Influence of Bio-Oil Phospholipid on the Hydrodeoxygenation Activity of NiMoS/Al2O3 Catalyst

 ,
,
Abstract
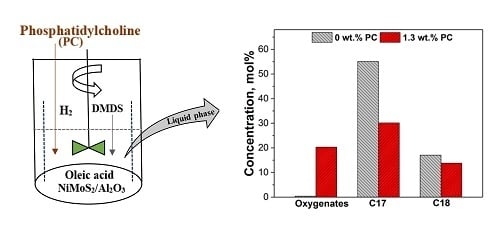
1. Introduction
2. Results and Discussions
2.1. HDO Activity of NiMo/Al2O3
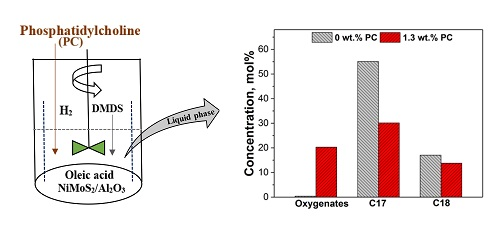
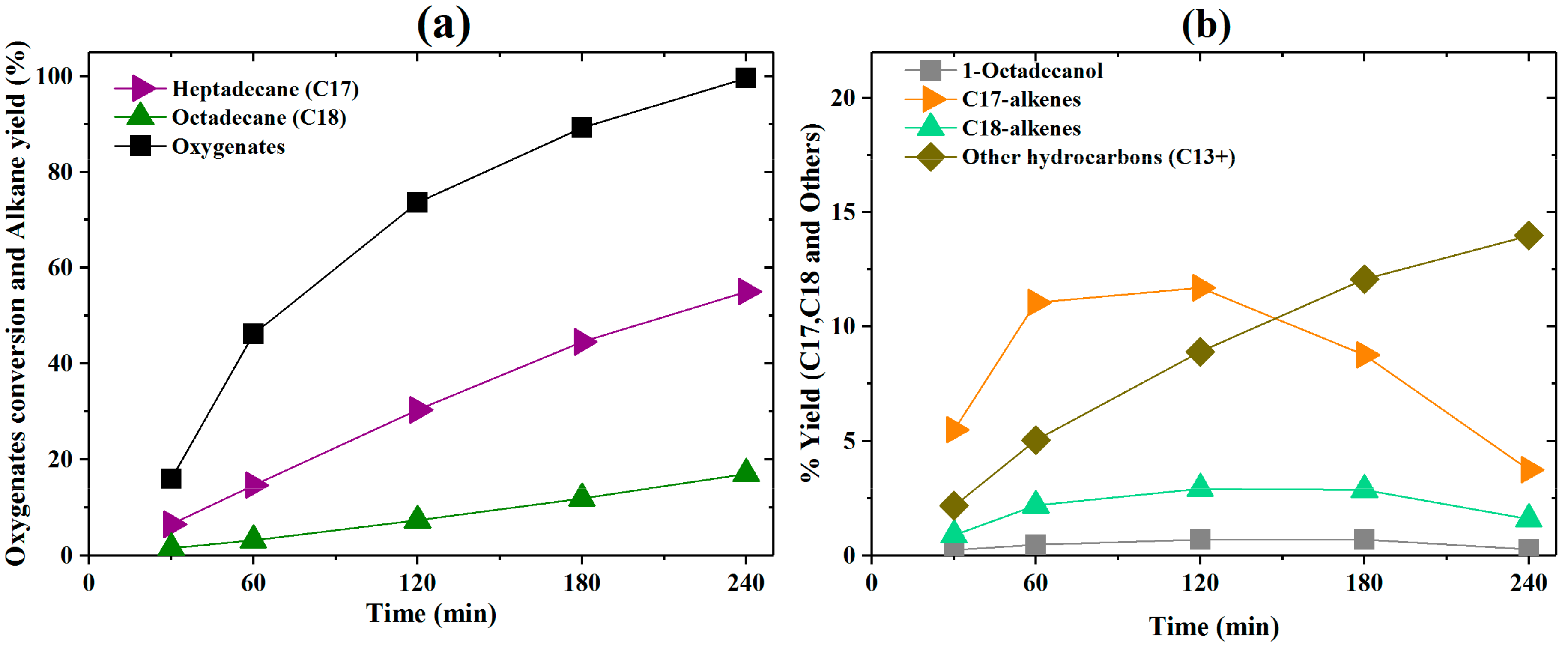
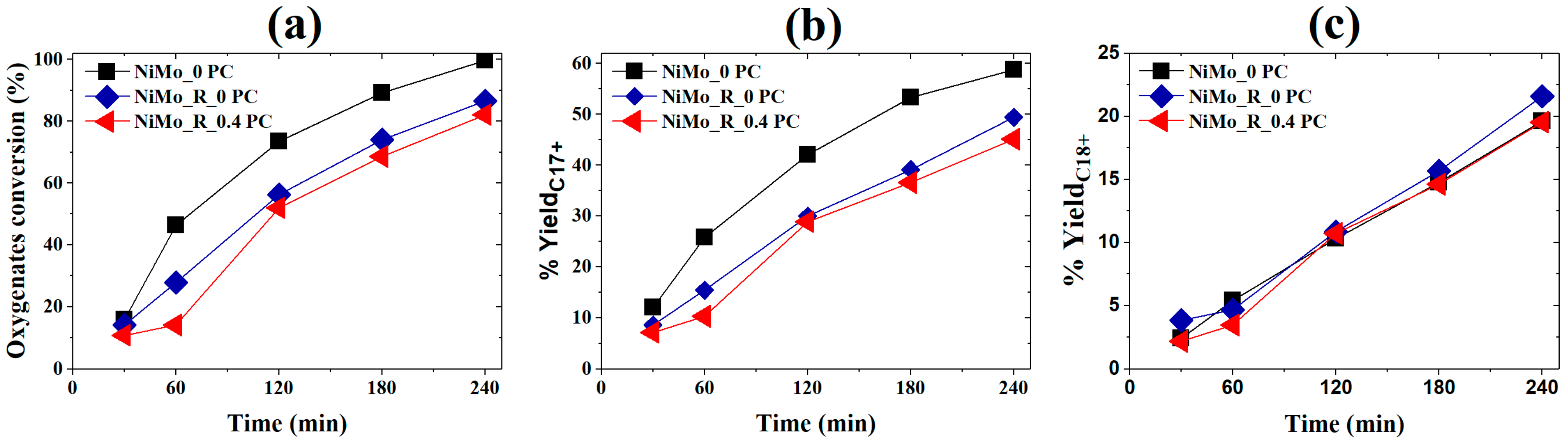
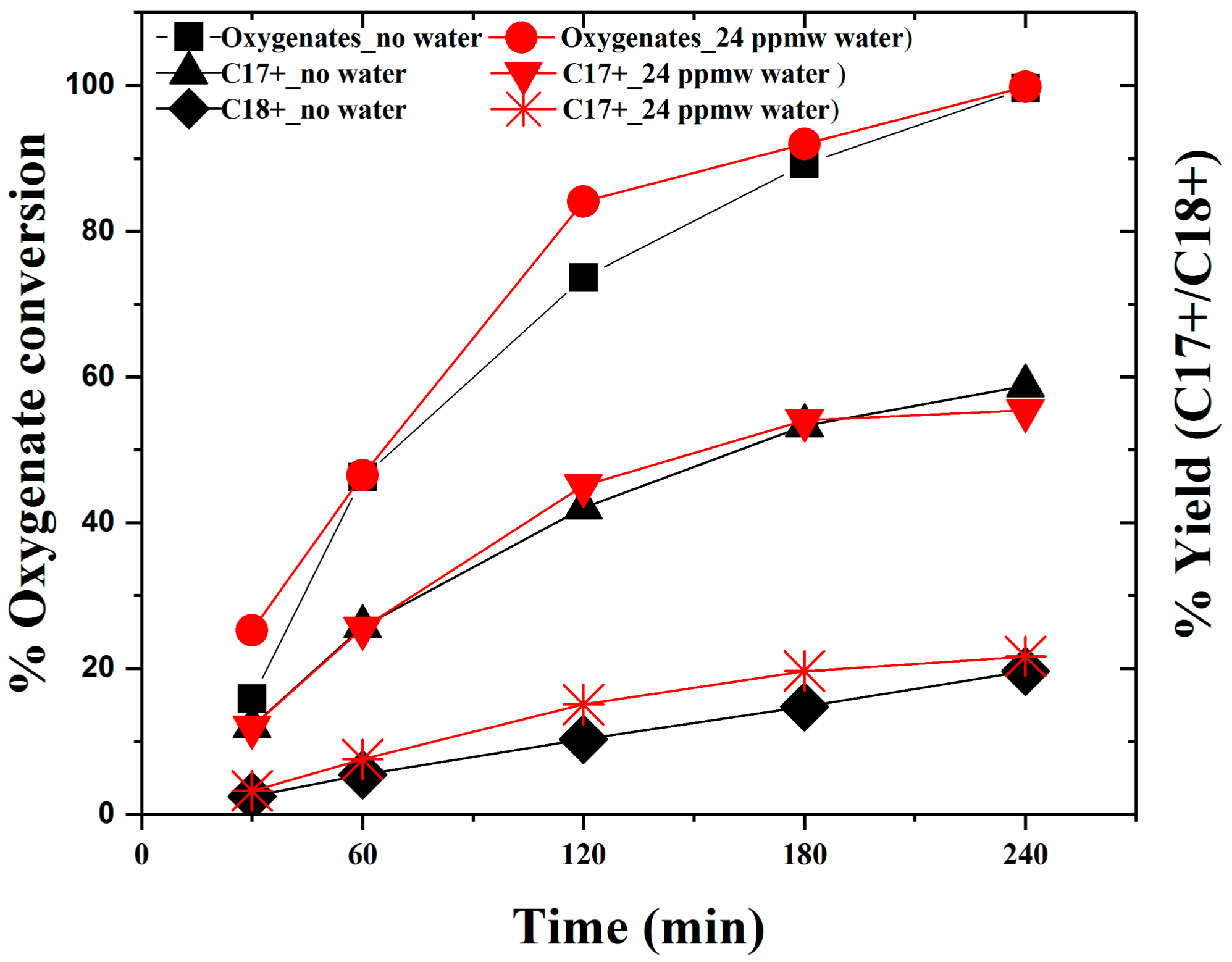
2.1.1. Activity Measurements without Phospholipid
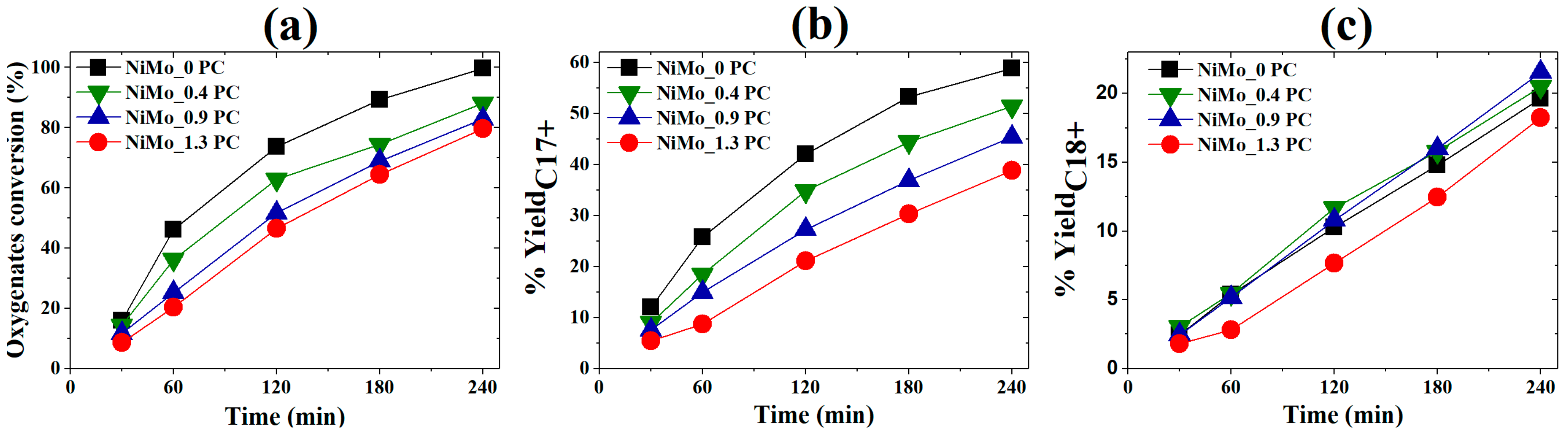
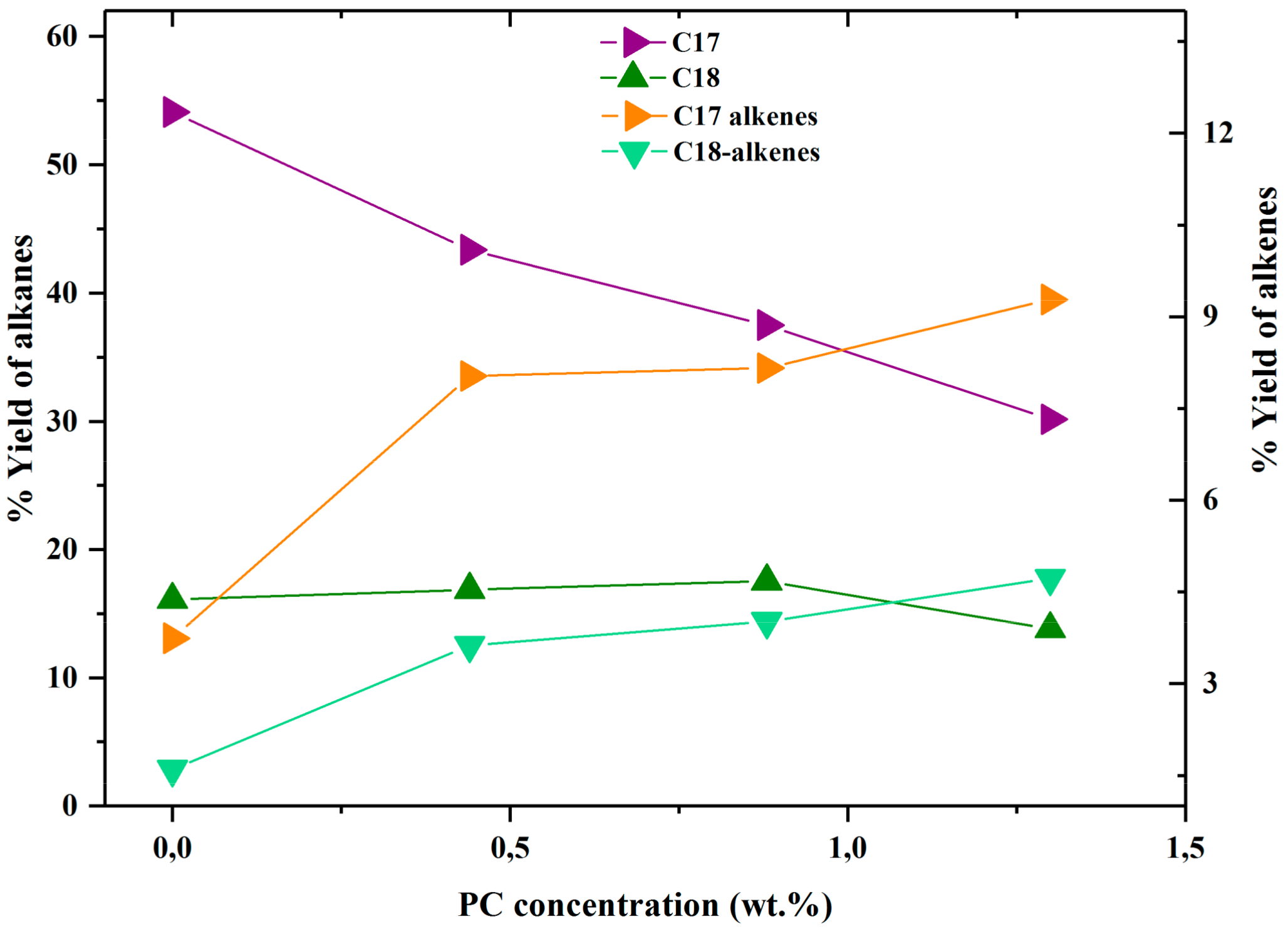
2.1.2. Activity Measurements with Phospholipid
2.2. Catalyst Characterization
2.2.1. Textural Properties of the Catalyst
2.2.2. ICP-SFMS on the Recovered Catalyst and Liquid Samples
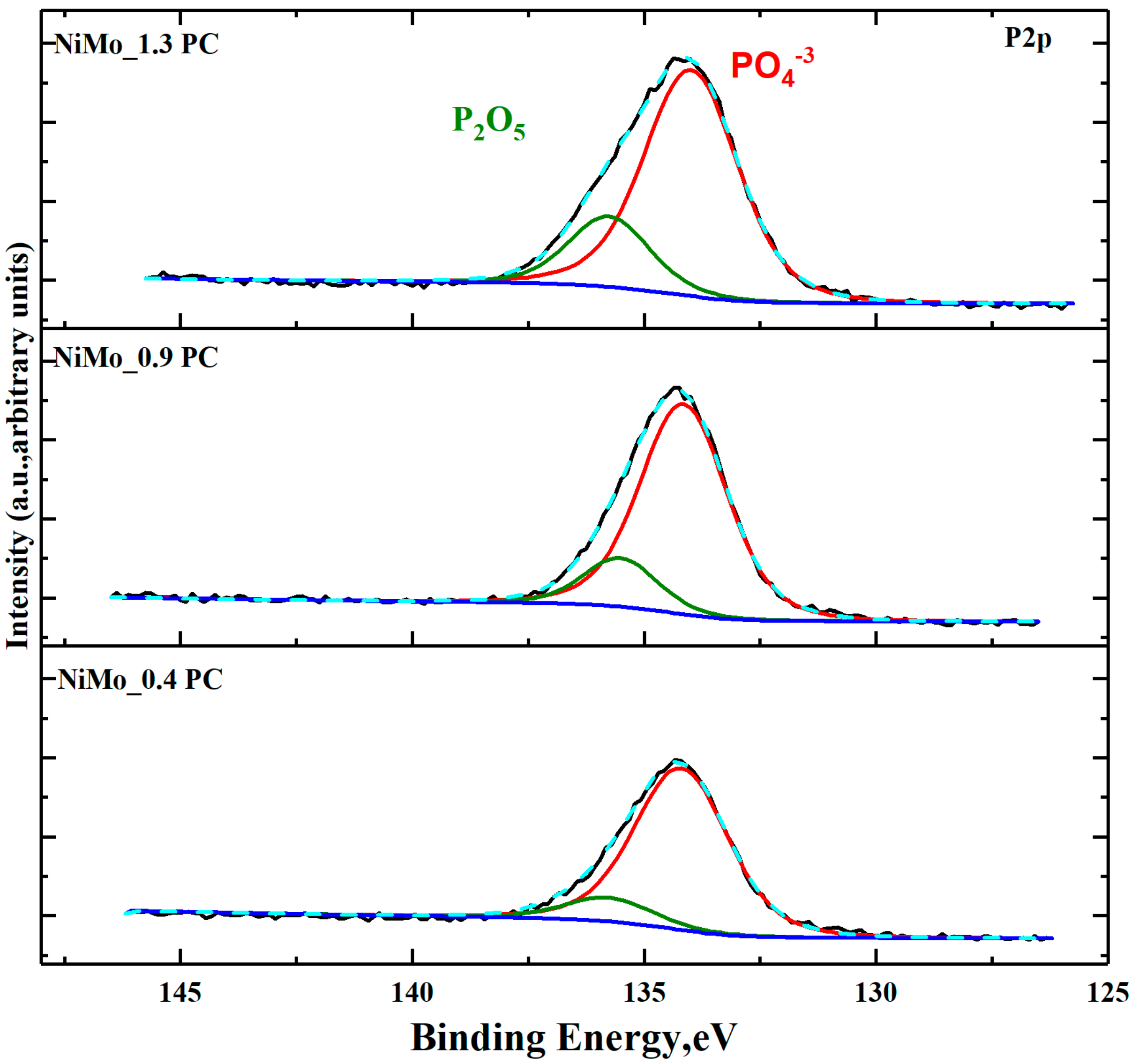
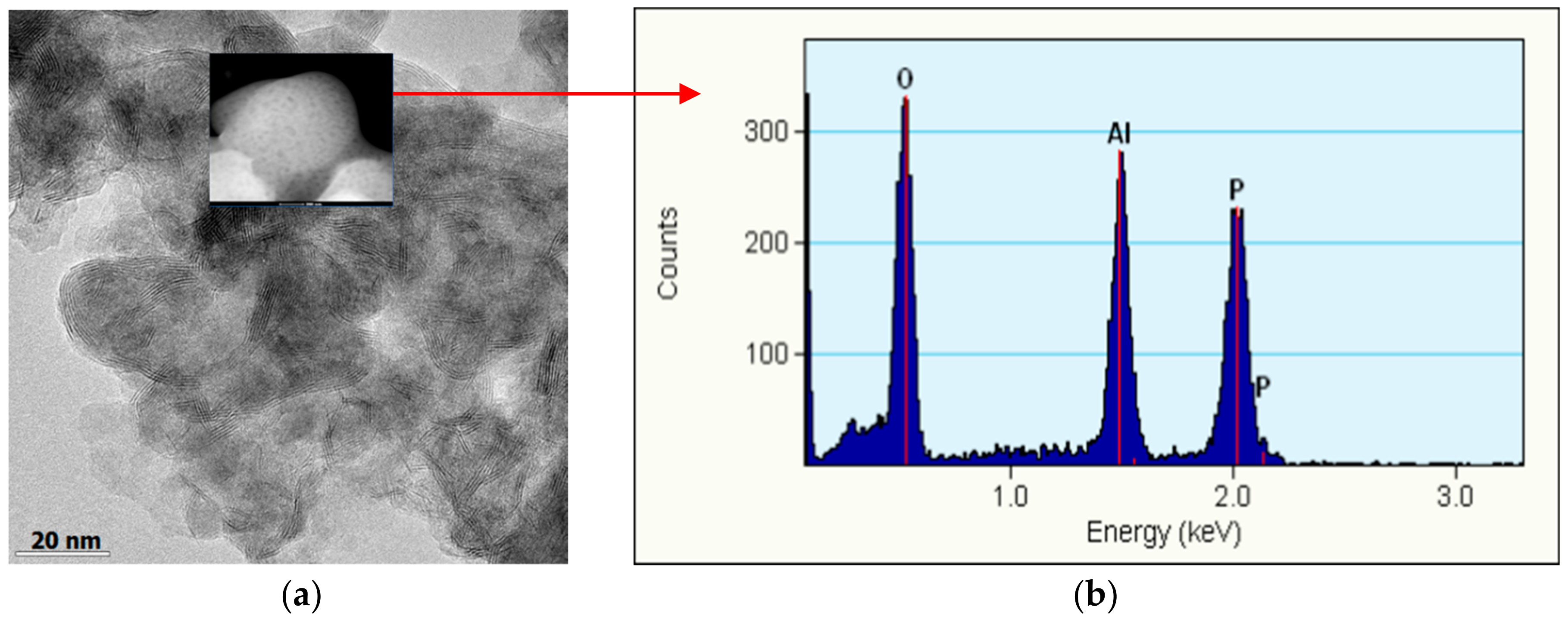
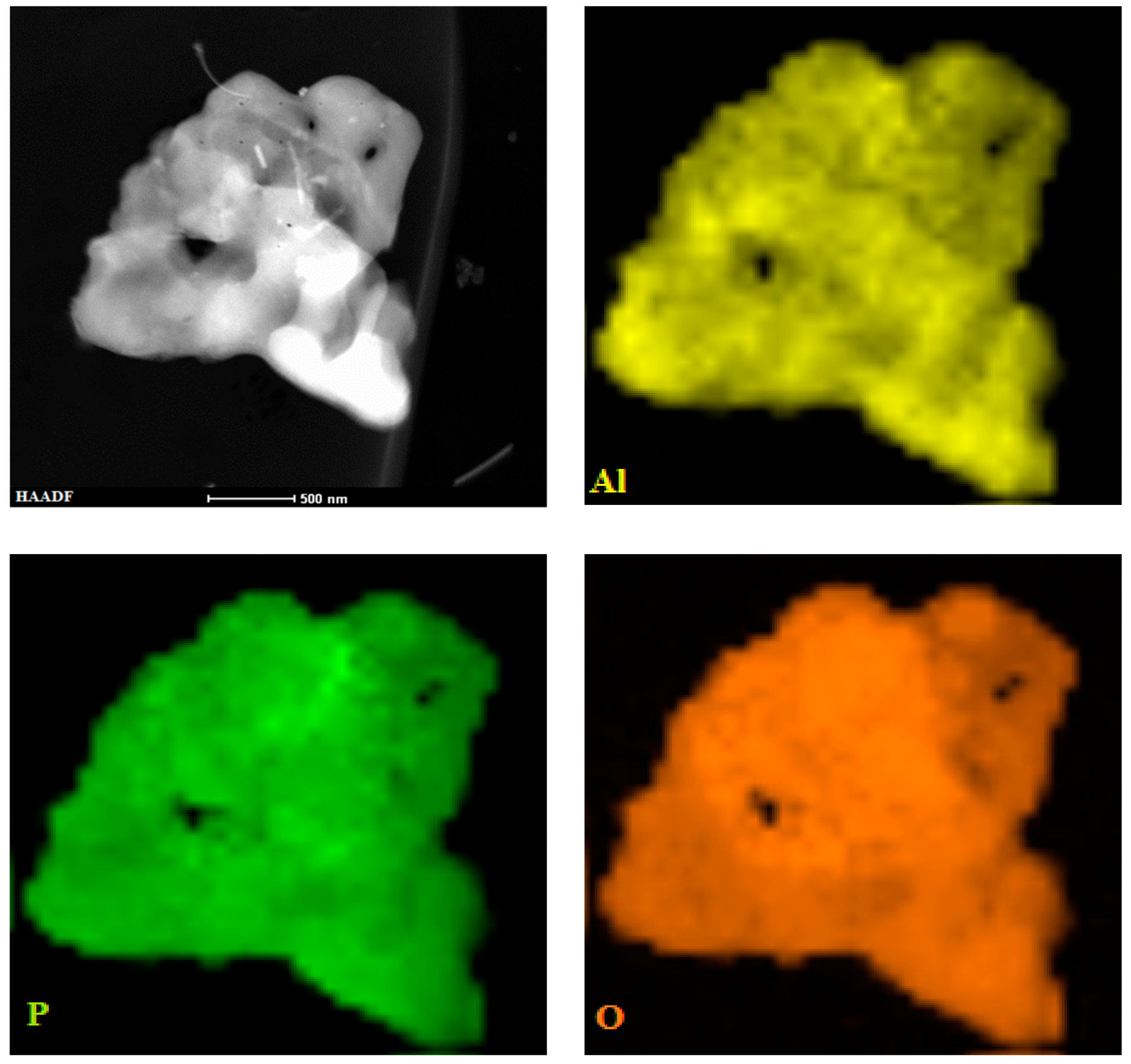
2.2.3. XPS and TEM Analysis
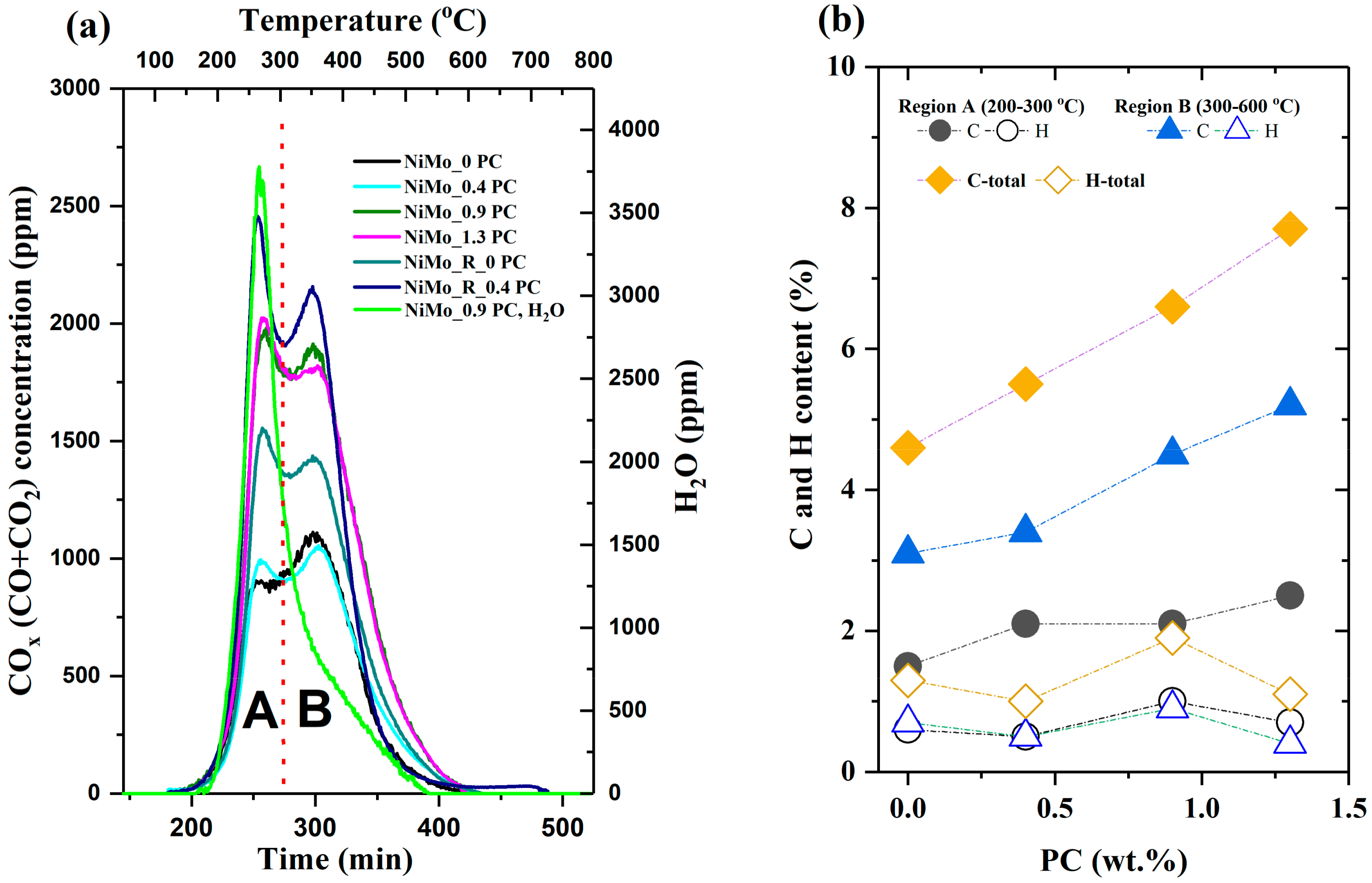
2.2.4. Temperature Programmed Oxidation (TPO) of Recovered Catalysts
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalytic Activity Measurements
3.2.1. Sulfidation
3.2.2. Hydrodeoxygenation of Oleic Acid

3.3. Product Analysis
3.4. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dauenhauer, P.J. Handbook of Plant-Based Biofuels; Ashok, P., Ed.; CRC Press: Boca Raton, FL, USA, 2010; Volume 3, pp. 386–387. ISBN 978-1-56022-175-3. [Google Scholar]
- Kubička, D.; Tukač, V. Hydrotreating of triglyceride-based feedstocks in refineries. Adv. Chem. Eng. 2012, 42, 141–194. [Google Scholar]
- Srifa, A.; Viriya-empikul, N.; Assabumrungrat, S.; Faungnawakij, K. Catalytic behaviors of Ni/γ-Al2O3 and Co/γ-Al2O3 during the hydrodeoxygenation of palm oil. Catal. Sci. Technol. 2015, 5, 3693–3705. [Google Scholar] [CrossRef]
- Kubička, D.; Kaluža, L. Deoxygenation of vegetable oils over sulfided Ni, Mo and NiMo catalysts. Appl. Catal. A Gen. 2010, 372, 199–208. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Study of the hydrodeoxygenation of carbonyl, carboylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalyst. Appl. Catal. A Gen. 1994, 109, 97–115. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Enol, O.İ.; Viljava, T.-R.; Krause, A.O.I. Hydrodeoxygenation of aliphatic esters on sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 catalyst: The effect of water. Catal. Today 2005, 106, 186–189. [Google Scholar]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28–52. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A.; Furimsky, E. Handbook of Spent Hydroprocessing Catalysts: Regeneration, Rejuvenation, Reclamation, Environment and Safety; Elsevier BV: Oxford, UK, 2010; ISBN 978-0-444-53556-6. [Google Scholar]
- Kalnes, T.N.; Koers, K.P.; Marker, T.; Shonnard, D.R. A technoeconomic and environmental life cycle comparison of green diesel to biodiesel and syndiesel. Environ. Prog. Sustain. Energy 2009, 28, 111–120. [Google Scholar] [CrossRef]
- Arora, P.; Ojagh, H.; Woo, J.; Grennfelt, E.L.; Olsson, L.; Creaser, D. Investigating the effect of Fe as a poison for catalytic HDO over sulfided NiMo alumina catalysts. Appl. Catal. B Environ. 2018, 227, 240–251. [Google Scholar] [CrossRef]
- Laakkonen, M.; Myllyoja, J.; Toukoniitty, B.; Hujanen, M.; Saastamoinen, A.; Toivo, A. Process for Manufacture of Liquid Fuel Componenst from Renewable Sources. U.S. Patent 13,646,250, 4 November 2013. [Google Scholar]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Beckman, D.; Elliott, D.C. Comparisons of the yields and properties of the oil products from direct thermochemical biomass liquefaction processes. Can. J. Chem. Eng. 1985, 63, 99–104. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Deactivation of Ni-MoS2 by bio-oil impurities during hydrodeoxygenation of phenol and octanol. Appl. Catal. A Gen. 2016, 523, 159–170. [Google Scholar] [CrossRef]
- Kubička, D.; Horáček, J. Deactivation of HDS catalysts in deoxygenation of vegetable oils. Appl. Catal. A Gen. 2011, 394, 9–17. [Google Scholar] [CrossRef]
- Popov, S.; Kumar, S. Renewable fuels via catalytic hydrodeoxygenation of lipid-based feedstocks. Biofuels 2013, 4, 219–239. [Google Scholar] [CrossRef]
- Dijkstra, A.J. About water degumming and the hydration of non-hydratable phosphatides. Eur. J. Lipid Sci. Technol. 2017, 119, 1600496. [Google Scholar] [CrossRef]
- Paisan, S.; Chetpattananondh, P.; Chongkhong, S. Assessment of water degumming and acid degumming of mixed algal oil. J. Environ. Chem. Eng. 2017, 5, 5115–5123. [Google Scholar] [CrossRef]
- Kiatkittipong, W.; Phimsen, S.; Kiatkittipong, K.; Wongsakulphasatch, S.; Laosiripojana, N.; Assabumrungrat, S. Diesel-like hydrocarbon production from hydroprocessing of relevant refining palm oil. Fuel Process. Technol. 2013, 116, 16–26. [Google Scholar] [CrossRef]
- Bailey, D.S.; Northcote, D.H. Phospholipid composition of the plasma membrane of the green alga, hydrodictyon africanum. Biochem. J. 1976, 156, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Pan, Q.; Ding, Y.; Jiang, L. Rapid determination of phospholipid content of vegetable oils by FTIR spectroscopy combined with partial least-square regression. Food Chem. 2014, 147, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Fitz, C.W.; Rase, H.F. Effects of phosphorus on nickel-molybdenum hydrodesulfurization/hydrodenitrogenation catalysts of varying metals content. Ind. Eng. Chem. Prod. Res. Dev. 1983, 22, 40–44. [Google Scholar] [CrossRef]
- Chadwick, D.; Aitchison, D.W.; Badilla-Ohlbaum, R.; Josefsson, L. Influence of phosphorus on the HDS activity of Ni-Mo/γ-Al2O3 catalysts. Stud. Surf. Sci. Catal. 1983, 323–332. [Google Scholar]
- Bouwens, S.M.A.M.; Vissers, J.P.R.; de Beer, V.H.J.; Prins, R. Phosphorus poisoning of molybdenum sulfide hydrodesulfurization catalysts supported on carbon and alumina. J. Catal. 1988, 112, 401–410. [Google Scholar] [CrossRef]
- Rayo, P.; Ramírez, J.; Torres-Mancera, P.; Marroquín, G.; Maity, S.K.; Ancheyta, J. Hydrodesulfurization and hydrocracking of maya crude with P-modified NiMo/Al2O3 catalysts. Fuel 2012, 100, 34–42. [Google Scholar] [CrossRef]
- Eijsbouts, S.; Van Gestel, J.N.M.; Van Veen, J.N.M.; De Beer, V.H.J.; Prins, R. The effect of phosphate on the hydrodenitrogenation activity and selectivity of alumina-supported sulfided Mo, Ni, and Ni-Mo catalysts. J. Catal. 1991, 131, 412–432. [Google Scholar] [CrossRef]
- Mangnus, P.J.; van Langeveld, A.D.; de Beer, V.H.J.; Moulijn, J.A. Influence of phosphate on the structure of sulfided alumina supported cobalt-molybdenum catalysts. Appl. Catal. 1991, 68, 161–177. [Google Scholar] [CrossRef]
- Zhou, T.; Yin, H.; Liu, Y.; Han, S.; Chai, Y.; Liu, C. Effect of phosphorus content on the active phase structure of NiMoP/Al2O3 catalyst. J. Fuel Chem. Technol. 2010, 38, 69–74. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Furimsky, E.; Massoth, F.E. Deactivation of hydroprocessing catalysts. Catal. Today 1999, 52, 381–495. [Google Scholar] [CrossRef]
- Diez, F.; Gates, B.C.; Miller, J.T.; Sajkowski, D.J.; Kukes, S.G. Deactivation of a nickel-molybdenum/.gamma.-alumina catalyst: Influence of coke on the hydroprocessing activity. Ind. Eng. Chem. Res. 1990, 29, 1999–2004. [Google Scholar] [CrossRef]
- Simakova, I.; Simakova, O.; Mäki-Arvela, P.; Murzin, D.Y. Decarboxylation of fatty acids over Pd supported on mesoporous carbon. Catal. Today 2010, 150, 28–31. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Influence of water in the deactivation of a sulfided NiMoγ-Al2O3 catalyst during hydrodeoxygenation. J. Catal. 1994, 146, 281–291. [Google Scholar] [CrossRef]
- Froment, G.; Delmon, B.F. Catalyst Deactivation, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1994; ISBN 9780080887388. [Google Scholar]
- Badawi, M.; Paul, J.F.; Cristol, S.; Payen, E.; Romero, Y.; Richard, F.; Brunet, S.; Lambert, D.; Portier, X.; Popov, A.; et al. Effect of water on the stability of Mo and CoMo hydrodeoxygenation catalysts: A combined experimental and dft study. J. Catal. 2011, 282, 155–164. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Viljava, T.-R.; Krause, A.O.I. Effect of sulphiding agents on the hydrodeoxygenation of aliphatic esters on sulphided catalysts. Appl. Catal. A Gen. 2007, 326, 236–244. [Google Scholar] [CrossRef]
- Coumans, A.E.; Hensen, E.J.M. A model compound (methyl oleate, oleic acid, triolein) study of triglycerides hydrodeoxygenation over alumina-supported nimo sulfide. Appl. Catal. B Environ. 2017, 201, 290–301. [Google Scholar] [CrossRef]
- Ryymin, E.-M.; Honkela, M.L.; Viljava, T.-R.; Krause, A.O.I. Insight to sulfur species in the hydrodeoxygenation of aliphatic esters over sulfided NiMo/γ-Al2O3 catalyst. Appl. Catal. A Gen. 2009, 358, 42–48. [Google Scholar] [CrossRef]
- Dupont, C.; Lemeur, R.; Daudin, A.; Raybaud, P. Hydrodeoxygenation pathways catalyzed by MoS2 and NiMoS active phases: A DFT study. J. Catal. 2011, 279, 276–286. [Google Scholar] [CrossRef]
- Donnis, B.; Egeberg, R.G.; Blom, P.; Knudsen, K.G. Hydroprocessing of bio-oils and oxygenates to hydrocarbons. Understanding the Reaction Routes. Top. Catal. 2009, 52, 229–240. [Google Scholar] [CrossRef]
- Kubička, D. Future refining catalysis-introduction of biomass feedstocks. Collect. Czechoslov. Chem. Commun. 2008, 73, 1015–1044. [Google Scholar] [CrossRef]
- Ojagh, H.; Creaser, D.; Tamm, S.; Arora, P.; Nyström, S.; Grennfelt, E.L.; Olsson, L. Effect of dimethyl disulfide on activity of NiMo based catalysts used in hydrodeoxygenation of oleic acid. Ind. Eng. Chem. Res. 2017, 56, 5547–5557. [Google Scholar] [CrossRef]
- Changi, S.; Matzger, A.J.; Savage, P.E. Kinetics and pathways for an algal phospholipid (1,2-dioleoyl-sn-glycero-3-phosphocholine) in high-temperature (175–350 °C) water. Green Chem. 2012, 14, 2856–2867. [Google Scholar] [CrossRef]
- de Koning, A.J.; McMullan, K.B. Hydrolysis of phospholipids with hydrochloric acid. Biochim. Biophys. Acta, Lipids Lipid Metab. 1965, 106, 519–526. [Google Scholar] [CrossRef]
- Qu, L.; Prins, R. Hydrogenation of cyclohexene over in situ fluorinated NiMoS catalysts supported on alumina and silica-alumina. J. Catal. 2002, 207, 286–295. [Google Scholar] [CrossRef]
- De Klerk, A. Oligomerization of 1-hexene and 1-octene over solid acid catalysts. Ind. Eng. Chem. Res. 2005, 44, 3887–3993. [Google Scholar] [CrossRef]
- Iwamoto, R.; Grimblot, J. Influence of phosphorus on the properties of alumina-based hydrotreating catalysts. Adv. Catal. 1999, 44, 417–503. [Google Scholar]
- Liu, G.; Robertson, A.W.; Li, M.M.-J.; Kuo, W.C.H.; Darby, M.T.; Muhieddine, M.H.; Lin, Y.-C.; Suenaga, K.; Stamatakis, M.; Warner, J.H.; et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, S.; Sundaramurthy, V.; Dalai, A.K.; Adjaye, J. Phosphorus promoted trimetallic NiMoW/γ-Al2O3 sulfide catalysts in gas oil hydrotreating. J. Mol. Catal. A Chem. 2008, 291, 30–37. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A. Effect of initial coking on hydrotreating catalyst functionalities and properties. Appl. Catal. A Gen. 1997, 159, 259–267. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A. Influence of catalyst acidity and feedstock quality on hydrotreating catalyst deactivation by coke deposition. Pet. Sci. Technol. 2001, 19, 697–710. [Google Scholar] [CrossRef]
- Gong, S.; Shinozaki, A.; Shi, M.; Qian, E.W. Hydrotreating of jatropha oil over alumina based catalysts. Energy Fuels 2012, 26, 2394–2399. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | roxygenates (mmol/g/h) | rC17+ (mmol/g/h) | rC18+ (mmol/g/h) |
|---|---|---|---|
| NiMo_0 PC | 59.1 | 65.8 | 13.8 |
| NiMo_0.4 PC | 46.3 | 47.3 | 13.9 |
| NiMo_0.9 PC | 32.2 | 38.2 | 13.2 |
| NiMo_1.3 PC | 27.6 | 23.8 | 7.6 |
| NiMo_Choline | 45.9 | 39.5 | 14.9 |
| NiMo_R_0 PC | 35.7 | 39.4 | 11.9 |
| NiMo_R_0.4 PC | 18.0 | 26.3 | 8.8 |
| Catalyst | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Average Pore Size (Å) |
|---|---|---|---|
| Synthesized | 143 | 0.33 | 62.3 |
| NiMo_0 PC | 126 | 0.27 | 62.1 |
| NiMo_0.4 PC | 105 | 0.22 | 55.2 |
| Catalyst | C, wt.% | H, wt.% | S, wt.% | N, wt.% |
|---|---|---|---|---|
| Sulfided | 0.4 | 1.3 | 9.22 | <0.05 |
| NiMo_0 PC | 2.96 | 1.57 | 7.30 | <0.05 |
| NiMo_0.4 PC | 5.34 | 1.65 | 7.39 | 0.36 |
| NiMo_0.9 PC | 5.90 | 1.3 | 7.40 | 0.30 |
| NiMo_1.3 PC | 7.74 | 1.73 | 7.47 | 0.37 |
| NiMo_R_0 PC | 7.26 | 1.87 | 7.12 | <0.05 |
| NiMo_R_0.4 PC | 6.31 | 1.42 | 7.05 | 0.26 |
| Catalyst | P Added in Liquid, ppm | Theoretical Maximum P in Catalyst, wt.% | Actual P in Catalyst (ICP), wt.% | Atomic Ratio of Ni/(Ni +Mo) | P Remaining in Liquid Phase (ICP) ppm |
|---|---|---|---|---|---|
| As Synthesized | - | - | - | 0.37 | - |
| NiMo_0.9 PC | 532 | 12.16 | 1.27 | 0.41 | 11.1 |
| NiMo_0.4 PC | 179 | 4.09 | 1.98 | 0.39 | 8.37 |
| NiMo_R_0.4 PC * | 179 | 8.18 | 3.18 | 0.39 | 10.6 |
| PC in Feed (wt.%) | Catalyst (NiMo/Al2O3) |
|---|---|
| 0 | NiMo_0 PC |
| 0.44 | NiMo_0.4 PC |
| 0.88 | NiMo_0.9 PC |
| 1.3 | NiMo_1.3 PC |
| 0 | NiMo_R_0 PC * |
| 0.44 | NiMo_R_0.4 PC * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdus Salam, M.; Creaser, D.; Arora, P.; Tamm, S.; Lind Grennfelt, E.; Olsson, L. Influence of Bio-Oil Phospholipid on the Hydrodeoxygenation Activity of NiMoS/Al2O3 Catalyst. Catalysts 2018, 8, 418. https://doi.org/10.3390/catal8100418
Abdus Salam M, Creaser D, Arora P, Tamm S, Lind Grennfelt E, Olsson L. Influence of Bio-Oil Phospholipid on the Hydrodeoxygenation Activity of NiMoS/Al2O3 Catalyst. Catalysts. 2018; 8(10):418. https://doi.org/10.3390/catal8100418
Chicago/Turabian StyleAbdus Salam, Muhammad, Derek Creaser, Prakhar Arora, Stefanie Tamm, Eva Lind Grennfelt, and Louise Olsson. 2018. "Influence of Bio-Oil Phospholipid on the Hydrodeoxygenation Activity of NiMoS/Al2O3 Catalyst" Catalysts 8, no. 10: 418. https://doi.org/10.3390/catal8100418
APA StyleAbdus Salam, M., Creaser, D., Arora, P., Tamm, S., Lind Grennfelt, E., & Olsson, L. (2018). Influence of Bio-Oil Phospholipid on the Hydrodeoxygenation Activity of NiMoS/Al2O3 Catalyst. Catalysts, 8(10), 418. https://doi.org/10.3390/catal8100418