Z isomerization of polarized alkenes. Chem. Eur. J. 2018, 24, 4228–4233. [Google Scholar] [CrossRef] [PubMed]
- Sagebiel, S.; Stricker, L.; Engel, S.; Ravoo, B.J. Self-assembly of colloidal molecules that respond to light and a magnetic field. Chem. Commun. 2017, 53, 9296–9299. [Google Scholar] [CrossRef] [PubMed]
- Billiet, S.; De Bruycker, K.; Driessen, F.; Goossens, H.; van Speybroeck, V.; Winne, J.M.; Du Prez, F.E. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems. Nat. Chem. 2014, 6, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.M.; Losada, J.; Maestro, A.; Rodríguez-Ferrer, P.; Pedrosa, R. Supported and unsupported chiral squaramides as organocatalysts fpr steroselective Miachel additions: Synthesis of eantiopure chromenes and spirochromanes. J. Org. Chem. 2017, 82, 8444–8454. [Google Scholar] [CrossRef] [PubMed]
- Osorio-planes, L.; Rodriguez-Escrich, C.; Pericás, M.A. Removing the superfluous: A supportedsquaramide catalyst with a minimalistic linker applied to the enantioselective flow synthesis of pyranonaphthoquinones. Cat. Sci. Technol. 2016, 6, 4686–4689. [Google Scholar] [CrossRef]
- Neves, C.M.S.S.; Silva, A.M.S.; Fernandes, A.M.; Coutinho, J.A.P.; Freire, M.G. Toward an understanding of the mechanisms behind the formation of liquid-liquid systems formed by two ionic liquids. J. Phys. Chem. Lett. 2017, 8, 3015–3019. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.D.; Singh, S. (L)-Prolinamide imidazolium hexafluorophosphate ionic liquid as an efficient reusable organocatalyst for direct asymmetric aldol reaction in solvent-free condition. RSC Adv. 2016, 6, 100459–100466. [Google Scholar] [CrossRef]
- Tukhvatshin, R.S.; Kucherenko, A.S.; Nelyubina, Y.V.; Zlotin, G. Tertiary amine-derived ionic liquid supported squaramide as a recyclable organocatalyst for noncovalent “on water” catalysis. ACS Catal. 2017, 7, 2981–2989. [Google Scholar] [CrossRef]
- Javle, B.R.; Kinage, A.K. Chiral amino-acid-amide based ionic liquids as a stereoselective organocatalyst in asymmetric transfer hydrogenation of acetophenone at room-temperature. Chem. Sel. 2018, 3, 2623–2625. [Google Scholar] [CrossRef]
- Arceo, E.; Montroni, E.; Melchiorre, P. Photo-organocatalysis of atom-transfer radical additions to alkenes. Angew. Chem. Int. Ed. 2014, 53, 12064–12068. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.G. Organometallic chemistry: C–H activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Hager, D.; MacMillan, D.W.C. Activation of C–H Bonds via the merger of photoredox and organocatalysis: A coupling of benzylic ethers with schiff bases. J. Am. Chem. Soc. 2014, 136, 16986–16989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Liu, Q.; Yang, Z.; Huang, Y. Synthesis of a,b-unsaturated carbonyl compounds via a visible-light-promoted organocatalytic aerobic oxidation. Chem. Commun. 2013, 49, 11662–11664. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233, 351–371. [Google Scholar] [CrossRef]
- Stracke, F.; Heupel, M.; Thiel, E. Singlet molecular oxygen photosensitized by Rhodamine dyes: Correlation with photophysical properties of the sensitizers. J. Photochem. Photobiol. A 1999, 126, 51–58. [Google Scholar] [CrossRef]
- Srivastava, V.P.; Yadav, A.K.; Yadav, L.D.S. Eosin Y catalyzed visible-light-driven aerobic oxidative cyclization of thioamides to 1,2,4-thiadiazoles. Synlett 2013, 24, 465–470. [Google Scholar] [CrossRef]
- Hari, D.P.; König, B. Eosin Y catalyzed visible light oxidative C–C and C–P bond formation. Org. Lett. 2011, 13, 3852–3855. [Google Scholar] [CrossRef] [PubMed]












































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, V.d.G.; Cardoso, M.F.d.C.; Forezi, L.d.S.M. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. https://doi.org/10.3390/catal8120605
Oliveira VdG, Cardoso MFdC, Forezi LdSM. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts. 2018; 8(12):605. https://doi.org/10.3390/catal8120605
Chicago/Turabian StyleOliveira, Vanessa da Gama, Mariana Filomena do Carmo Cardoso, and Luana da Silva Magalhães Forezi. 2018. "Organocatalysis: A Brief Overview on Its Evolution and Applications" Catalysts 8, no. 12: 605. https://doi.org/10.3390/catal8120605
APA StyleOliveira, V. d. G., Cardoso, M. F. d. C., & Forezi, L. d. S. M. (2018). Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts, 8(12), 605. https://doi.org/10.3390/catal8120605