First-Principles Modeling in Heterogeneous Electrocatalysis



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
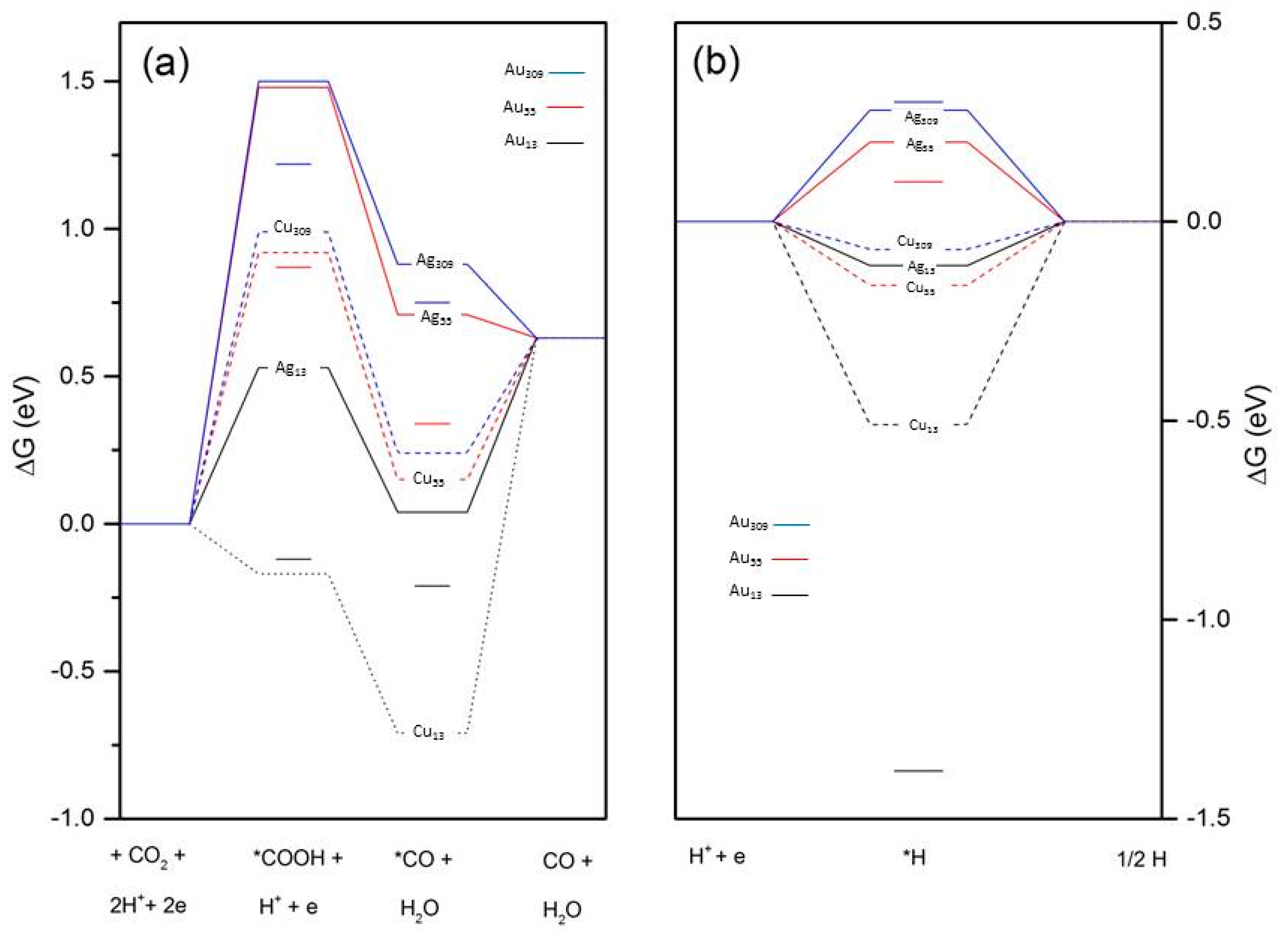{kind=link}
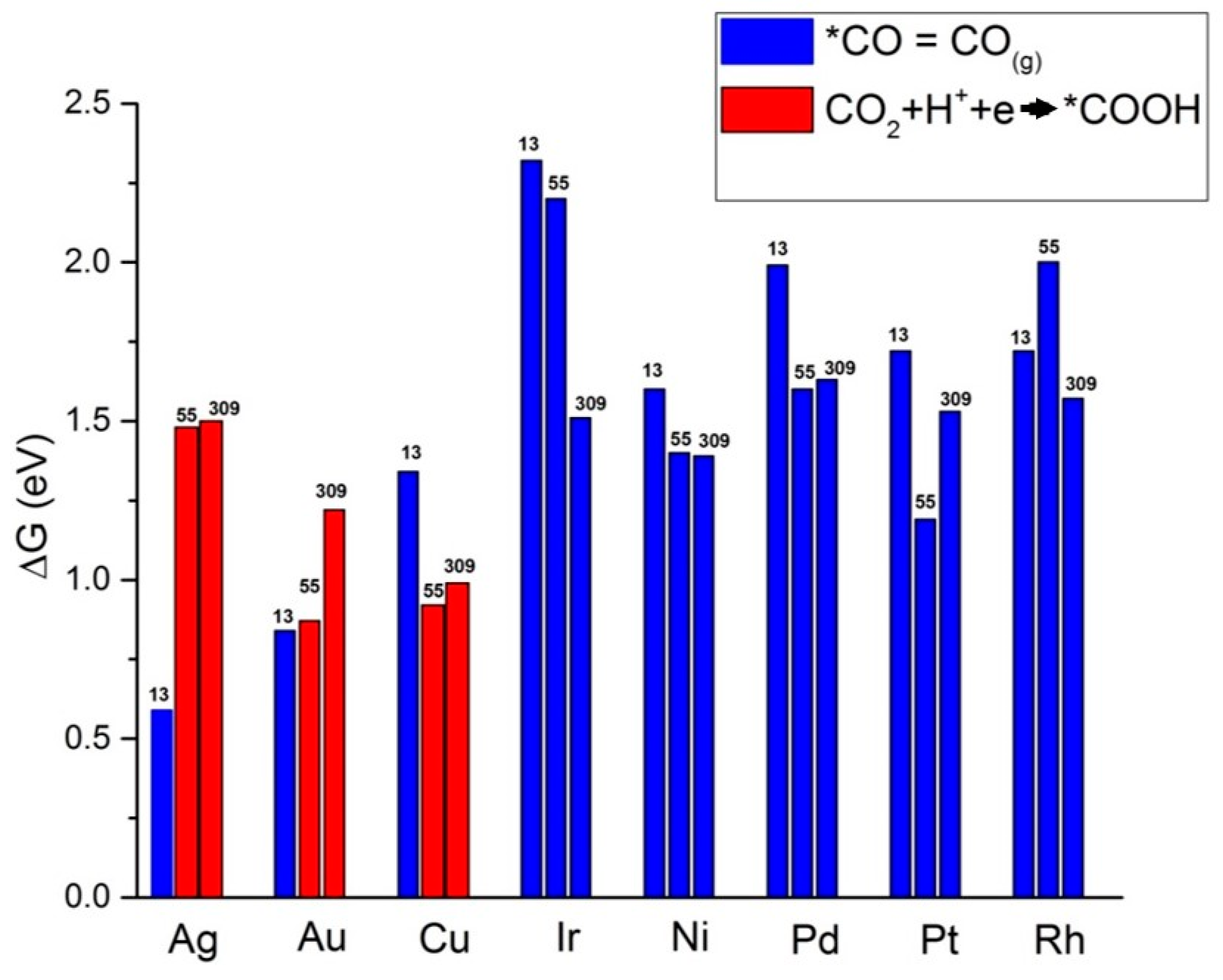{kind=link}
{kind=link}
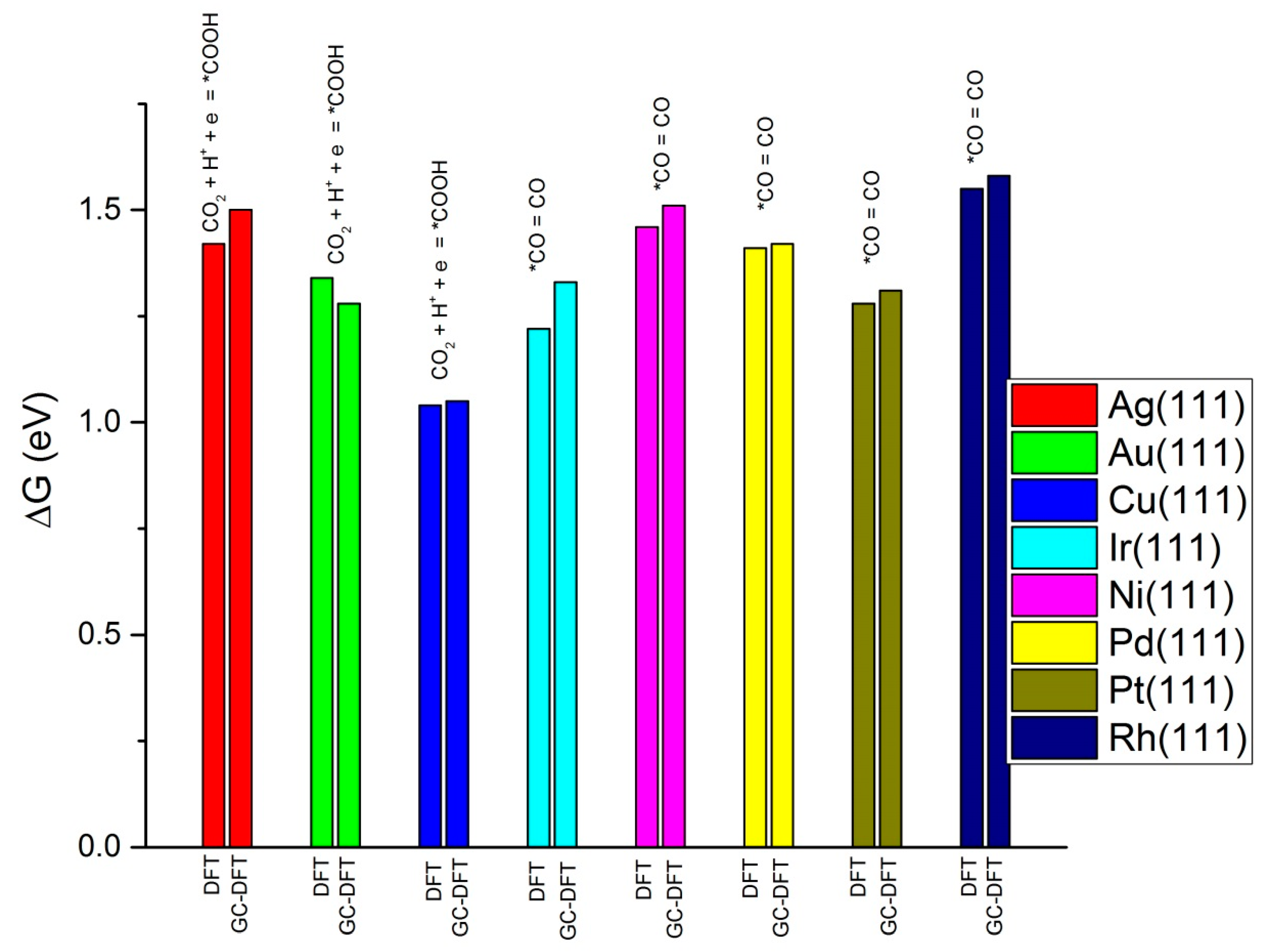{kind=link}
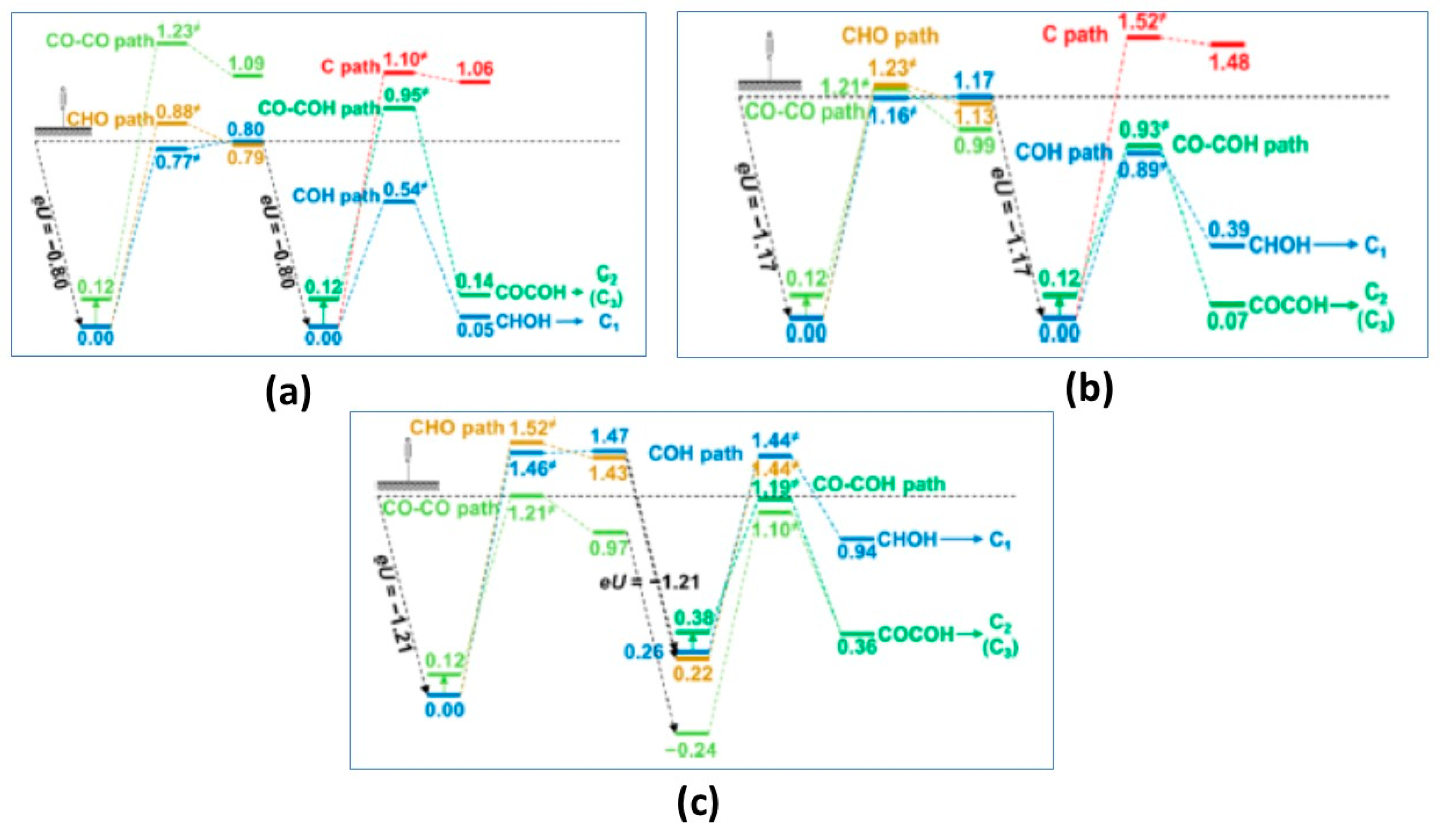{kind=link}
1. Introduction
2. Computational Hydrogen Electrode Approach
3. CHE Approach to Electro-Catalytic Reactions on the Active Sites of Nanoparticles
3.1. CO2RR on Ligand Protected Au25 Cluster
3.2. CO2RR vs HER on Metal Nanoparticles
3.3. Water Splitting on Organometallic Complexes
4. Beyond the Computational Hydrogen Electrode Method: The Grand Canonical Density Functional Theory Approach
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqeous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Hori, Y. Modern Aspects of Electrochemistry; Springer: New York, NY, USA, 2008. [Google Scholar]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley: Wienheim, Germany, 2010. [Google Scholar]
- Ganesh, I. Conversion of carbon dioxide into methanol-A potential liquid fuel: Fundamental challenges and opportunity. Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Guo, C.; Liu, X.; Liu, J.; Vasileff, A.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Emerging two-dimensional nanomaterials for electrocatalysis. Chem. Rev. 2018, 118, 6337–6408. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Koper, M.T.M. First-principles computational electrochemistry; achievements and challenges. Electrochim. Acta 2012, 84, 3–11. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Michel, C.; Schwiedernoch, R.; Sautet, P. Impacts of electrode potentials and solvents on the electroreduction of CO2: A comparison of theoretical approaches. Phys. Chem. Chem. Phys. 2015, 17, 13949–13963. [Google Scholar] [CrossRef] [PubMed]
- Letchworth-Weaver, K.; Arias, T.A. Joint density functional theory of the electrode-electrolyte interface: Application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge. Phys. Rev. B 2012, 86, 075140. [Google Scholar] [CrossRef]
- Sundararaman, R.; Goddard, W.A.; Arias, T.A. Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry. J. Chem. Phys. 2017, 146, 114104. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Gubbins, K.E.; Moore, J.D. Molecular modeling of matter: Impact and prospects in engineering. Ind. Eng. Chem. Res. 2010, 49, 3026–3046. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using vasp: Density-functional theory and beyond. J. Comp. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Shen, V.K.; Siderius, D.W.; Krekelberg, W.P.; Hatch, H.W. Nist Standard Reference Simulation Website; Nist Standard Reference Database 173; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017.
- Daniel, M.-C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Sardar, R.; Funston, A.M.; Mulvaney, P.; Murray, R.W. Gold nanoparticles: Past, present, and future. Langmuir 2009, 25, 13840–13851. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.; Kwon, Y.; Koper, M.T.M. The promoting effect of adsorbed carbon monoxide on the oxidation of alcohols on a gold catalyst. Nat. Chem. 2011, 4, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Jin, R. Quantum sized, thiolate-protected gold nanoclusters. Nanoscale 2010, 2, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.F.; Fields-Zinna, C.A.; Murray, R.W. The story of a monodisperse gold nanoparticle: Au25L18. Acc. Chem. Res. 2010, 43, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Heaven, M.W.; Dass, A.; White, P.S.; Holt, K.M.; Murray, R.W. Crystal structure of the gold nanoparticle [N(C8H17)4][Au25(SCH2CH2Ph)18]. J. Am. Chem. Soc. 2008, 130, 3754–3755. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Aikens, C.M.; Hollander, F.J.; Schatz, G.C.; Jin, R. Correlating the crystal structure of a thiol-protected Au25 cluster and optical properties. J. Am. Chem. Soc. 2008, 130, 5883–5885. [Google Scholar] [CrossRef] [PubMed]
- Akola, J.; Walter, M.; Whetten, R.L.; Häkkinen, H.; Grönbeck, H. On the structure of thiolate-protected Au25. J. Am. Chem. Soc. 2008, 130, 3756–3757. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qian, H.; Jin, R. Catalysis opportunities of atomically precise gold nanoclusters. J. Mater. Chem. 2011, 21, 6793–6799. [Google Scholar] [CrossRef]
- Zhu, Y.; Qian, H.; Drake, B.A.; Jin, R. Atomically precise Au25(SR)18 nanoparticles as catalysts for the selective hydrogenation of α,β-unsaturated ketones and aldehydes. Angew. Chem. Int. Ed. 2010, 49, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, S. Oxygen electroreduction catalyzed by gold nanoclusters: Strong core size effects. Angew. Chem. Int. Ed. 2009, 48, 4386–4389. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, D.; Alfonso, D.; Matranga, C.; Qian, H.; Jin, R. Experimental and computational investigation of au25 clusters and CO2: A unique interaction and enhanced electrocatalytic activity. J. Am. Chem. Soc. 2012, 134, 10237–10243. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, D.R.; Kauffman, D.; Matranga, C. Active sites of ligand-protected Au25 nanoparticle catalysts for CO2 electroreduction to CO. J. Chem. Phys. 2016, 144, 184705. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Hansen, H.A.; Varley, J.B.; Peterson, A.A.; Norskov, J. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Zhao, S.; Austin, N.; Li, M.; Song, Y.; House, S.D.; Bernhard, S.; Yang, J.C.; Mpourmpakis, G.; Jin, R. Influence of atomic-level morphology on catalysis: The case of sphere and rod-like gold nanoclusters for CO2 electroreduction. ACS Catal. 2018, 8, 4996–5001. [Google Scholar] [CrossRef]
- Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse au nanoparticles for selective electrocatalytic reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Roldan Cuenya, B. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef] [PubMed]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.; Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhou, H.; Wang, J.; Miao, S.; Yang, F.; Wang, G.; Wang, J.; Bao, X. Size-dependent electrocatalytic reduction of CO2 over Pd nanoparticles. J. Am. Chem. Soc. 2015, 137, 4288–4291. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, D.R.; Kauffman, D.R. Assessment of trends in the electrochemical CO2 reduction and H2 evolution reactions on metal nanoparticles. MRS Commun. 2017, 7, 601–606. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, W.J.; Zhang, H.; Lv, H.; Li, Q.; Michalski, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Yeom, M.S.; Jung, Y. Active sites of Au and Ag nanoparticle catalysts for CO2 electroreduction to CO. ACS Catal. 2015, 5, 5089–5096. [Google Scholar] [CrossRef]
- Yan, K.; Kim, S.K.; Khorshidi, A.; Guduru, P.R.; Peterson, A.A. High elastic strain directly tunes the hydrogen evolution reaction on tungsten carbide. J. Phys. Chem. C 2017, 121, 6177–6183. [Google Scholar] [CrossRef]
- Yan, K.; Maark, T.A.; Khorshidi, A.; Sethuraman, V.A.; Peterson, A.A.; Guduru, P.R. The influence of elastic strain on catalytic activity in the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2016, 55, 6175–6181. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Ghosh, S.; Pradhan, B.; Dalui, A.; Shrestha, L.K.; Acharya, S.; Ariga, K. Two-dimensional (2D) nanomaterials towards electrochemical nanoarchitectonics in energy-related applications. Bull. Chem. Soc. Jpn. 2017, 90, 627–648. [Google Scholar] [CrossRef]
- Helm, M.L.; Stewart, M.P.; Bullock, R.M.; DuBois, M.R.; DuBois, D.L. A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s-1 for H2 production. Science 2011, 333, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, D.R.; Alfonso, D.; Tafen, D.N.; Lekse, J.; Wang, C.; Deng, X.; Lee, J.; Jang, H.; Lee, J.-S.; Kumar, S.; et al. Electrocatalytic oxygen evolution with an atomically precise nickel catalyst. ACS Catal. 2016, 6, 1225–1234. [Google Scholar] [CrossRef]
- Tafen, D.N.; Kauffman, D.R.; Alfonso, D.R. Electrocatalytic oxygen evolution with pure and substituted M6(SR)12 (M = Pd, Fe, Rh) complexes. Comp. Mater. Sci. 2018, 150, 283–290. [Google Scholar] [CrossRef]
- Bajdich, M.; García-Mota, M.; Vojvodic, A.; Nørskov, J.K.; Bell, A.T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 2013, 135, 13521–13530. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Keith, J.A.; Carter, E.A. Water oxidation on pure and doped hematite (0001) surfaces: Prediction of Co and Ni as effective dopants for electrocatalysis. J. Am. Chem. Soc. 2012, 134, 13296–13309. [Google Scholar] [CrossRef] [PubMed]
- García-Mota, M.; Vojvodic, A.; Metiu, H.; Man, I.C.; Su, H.-Y.; Rossmeisl, J.; Nørskov, J.K. Tailoring the activity for oxygen evolution electrocatalysis on rutile TiO2 by transition-metal substitution. ChemCatChem 2011, 3, 1607–1611. [Google Scholar] [CrossRef]
- Siahrostami, S.; Verdaguer-Casadevall, A.; Karamad, M.; Deiana, D.; Malacrida, P.; Wickman, B.; Escudero-Escribano, M.; Paoli, E.A.; Frydendal, R.; Hansen, T.W.; et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 2013, 12, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Xue, Y.; Qu, L.; Choi, H.-J.; Baek, J.-B. Metal-free catalysts for oxygen reduction reaction. Chem. Rev. 2015, 115, 4823–4892. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.C.; Nilekar, A.U.; Xu, Y.; Mavrikakis, M. Partial and complete reduction of O2 by hydrogen on transition metal surfaces. Surf. Sci. 2010, 604, 1565–1575. [Google Scholar] [CrossRef]
- Jirkovský, J.S.; Panas, I.; Ahlberg, E.; Halasa, M.; Romani, S.; Schiffrin, D.J. Single atom hot-spots at Au–Pd nanoalloys for electrocatalytic H2O2 production. J. Am. Chem. Soc. 2011, 133, 19432–19441. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Siahrostami, S.; Li, G.-L.; Zhang, Y.; Chakthranont, P.; Studt, F.; Jaramillo, T.F.; Zheng, X.; Nørskov, J.K. Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 2017, 8, 701. [Google Scholar] [CrossRef] [PubMed]
- Filhol, J.-S.; Neurock, M. Elucidation of the electrochemical activation of water over Pd by first principles. Angew. Chem. Int. Ed. 2006, 45, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Otani, M.; Sugino, O. First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach. Phys. Rev. B 2006, 73, 115407. [Google Scholar] [CrossRef]
- Skulason, E.; Tripkovic, V.; Bjorketum, M.E.; Gudmunsdottir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jonsson, H.; Norskov, J.J. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar]
- Rossmeisl, J.; Skúlason, E.; Björketun, M.E.; Tripkovic, V.; Nørskov, J.K. Modeling the electrified solid–liquid interface. Chem. Phys. Lett. 2008, 466, 68–71. [Google Scholar] [CrossRef]
- Goodpaster, J.D.; Bell, A.T.; Head-Gordon, M. Identification of possible pathways for C–C bond formation during electrochemical reduction of CO2: New theoretical insights from an improved electrochemical model. J. Phys. Chem. Lett. 2016, 7, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, R.; Letchworth-Weaver, K.; Schwarz, K.A.; Gunceler, D.; Ozhabes, Y.; Arias, T.A. JDFTx: Software for joint density-functional theory. SoftwareX 2017, 6, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, R.; Goddard, W.A. The charge-asymmetric nonlocally determined local-electric (candle) solvation model. J. Chem. Phys. 2015, 142, 064107. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Cheng, T.; Goddard, W.A.; Sundararaman, R. Mechanistic explanation of the pH dependence and onset potentials for hydrocarbon products from electrochemical reduction of CO on Cu (111). J. Am. Chem. Soc. 2016, 138, 483–486. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfonso, D.R.; Tafen, D.N.; Kauffmann, D.R. First-Principles Modeling in Heterogeneous Electrocatalysis. Catalysts 2018, 8, 424. https://doi.org/10.3390/catal8100424
Alfonso DR, Tafen DN, Kauffmann DR. First-Principles Modeling in Heterogeneous Electrocatalysis. Catalysts. 2018; 8(10):424. https://doi.org/10.3390/catal8100424
Chicago/Turabian StyleAlfonso, Dominic R., De Nyago Tafen, and Douglas R. Kauffmann. 2018. "First-Principles Modeling in Heterogeneous Electrocatalysis" Catalysts 8, no. 10: 424. https://doi.org/10.3390/catal8100424
APA StyleAlfonso, D. R., Tafen, D. N., & Kauffmann, D. R. (2018). First-Principles Modeling in Heterogeneous Electrocatalysis. Catalysts, 8(10), 424. https://doi.org/10.3390/catal8100424